Bicyclic Boronates as Potent Inhibitors of AmpC, the Class C β-Lactamase from Escherichia coli

 , , , , , and
, , , , , and
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Enzyme Production
2.3. Kinetics
2.4. Antimicrobial Susceptibility Testing
2.5. Crystallization Experiments, X-Ray Data Collection and Processing
3. Results and Discussion
3.1. Kinetic Studies of AmpCEC Inhibition by Bicyclic Boronates
3.2. Microbiology Experiments Confirm Potential of (Bi)cyclic Boronates to Inhibit AmpCEC in Cells
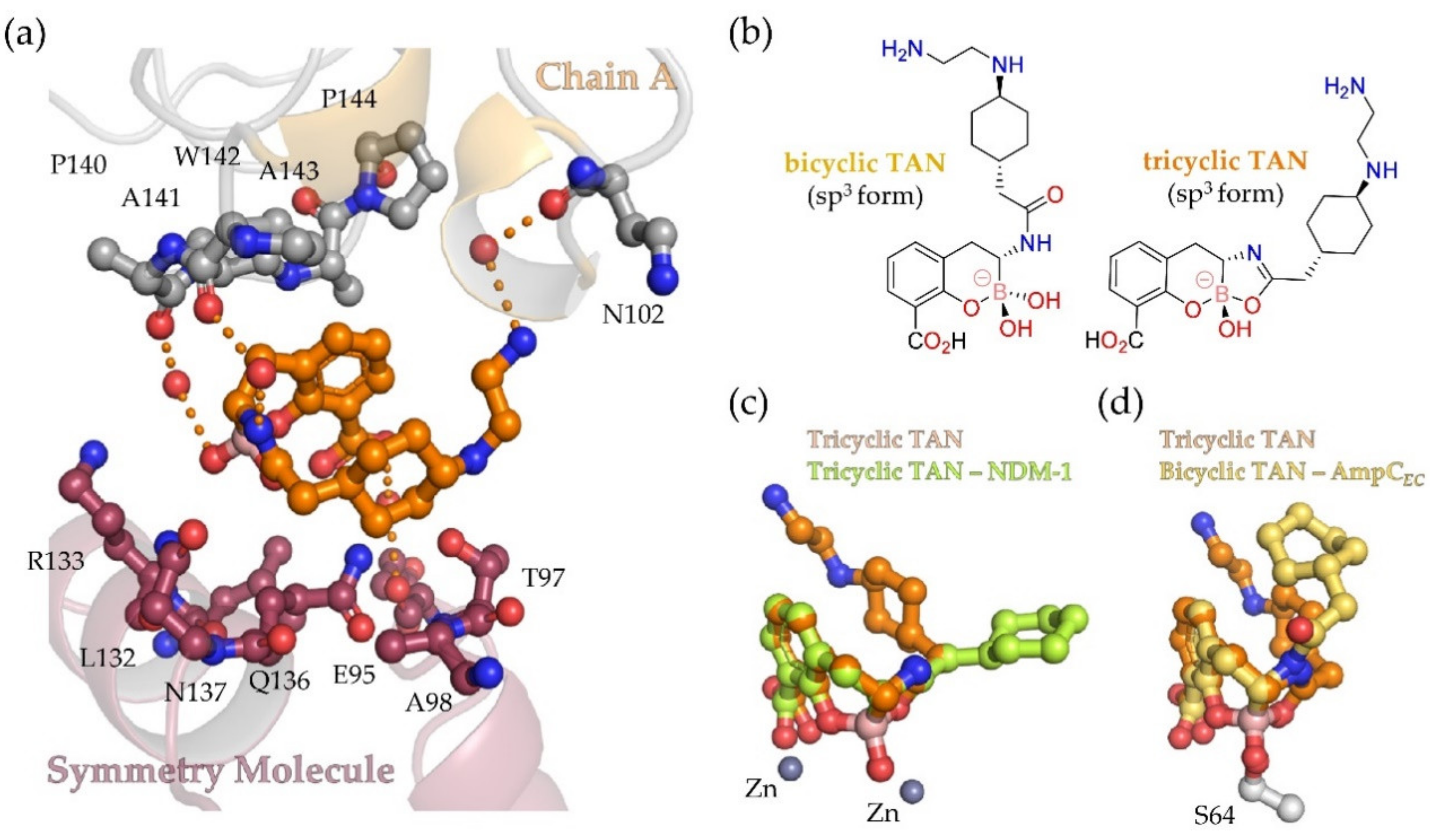
3.3. AmpCEC Crystal Structures with Bicyclic Boronates Give New Insights into Structural Basis of Inhibition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Versporten, A.; Bolokhovets, G.; Ghazaryan, L.; Abilova, V.; Pyshnik, G.; Spasojevic, T.; Korinteli, I.; Raka, L.; Kambaralieva, B.; Cizmovic, L.; et al. Antibiotic use in eastern Europe: A cross-national database study in coordination with the WHO Regional Office for Europe. Lancet Infect. Dis. 2014, 14, 381–387. [Google Scholar] [CrossRef]
- Bush, K. Past and Present Perspectives on β-Lactamases. Antimicrob. Agents Chemother. 2018, 62, e01076-18. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Bonomo, R.A. Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A. Updated Functional Classification of β-Lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Leigh, D.A.; Bradnock, K.; Marriner, J.M. Augmentin (amoxycillin and clavulanic acid) therapy in complicated infections due to β-lactamase producing bacteria. J. Antimicrob. Chemother. 1981, 7, 229–236. [Google Scholar] [CrossRef]
- Reading, C.; Cole, M. Clavulanic acid: A β-lactamase-inhiting β-lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 1977, 11, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Benson, J.M.; Nahata, M.C. Sulbactam/ampicillin, a new β-lactamase inhibitor/β-lactam antibiotic combination. Drug Intell. Clin. Pharm. 1988, 22, 534–541. [Google Scholar] [CrossRef]
- Gutmann, L.; Kitzis, M.D.; Yamabe, S.; Acar, J.F. Comparative evaluation of a new β-lactamase inhibitor, YTR 830, combined with different β-lactam antibiotics against bacteria harboring known β-lactamases. Antimicrob. Agents Chemother. 1986, 29, 955–957. [Google Scholar] [CrossRef]
- Blizzard, T.A.; Chen, H.; Kim, S.; Wu, J.; Bodner, R.; Gude, C.; Imbriglio, J.; Young, K.; Park, Y.-W.; Ogawa, A.; et al. Discovery of MK-7655, a β-lactamase inhibitor for combination with Primaxin®. Bioorg. Med. Chem. Lett. 2014, 24, 780–785. [Google Scholar] [CrossRef]
- Wang, D.Y.; Abboud, M.I.; Markoulides, M.S.; Brem, J.; Schofield, C.J. The road to avibactam: The first clinically useful non-β-lactam working somewhat like a β-lactam. Future Med. Chem. 2016, 8, 1063–1084. [Google Scholar] [CrossRef]
- Ehmann, D.E.; Jahic, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Durand-Réville, T.F.; Lahiri, S.; Thresher, J.; Livchak, S.; Gao, N.; et al. Kinetics of avibactam inhibition against Class A, C, and D β-lactamases. J. Biol. Chem. 2013, 288, 27960–27971. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.D.; Walkup, G.K.; Whiteaker, J.D.; Palmer, T.; McCormack, K.; Tanudra, M.A.; Nash, T.J.; Thresher, J.; Johnstone, M.R.; Hajec, L.; et al. Selection and molecular characterization of ceftazidime/avibactam-resistant mutants in Pseudomonas aeruginosa strains containing derepressed AmpC. J. Antimicrob. Chemother. 2015, 70, 1650–1658. [Google Scholar] [CrossRef] [PubMed]
- Shields, R.K.; Chen, L.; Cheng, S.; Chavda, K.D.; Press, E.G.; Snyder, A.; Pandey, R.; Doi, Y.; Kreiswirth, B.N.; Nguyen, M.H.; et al. Emergence of Ceftazidime-Avibactam Resistance Due to Plasmid-Borne blaKPC-3 Mutations during Treatment of Carbapenem-Resistant Klebsiella pneumoniae Infections. Antimicrob. Agents Chemother. 2017, 61, 1–11. [Google Scholar] [PubMed]
- Abboud, M.I.; Damblon, C.; Brem, J.; Smargiasso, N.; Mercuri, P.; Gilbert, B.; Rydzik, A.M.; Claridge, T.D.; Schofield, C.J.; Frere, J.M. Interaction of Avibactam with Class B Metallo-β-Lactamases. Antimicrob. Agents Chemother. 2016, 60, 5655–5662. [Google Scholar] [CrossRef]
- Pogue, J.M.; Bonomo, R.A.; Kaye, K.S. Ceftazidime/Avibactam, Meropenem/Vaborbactam, or Both? Clinical and Formulary Considerations. Clin. Infect. Dis. 2019, 68, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Parkova, A.; Lucic, A.; Krajnc, A.; Brem, J.; Calvopiña, K.; Langley, G.W.; McDonough, M.A.; Trapencieris, P.; Schofield, C.J. Broad Spectrum β-Lactamase Inhibition by a Thioether Substituted Bicyclic Boronate. ACS Infect. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Krajnc, A.; Lang, P.A.; Panduwawala, T.D.; Brem, J.; Schofield, C.J. Will morphing boron-based inhibitors beat the β-lactamases? Curr. Opin. Chem. Biol. 2019, 50, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M.; et al. Discovery of a Cyclic Boronic Acid β-Lactamase Inhibitor (RPX7009) with Utility vs Class A Serine Carbapenemases. J. Med. Chem. 2015, 58, 3682–3692. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, S.C.J.; Rybak, M.J. Meropenem and Vaborbactam: Stepping up the Battle against Carbapenem-resistant Enterobacteriaceae. Pharmacotherapy 2018, 38, 444–461. [Google Scholar] [CrossRef]
- Brem, J.; Cain, R.; Cahill, S.; McDonough, M.A.; Clifton, I.J.; Jimenez-Castellanos, J.C.; Avison, M.B.; Spencer, J.; Fishwick, C.W.; Schofield, C.J. Structural basis of metallo-β-lactamase, serine-β-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat. Commun. 2016, 7, 12406. [Google Scholar] [CrossRef]
- Cahill, S.T.; Cain, R.; Wang, D.Y.; Lohans, C.T.; Wareham, D.W.; Oswin, H.P.; Mohammed, J.; Spencer, J.; Fishwick, C.W.; McDonough, M.A.; et al. Cyclic Boronates Inhibit All Classes of β-Lactamases. Antimicrob. Agents Chemother. 2017, 61, 1–13. [Google Scholar] [CrossRef]
- Geibel, B.; Dowell, J.; Dickerson, D.; Henkel, T. 1401. A Randomized, Double-Blind, Placebo-Controlled Study of the Safety and Pharmacokinetics of Single and Repeat Doses of VNRX-5133 in Healthy Subjects. Open Forum Infect. Dis. 2018, 5 (Suppl. 1), S431. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, C.; Wang, Q.; Wang, Z.; Liang, X.; Zhang, F.; Zhang, Y.; Meng, H.; Chen, H.; Li, S.; et al. In vitro activity of the novel β-lactamase inhibitor taniborbactam (VNRX-5133), in combination with cefepime or meropenem, against MDR Gram-negative bacterial isolates from China. J. Antimicrob Chemother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Krajnc, A.; Brem, J.; Hinchliffe, P.; Calvopina, K.; Panduwawala, T.D.; Lang, P.A.; Kamps, J.; Tyrrell, J.M.; Widlake, E.; Saward, B.G.; et al. Bicyclic Boronate VNRX-5133 Inhibits Metallo- and Serine-β-Lactamases. J. Med. Chem. 2019, 62, 8544–8556. [Google Scholar] [CrossRef] [PubMed]
- Hamrick, J.C.; Docquier, J.D.; Uehara, T.; Myers, C.L.; Six, D.A.; Chatwin, C.L.; John, K.J.; Vernacchio, S.F.; Cusick, S.M.; Trout, R.E.L.; et al. VNRX-5133 (Taniborbactam), a Broad-Spectrum Inhibitor of Serine- and Metallo-β-Lactamases, Restores Activity of Cefepime in Enterobacterales and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2020, 64, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Trout, R.E.L.; Chu, G.-H.; McGarry, D.; Jackson, R.W.; Hamrick, J.C.; Daigle, D.M.; Cusick, S.M.; Pozzi, C.; De Luca, F.; et al. Discovery of Taniborbactam (VNRX-5133): A Broad-Spectrum Serine- and Metallo-β-lactamase Inhibitor for Carbapenem-Resistant Bacterial Infections. J. Med. Chem. 2020, 63, 2789–2801. [Google Scholar] [CrossRef]
- Jacoby, G.A. AmpC β-Lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182. [Google Scholar] [CrossRef]
- Jaurin, B.; Grundström, T.; Edlund, T.; Normark, S. The E. coli β-lactamase attenuator mediates growth rate-dependent regulation. Nature 1981, 290, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.C.; Elisha, B.G. Molecular Basis of AmpC Hyperproduction in Clinical Isolates of Escherichia coli. Antimicrob. Agents Chemother. 1999, 43, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Vila, J.; Sáez-López, E.; Johnson, J.R.; Römling, U.; Dobrindt, U.; Cantón, R.; Giske, C.G.; Naas, T.; Carattoli, A.; Martínez-Medina, M.; et al. Escherichia coli: An old friend with new tidings. FEMS Microbiol. Rev. 2016, 40, 437–463. [Google Scholar] [CrossRef] [PubMed]
- Tooke, C.L.; Hinchliffe, P.; Krajnc, A.; Mulholland, A.J.; Brem, J.; Schofield, C.J.; Spencer, J. Cyclic boronates as versatile scaffolds for KPC-2 β-lactamase inhibition. RCS Med. Chem. 2020, 11, 491–496. [Google Scholar] [CrossRef]
- Cahill, S.T.; Tyrrell, J.M.; Navratilova, I.H.; Calvopina, K.; Robinson, S.W.; Lohans, C.T.; McDonough, M.A.; Cain, R.; Fishwick, C.W.G.; Avison, M.B.; et al. Studies on the inhibition of AmpC and other β-lactamases by cyclic boronates. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Calvopiña, K.; Hinchliffe, P.; Brem, J.; Heesom, K.J.; Johnson, S.; Cain, R.; Lohans, C.T.; Fishwick, C.W.G.; Schofield, C.J.; Spencer, J.; et al. Structural/mechanistic insights into the efficacy of nonclassical β-lactamase inhibitors against extensively drug resistant Stenotrophomonas maltophilia clinical isolates. Mol. Microbiol. 2017, 106, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.J. Beta-lactamase inhibitors. PCT Patent WO 2010/130708 A1, 18 November 2010. [Google Scholar]
- Reddy, R.K. Boronic acid derivatives and therapeutic uses thereof. PCT Patent WO 2016003929 A1, 7 January 2016. [Google Scholar]
- Van Berkel, S.S.; Brem, J.; Rydzik, A.M.; Salimraj, R.; Cain, R.; Verma, A.; Owens, R.J.; Fishwick, C.W.G.; Spencer, J.; Schofield, C.J. Assay platform for clinically relevant metallo-β-lactamases. J. Med. Chem. 2013, 56, 6945–6953. [Google Scholar] [CrossRef] [PubMed]
- Page, M.G. The kinetics of non-stoichiometric bursts of β-lactam hydrolysis catalysed by class C β-lactamases. Biochem. J. 1993, 295 Pt 1, 295–304. [Google Scholar] [CrossRef]
- Dubus, A.; Wilkin, J.M.; Raquet, X.; Normark, S.; Frère, J.M. Catalytic mechanism of active-site serine β-lactamases: Role of the conserved hydroxy group of the Lys-Thr(Ser)-Gly triad. Biochem. J. 1994, 301 Pt 2, 485–494. [Google Scholar] [CrossRef]
- Morrison, J.F.; Walsh, C.T. The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Rel. Areas Mol. Biol. 1988, 61, 201–301. [Google Scholar]
- Williams, J.W.; Morrison, J.F. The kinetics of reversible tight-binding inhibition. In Methods Enzymology; Academic Press: Cambridge, MA, USA, 1979; Volume 63, pp. 437–467. [Google Scholar]
- Copeland, R.A.; Basavapathruni, A.; Moyer, M.; Scott, M.P. Impact of enzyme concentration and residence time on apparent activity recovery in jump dilution analysis. Anal. Biochem. 2011, 416, 206–210. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40 Pt 4, 658–674. [Google Scholar] [CrossRef]
- Powers, R.A.; Caselli, E.; Focia, P.J.; Prati, F.; Shoichet, B.K. Structures of Ceftazidime and Its Transition-State Analogue in Complex with AmpC β-Lactamase: Implications for Resistance Mutations and Inhibitor Design. Biochemistry 2001, 40, 9207–9214. [Google Scholar] [CrossRef]
- Adams, P.D.; Grosse-Kunstleve, R.W.; Hung, L.W.; Ioerger, T.R.; McCoy, A.J.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; Sauter, N.K.; Terwilliger, T.C. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002, 58 Pt 11, 1948–1954. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 4, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.D.; Mangani, S.; Durand-Reville, T.; Benvenuti, M.; De Luca, F.; Sanyal, G.; Docquier, J.D. Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: Avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-lactamases. Antimicrob. Agents Chemother. 2013, 57, 2496–2505. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.D.; Johnstone, M.R.; Ross, P.L.; McLaughlin, R.E.; Olivier, N.B.; Alm, R.A. Avibactam and class C β-lactamases: Mechanism of inhibition, conservation of the binding pocket, and implications for resistance. Antimicrob. Agents Chemother. 2014, 58, 5704–5713. [Google Scholar] [CrossRef]
- Haenni, M.; Châtre, P.; Madec, J.-Y. Emergence of Escherichia coli producing extended-spectrum AmpC β-lactamases (ESAC) in animals. Front. Microbiol. 2014, 5, 53. [Google Scholar] [CrossRef]
- Inglis, S.R.; Woon, E.C.Y.; Thompson, A.L.; Schofield, C.J. Observations on the Deprotection of Pinanediol and Pinacol Boronate Esters via Fluorinated Intermediates. J. Org. Chem. 2010, 75, 468–471. [Google Scholar] [CrossRef]
- Hecker, S.J.; Reddy, K.R.; Lomovskaya, O.; Griffith, D.C.; Rubio-Aparicio, D.; Nelson, K.; Tsivkovski, R.; Sun, D.; Sabet, M.; Tarazi, Z.; et al. Discovery of Cyclic Boronic Acid QPX7728, an Ultrabroad-Spectrum Inhibitor of Serine and Metallo-β-lactamases. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Ebejer, J.-P.; Charlton, M.H.; Finn, P.W. Are the physicochemical properties of antibacterial compounds really different from other drugs? J. Cheminform. 2016, 8, 30. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Kiapp (μM) | (M−1 s−1) × 103 | koff (s−1) × 10−3 | t1/2 (min) | pIC50 |
|---|---|---|---|---|---|
| VAB | 19.1 ± 1.32 | 6.55 ± 0.46 | 0.69 ± 0.03 | 16.8 ± 0.7 | 6.32 ± 0.03 |
| TAN | 3.73 ± 0.50 | 85.6 ± 9.2 | 2.54 ± 0.51 | 4.55 ± 0.91 | 7.53 ± 0.02 |
| CB2 | 3.26 ± 0.40 | 81.0 ± 6.4 | 2.77 ± 0.27 | 4.18 ± 0.41 | 7.49 ± 0.01 |
| CB3 | 1.16 ± 0.14 | 224 ± 21 | 20.9 ± 12.8 | 0.55 ± 0.34 | 7.53 ± 0.02 |
| Minimum Inhibitory Concentration Ceftazidime (µg mL−1) | ||||||
|---|---|---|---|---|---|---|
| Strain | Plasmid | CAZ | CAZ (+ VAB) | CAZ (+ TAN) | CAZ (+ CB2) | CAZ (+ CB3) |
| DH5α | - | 1 | - | - | - | - |
| DH5α | pAD7 AmpCEC | 256 | 1 | 1 | 0.5 | 4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lang, P.A.; Parkova, A.; Leissing, T.M.; Calvopiña, K.; Cain, R.; Krajnc, A.; Panduwawala, T.D.; Philippe, J.; Fishwick, C.W.G.; Trapencieris, P.; et al. Bicyclic Boronates as Potent Inhibitors of AmpC, the Class C β-Lactamase from Escherichia coli. Biomolecules 2020, 10, 899. https://doi.org/10.3390/biom10060899
Lang PA, Parkova A, Leissing TM, Calvopiña K, Cain R, Krajnc A, Panduwawala TD, Philippe J, Fishwick CWG, Trapencieris P, et al. Bicyclic Boronates as Potent Inhibitors of AmpC, the Class C β-Lactamase from Escherichia coli. Biomolecules. 2020; 10(6):899. https://doi.org/10.3390/biom10060899
Chicago/Turabian StyleLang, Pauline A., Anete Parkova, Thomas M. Leissing, Karina Calvopiña, Ricky Cain, Alen Krajnc, Tharindi D. Panduwawala, Jules Philippe, Colin W. G. Fishwick, Peteris Trapencieris, and et al. 2020. "Bicyclic Boronates as Potent Inhibitors of AmpC, the Class C β-Lactamase from Escherichia coli" Biomolecules 10, no. 6: 899. https://doi.org/10.3390/biom10060899
APA StyleLang, P. A., Parkova, A., Leissing, T. M., Calvopiña, K., Cain, R., Krajnc, A., Panduwawala, T. D., Philippe, J., Fishwick, C. W. G., Trapencieris, P., Page, M. G. P., Schofield, C. J., & Brem, J. (2020). Bicyclic Boronates as Potent Inhibitors of AmpC, the Class C β-Lactamase from Escherichia coli. Biomolecules, 10(6), 899. https://doi.org/10.3390/biom10060899