Identification and Validation of VEGFR2 Kinase as a Target of Voacangine by a Systematic Combination of DARTS and MSI

 ,
,  ,
, 
Abstract
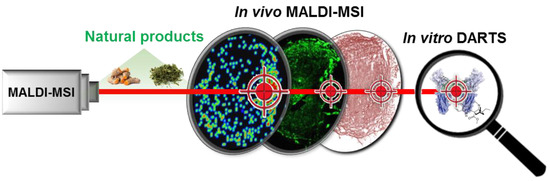
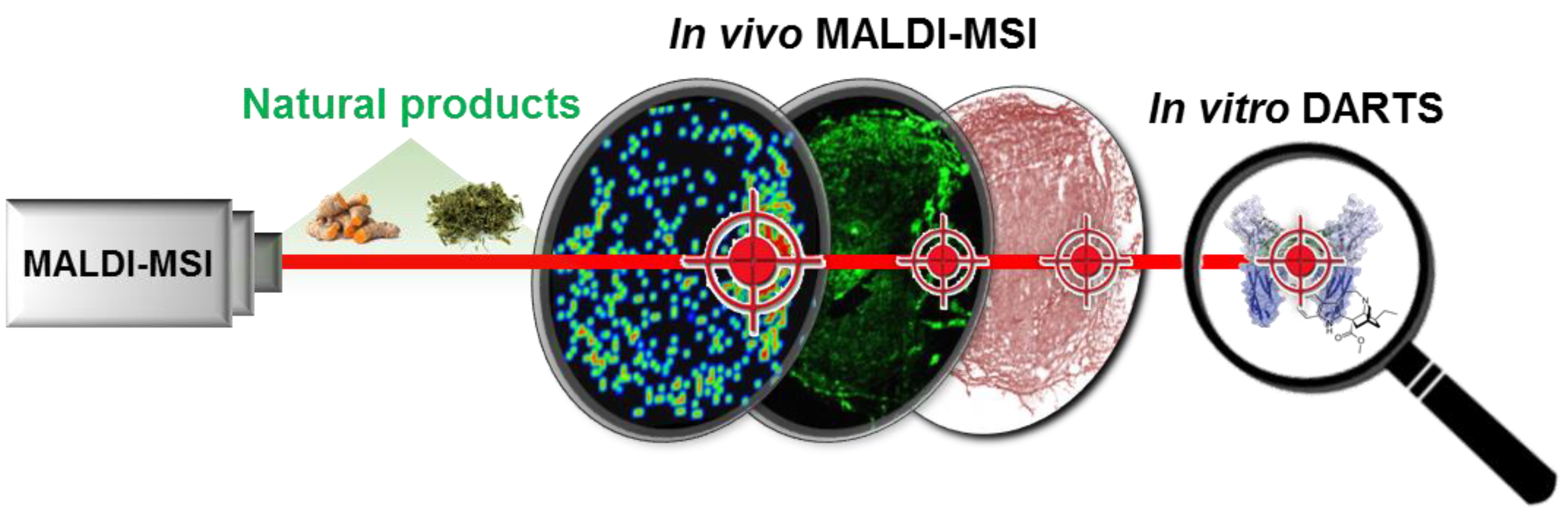
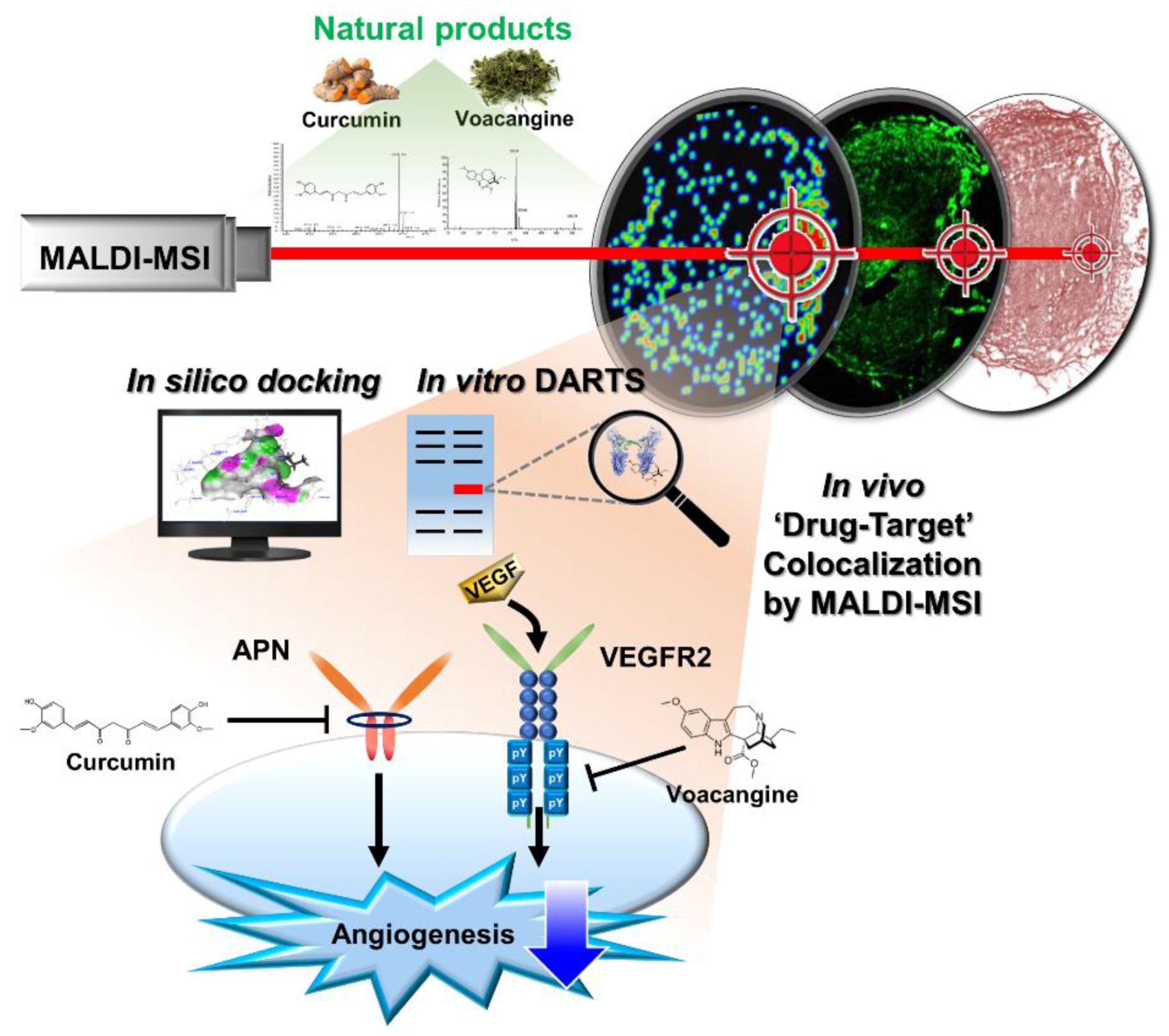
1. Introduction
2. Materials and Methods
2.1. Materials and General Methods
2.2. In Vivo Mouse Tumor Xenograft Assays
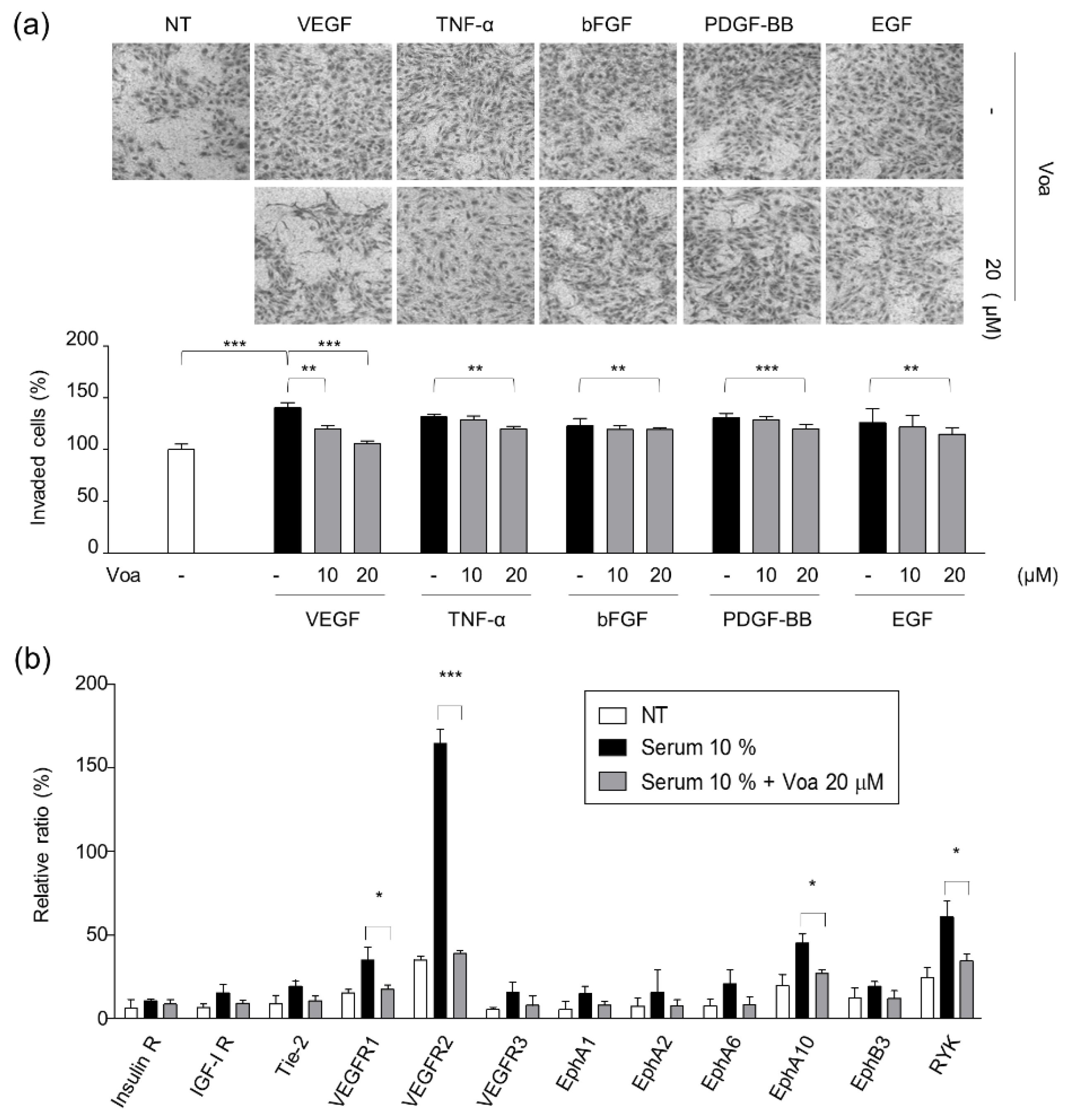
2.3. Growth Factor-Induced Chemoinvasion Assays
2.4. Drug Affinity Responsive Target Stability (DARTS) Assay
2.5. In Silico Docking Simulation
2.6. Immunoblotting Analysis
2.7. Cell Growth Condition and Cell Proliferation Assays
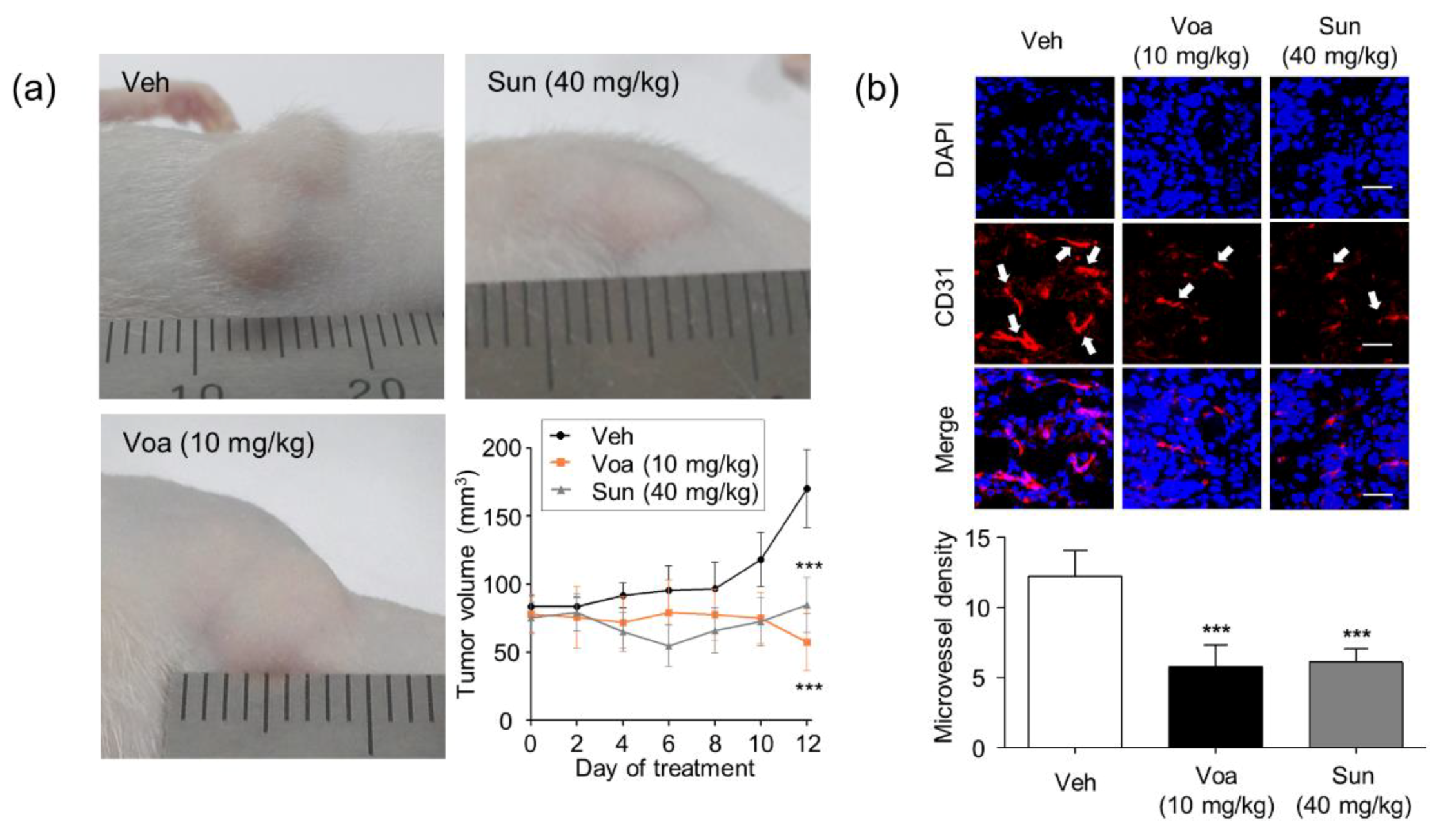
2.8. Quantification of Microvessel Density
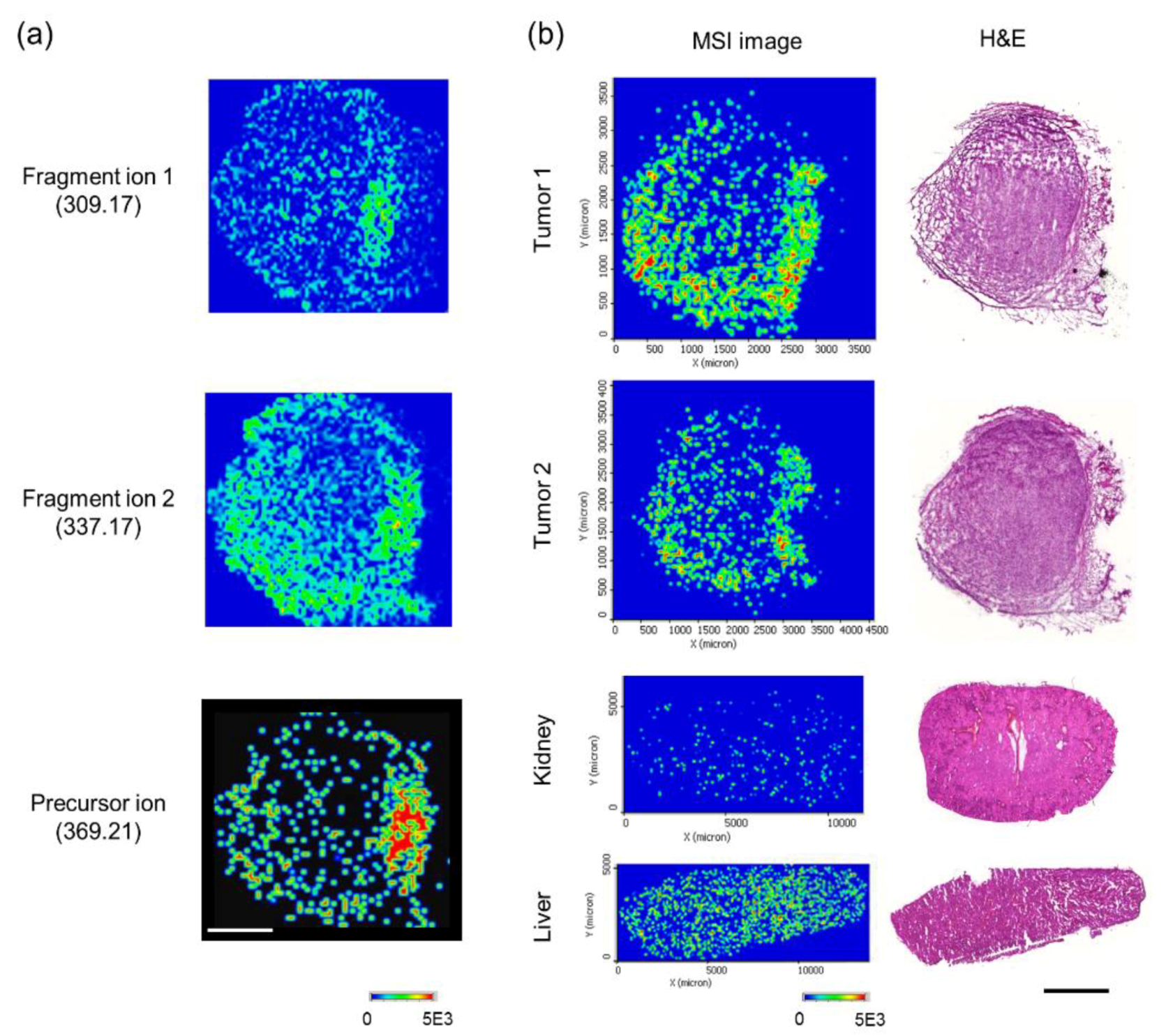
2.9. Compound Detection and Analysis of Drug Distribution Using MSI
2.10. Quantitation of the Precursor Compound
2.11. Histochemical Analysis of Protein Target in Tissues and Compound Colocalization
2.12. Statistical Analysis
3. Results and Discussions
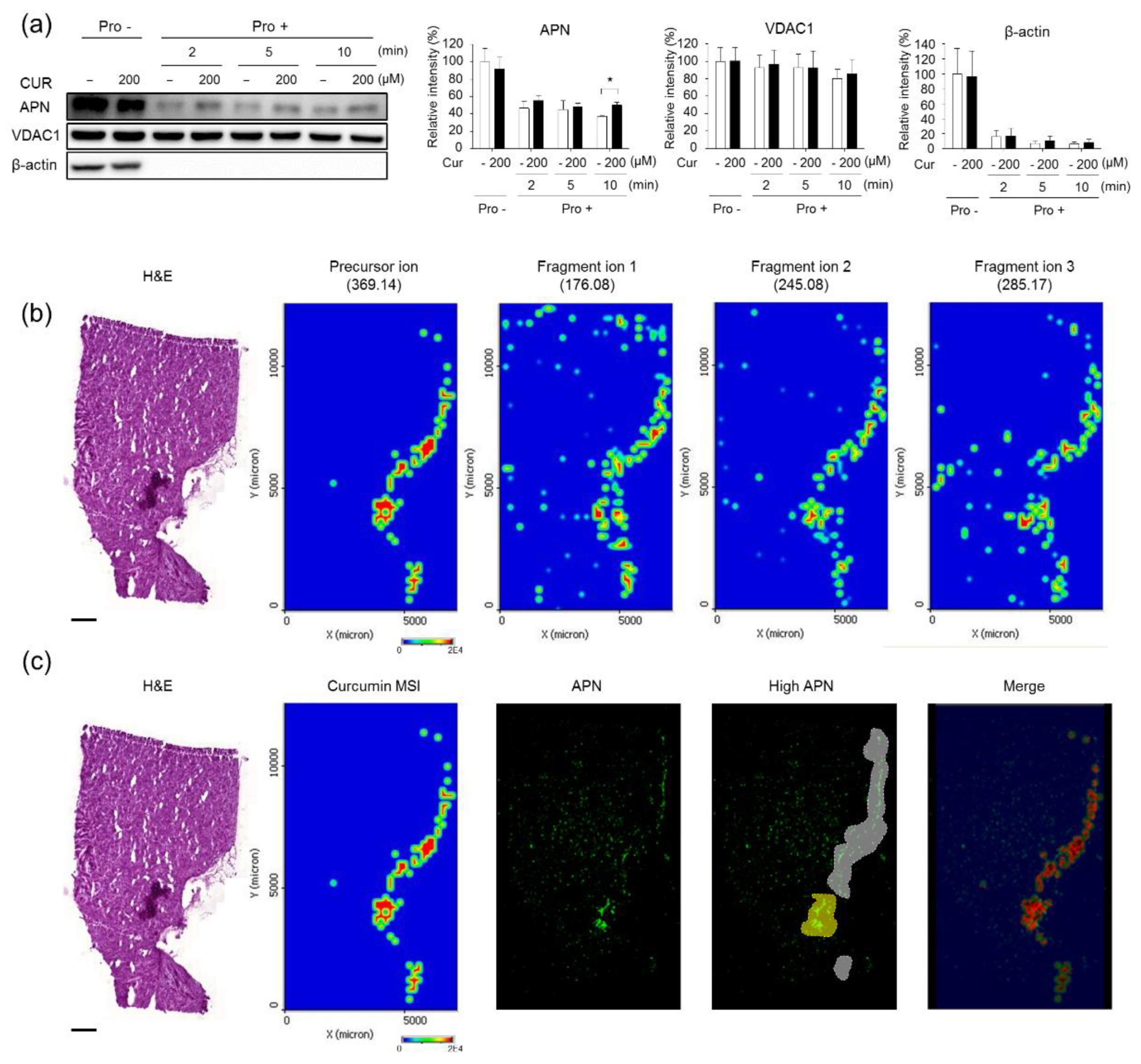
3.1. Validation of the Systematic Combination of DARTS and MSI for Natural Product-Target Protein Interaction
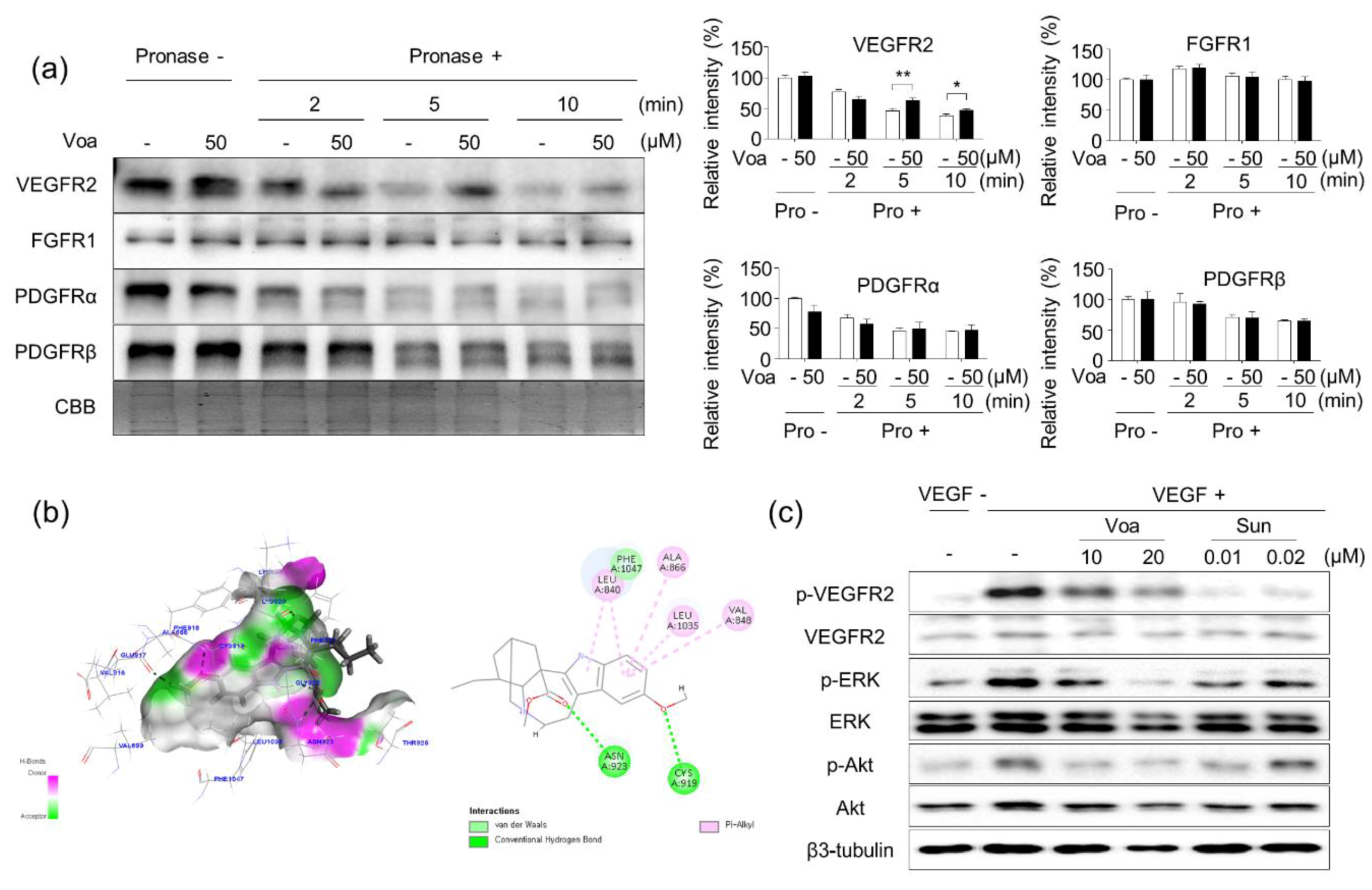
3.2. Identification of VEGFR2 as a Potent Target for Voacangine
3.3. Validation of In Vitro Direct Interaction between Voacangine and VEGFR2 by DARTS
3.4. Voacangine Inhibits Xenograft Tumor Growth and Angiogenesis In Vivo
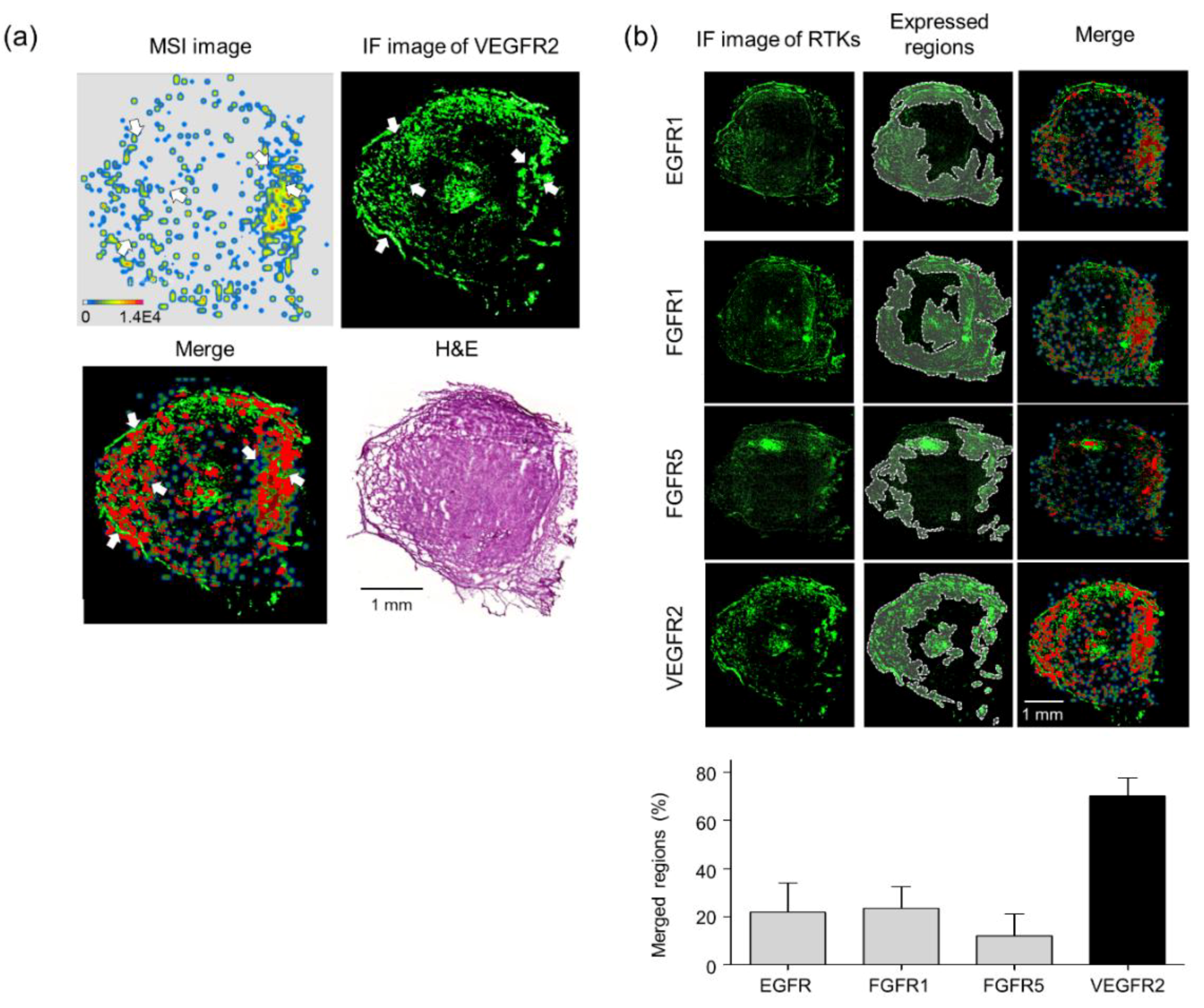
3.5. Validation of Target Interaction by Colocalization of Voacangine with VEGFR2 and Other RTKs
3.6. Discussions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Schenone, M.; Dančík, V.; Wagner, B.K.; Clemons, P.A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013, 9, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.B. Mechanism-based target identification and drug discovery in cancer research. Science 2000, 287, 1969–1973. [Google Scholar] [CrossRef]
- Drews, J. Drug discovery: A historical perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Zhang, H.; Wang, C.; Yao, S.C.L.; Yao, S.Q. Target identification of natural products and bioactive compounds using affinity-based probes. Nat. Prod. Rep. 2016, 33, 612–620. [Google Scholar] [CrossRef]
- Ziegler, S.; Pries, V.; Hedberg, C.; Waldmann, H. Target identification for small bioactive molecules: Finding the needle in the haystack. Angew. Chemie-Int. Ed. 2013, 52, 2744–2792. [Google Scholar] [CrossRef] [PubMed]
- Rix, U.; Superti-Furga, G. Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol. 2009, 5, 616–624. [Google Scholar] [CrossRef]
- Chang, J.; Kim, Y.; Kwon, H.J. Advances in identification and validation of protein targets of natural products without chemical modification. Nat. Prod. Rep. 2016, 33, 719–730. [Google Scholar] [CrossRef]
- Annis, D.A.; Nickbarg, E.; Yang, X.; Ziebell, M.R.; Whitehurst, C.E. Affinity selection-mass spectrometry screening techniques for small molecule drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 518–526. [Google Scholar] [CrossRef]
- Schirle, M.; Bantscheff, M.; Kuster, B. Mass spectrometry-based proteomics in preclinical drug discovery. Chem. Biol. 2012, 19, 72–84. [Google Scholar] [CrossRef]
- Molina, D.M.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Jung, G.; Wohlschlegel, J.A.; Huang, J. Target Identification using drug affinity responsive target stability (DARTS). Curr. Protoc. Chem. Biol. 2011, 3, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Hao, R.; Jonai, N.; Chin, R.M.; Aghajan, M.; Warburton, S.; Wang, J.; Wu, R.P.; Gomez, F.; Loo, J.A.; et al. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. USA 2009, 106, 21984–21989. [Google Scholar] [CrossRef]
- West, G.M.; Tang, L.; Fitzgerald, M.C. Thermodynamic analysis of protein stability and ligand binding using a chemical modification- and mass spectrometry-based strategy. Anal. Chem. 2008, 80, 4175–4185. [Google Scholar] [CrossRef] [PubMed]
- Pai, M.Y.; Lomenick, B.; Hwang, H.; Schiestl, R.; McBride, W.; Loo, J.A.; Huang, J. Drug affinity responsive target stability (DARTS) for small-molecule target identification. Methods Mol. Biol. 2015, 1263, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Groseclose, M.R.; Deutch, A.Y.; Caprioli, R.M. Imaging mass spectrometry of proteins and peptides: 3D volume reconstruction. Nat. Methods 2008, 5, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Stoeckli, M.; Chaurand, P.; Hallahan, D.E.; Caprioli, R.M. Imaging mass spectrometry: A new technology for the analysis of protein expression in mammalian tissues. Nat. Med. 2001, 7, 493–496. [Google Scholar] [CrossRef]
- Végvári, Á. Drug localizations in tissue by mass spectrometry imaging. Biomark. Med. 2015, 9, 869–876. [Google Scholar] [CrossRef]
- Marko-Varga, G.; Fehniger, T.E.; Rezeli, M.; Döme, B.; Laurell, T.; Végvári, Á. Drug localization in different lung cancer phenotypes by MALDI mass spectrometry imaging. J. Proteom. 2011, 74, 982–992. [Google Scholar] [CrossRef]
- Seeley, E.H.; Caprioli, R.M. MALDI imaging mass spectrometry of human tissue: Method challenges and clinical perspectives. Trends Biotechnol. 2011, 29, 136–143. [Google Scholar] [CrossRef]
- Cornett, D.S.; Reyzer, M.L.; Chaurand, P.; Caprioli, R.M. MALDI imaging mass spectrometry: Molecular snapshots of biochemical systems. Nat. Methods 2007, 4, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Fujimura, Y.; Hagihara, T.; Sasaki, M.; Yukihira, D.; Nagao, T.; Miura, D.; Yamaguchi, S.; Saito, K.; Tanaka, H.; et al. In situ label-free imaging for visualizing the biotransformation of a bioactive polyphenol. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Schulz, S.; Becker, M.; Groseclose, M.R.; Schadt, S.; Hopf, C. Advanced MALDI mass spectrometry imaging in pharmaceutical research and drug development. Curr. Opin. Biotechnol. 2019, 55, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Kim, J.H.; Cho, H.Y.; Yum, Y.N.; Kim, S.H.; Park, H.J.; Shim, B.S.; Choi, S.H.; Kwon, H.J. Irreversible inhibition of CD13/aminopeptidase N by the antiangiogenic agent curcumin. Chem. Biol. 2003, 10, 695–704. [Google Scholar] [CrossRef]
- Kim, Y.; Jung, H.J.; Kwon, H.J. A natural small molecule voacangine inhibits angiogenesis both in vitro and in vivo. Biochem. Biophys. Res. Commun. 2012, 417, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Mendel, D.B.; Douglas Laird, A.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar]
- Sun, L.; Liang, C.; Shirazian, S.; Zhou, Y.; Miller, T.; Cui, J.; Fukuda, J.Y.; Chu, J.Y.; Nematalla, A.; Wang, X.; et al. Discovery of 5-[5-fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2, 4-dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosin. J. Med. Chem. 2003, 46, 1116–1119. [Google Scholar] [CrossRef]
- Patyna, S.; Laird, A.D.; Mendel, D.B.; O’Farrell, A.M.; Liang, C.; Guan, H.; Vojkovsky, T.; Vasile, S.; Wang, X.; Chen, J.; et al. SU14813: A novel multiple receptor tyrosine kinase inhibitor with potent antiangiogenic and antitumor activity. Mol. Cancer Ther. 2006, 5, 1774–1782. [Google Scholar] [CrossRef]
- Adelaiye, R.; Ciamporcero, E.; Miles, K.M.; Sotomayor, P.; Bard, J.; Tsompana, M.; Conroy, D.; Shen, L.; Ramakrishnan, S.; Ku, S.Y.; et al. Sunitinib dose escalation overcomes transient resistance in clear cell renal cell carcinoma and is associated with epigenetic modifications. Mol. Cancer Ther. 2015, 14, 513–522. [Google Scholar] [CrossRef]
- Döme, B.; Paku, S.; Somlai, B.; Tmr, J. Vascularization of cutaneous melanoma involves vessel co-option and has clinical significance. J. Pathol. 2002, 197, 355–362. [Google Scholar] [CrossRef]
- Torok, S.; Vegvari, A.; Rezeli, M.; Fehniger, T.E.; Tovari, J.; Paku, S.; Laszlo, V.; Hegedus, B.; Rozsas, A.; Dome, B.; et al. Localization of sunitinib, its metabolites and its target receptors in tumour-bearing mice: A MALDI-MS imaging study. Br. J. Pharmacol. 2015, 172, 1148–1163. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling-In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis-dependent diseases. Semin. Oncol. 2001, 28, 536–542. [Google Scholar] [CrossRef]
- Flier, J.S.; Underhill, L.H.; Folkman, J. Clinical applications of research on angiogenesis. N. Engl. J. Med. 1995, 333, 1757–1763. [Google Scholar] [CrossRef]
- Ivy, S.P.; Wick, J.Y.; Kaufman, B.M. An overview of small-molecule inhibitors of VEGFR signaling. Nat. Rev. Clin. Oncol. 2009, 6, 569–579. [Google Scholar] [CrossRef]
- Sarkar, S.; Mazumdar, A.; Dash, R.; Sarkar, D.; Fisher, P.B.; Mandal, M. ZD6474, a dual tyrosine kinase inhibitor of EGFR and VEGFR-2, inhibits MAPK/ERK and AKT/PI3-K and induces apoptosis in breast cancer cells. Cancer Biol. Ther. 2010, 9, 592–603. [Google Scholar] [CrossRef]
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef]
- Emmenegger, U.; Shaked, Y.; Man, S.; Bocci, G.; Spasojevic, I.; Francia, G.; Kouri, A.; Coke, R.; Cruz-Munoz, W.; Ludeman, S.M.; et al. Pharmacodynamic and pharmacokinetic study of chronic low-dose metronomic cyclophosphamide therapy in mice. Mol. Cancer Ther. 2007, 6, 2280–2289. [Google Scholar] [CrossRef]
- Zhang, L.; Smith, K.M.; Chong, A.L.; Stempak, D.; Yeger, H.; Marrano, P.; Thorner, P.S.; Irwin, M.S.; Kaplan, D.R.; Baruchel, S. In vivo antitumor and antimetastatic activity of Sunitinib in preclinical Neuroblastoma mouse model. Neoplasia 2009, 11, 426–435. [Google Scholar] [CrossRef]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Cho, S.G.; Yi, Z.; Pang, X.; Rodriguez, M.; Wang, Y.; Sethi, G.; Aggarwal, B.B.; Liu, M. Thymoquinone inhibits tumor angiogenesis and tumor growth through suppressing AKT and extracellular signal-regulated kinase signaling pathways. Mol. Cancer Ther. 2008, 7, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Fischer, I.; Gagner, J.-P.; Law, M.; Newcomb, E.W.; Zagzag, D. Angiogenesis in Gliomas: Biology and Molecular Pathophysiology. Brain Pathol. 2006, 15, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.S.; Zea, A.H.; Rini, B.I.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Rock, E.P.; Goodman, V.; Jiang, J.X.; Mahjoob, K.; Verbois, S.L.; Morse, D.; Dagher, R.; Justice, R.; Pazdur, R. Food and Drug Administration Drug Approval Summary: Sunitinib Malate for the Treatment of Gastrointestinal Stromal Tumor and Advanced Renal Cell Carcinoma. Oncologist 2007, 12, 107–113. [Google Scholar] [CrossRef]
- Choueiri, T.K. Clinical treatment decisions for advanced renal cell cancer. JNCCN J. Natl. Compr. Cancer Netw. 2013, 11, 694–697. [Google Scholar] [CrossRef]
- Porta, C.; Sabbatini, R.; Procopio, G.; Paglino, C.; Galligioni, E.; Ortega, C. Primary resistance to tyrosine kinase inhibitors in patients with advanced renal cell carcinoma: State-of-the-science. Expert Rev. Anticancer Ther. 2012, 12, 1571–1577. [Google Scholar] [CrossRef]
- Heng, D.Y.; MacKenzie, M.J.; Vaishampayan, U.N.; Bjarnason, G.A.; Knox, J.J.; Tan, M.H.; Wood, L.; Wang, Y.; Kollmannsberger, C.; North, S.; et al. Primary anti-vascular endothelial growth factor (VEGF)-refractory metastatic renal cell carcinoma: Clinical characteristics, risk factors, and subsequent therapy. Ann. Oncol. 2012, 23, 1549–1555. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quantitation | Pixels | Merged Regions/Cur-MSI (%) |
|---|---|---|
| MALDI-MSI (curcumin) | 44,408 | 100 |
| Merged regions (high curcumin and high APN) | 32,264 | 72.65 |
| Quantitation | Pixels | IF/Voa-MSI (%) | |
|---|---|---|---|
| MALDI-MSI (voacangine) | 71,406 | 100 | |
| Merged regions (red) | EGFR1 | 15,572 | 21 |
| FGFR1 | 16,745 | 23 | |
| FGFR5 | 8616 | 12 | |
| VEGFR2 | 50,039 | 70 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Sugihara, Y.; Kim, T.Y.; Cho, S.M.; Kim, J.Y.; Lee, J.Y.; Yoo, J.S.; Song, D.; Han, G.; Rezeli, M.; et al. Identification and Validation of VEGFR2 Kinase as a Target of Voacangine by a Systematic Combination of DARTS and MSI. Biomolecules 2020, 10, 508. https://doi.org/10.3390/biom10040508
Kim Y, Sugihara Y, Kim TY, Cho SM, Kim JY, Lee JY, Yoo JS, Song D, Han G, Rezeli M, et al. Identification and Validation of VEGFR2 Kinase as a Target of Voacangine by a Systematic Combination of DARTS and MSI. Biomolecules. 2020; 10(4):508. https://doi.org/10.3390/biom10040508
Chicago/Turabian StyleKim, Yonghyo, Yutaka Sugihara, Tae Young Kim, Sung Min Cho, Jin Young Kim, Ju Yeon Lee, Jong Shin Yoo, Doona Song, Gyoonhee Han, Melinda Rezeli, and et al. 2020. "Identification and Validation of VEGFR2 Kinase as a Target of Voacangine by a Systematic Combination of DARTS and MSI" Biomolecules 10, no. 4: 508. https://doi.org/10.3390/biom10040508
APA StyleKim, Y., Sugihara, Y., Kim, T. Y., Cho, S. M., Kim, J. Y., Lee, J. Y., Yoo, J. S., Song, D., Han, G., Rezeli, M., Welinder, C., Appelqvist, R., Marko-Varga, G., & Kwon, H. J. (2020). Identification and Validation of VEGFR2 Kinase as a Target of Voacangine by a Systematic Combination of DARTS and MSI. Biomolecules, 10(4), 508. https://doi.org/10.3390/biom10040508