The Influence of Varying Fluorination Patterns on the Thermodynamics and Kinetics of Benzenesulfonamide Binding to Human Carbonic Anhydrase II

Abstract
1. Introduction
2. Materials and Methods
2.1. Calculation of Interface Areas
2.2. Protein Expression and Purification
2.3. Isothermal Titration Calorimetry
2.4. Isothermal Titration Calorimetry (ITC) Data Analysis
2.5. Macromolecular Crystallography
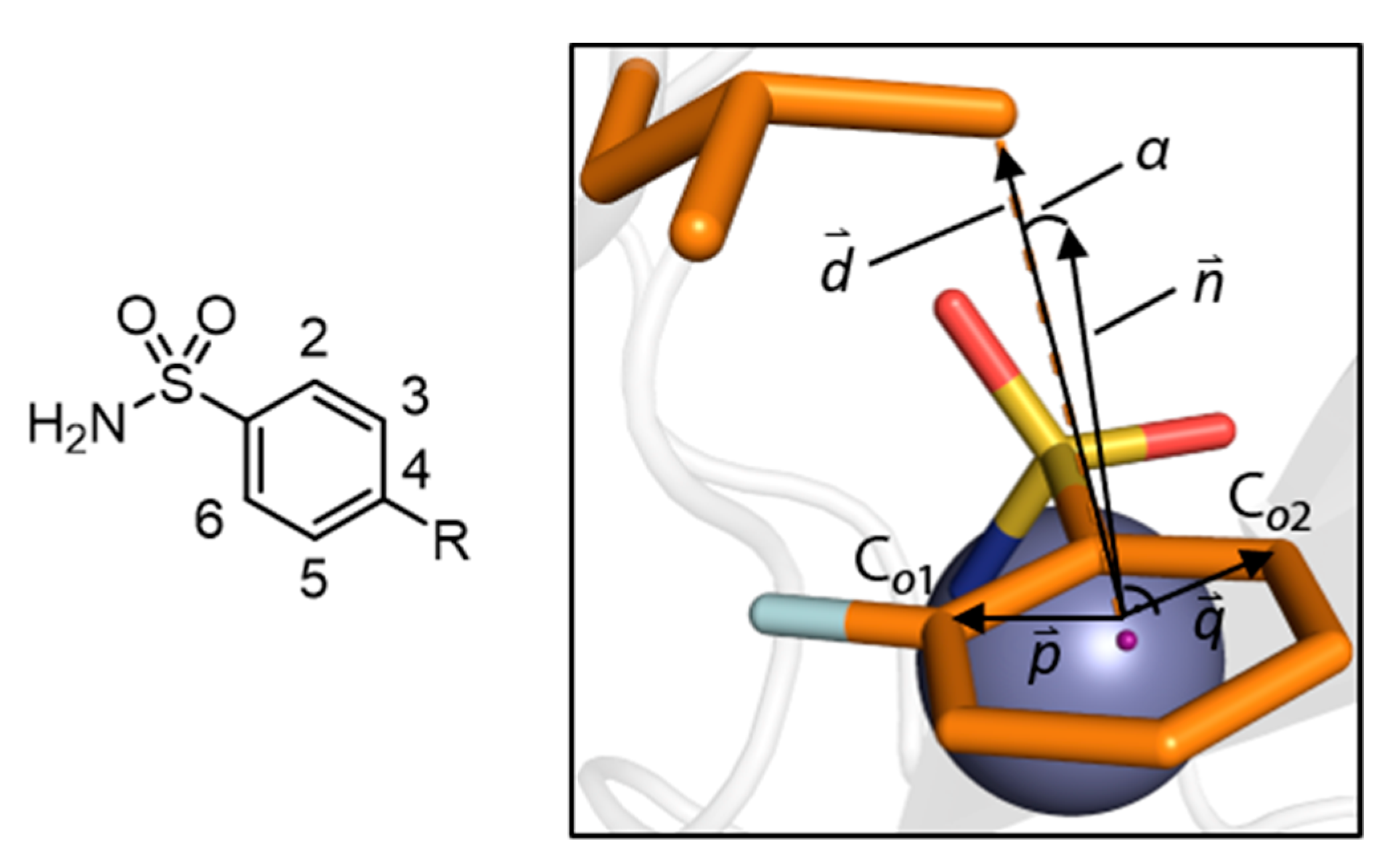
2.6. Calculation of the Deviation Angle α
2.7. pKa Measurements
2.8. Associated Content
PDB Accession Codes
3. Results
3.1. Crystallographic Data
3.2. Movement of Thr200
3.3. Accommodation of a Fluorine Atom in a Hydrophobic Pocket
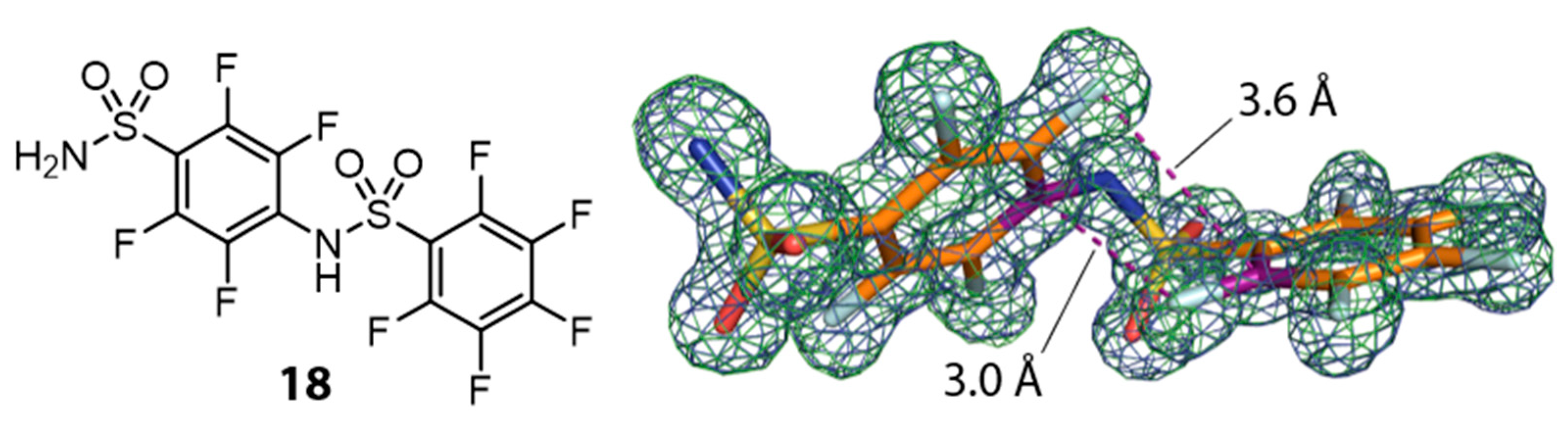
3.4. An Unexpected Dimerization Product
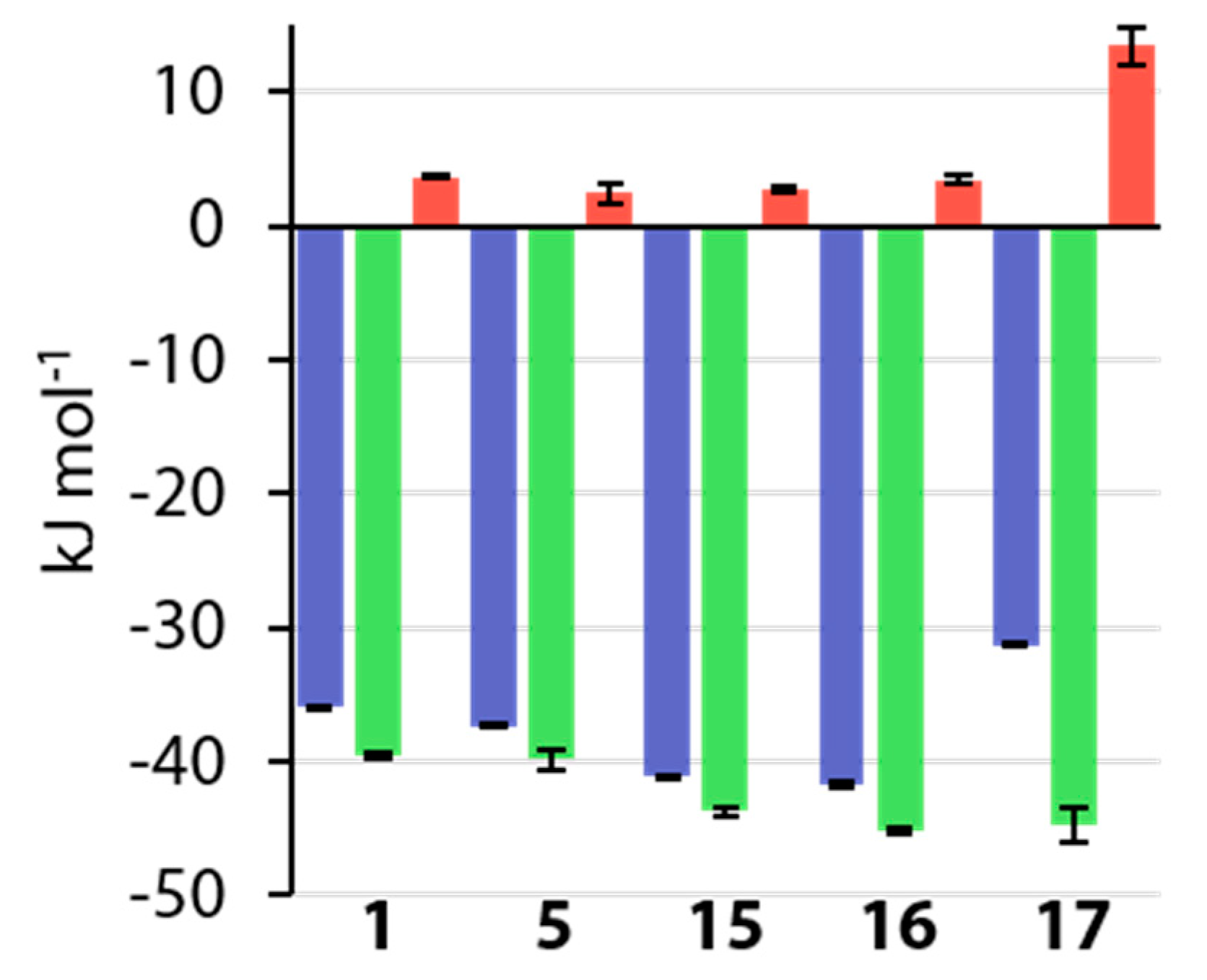
3.5. Thermodynamic Results
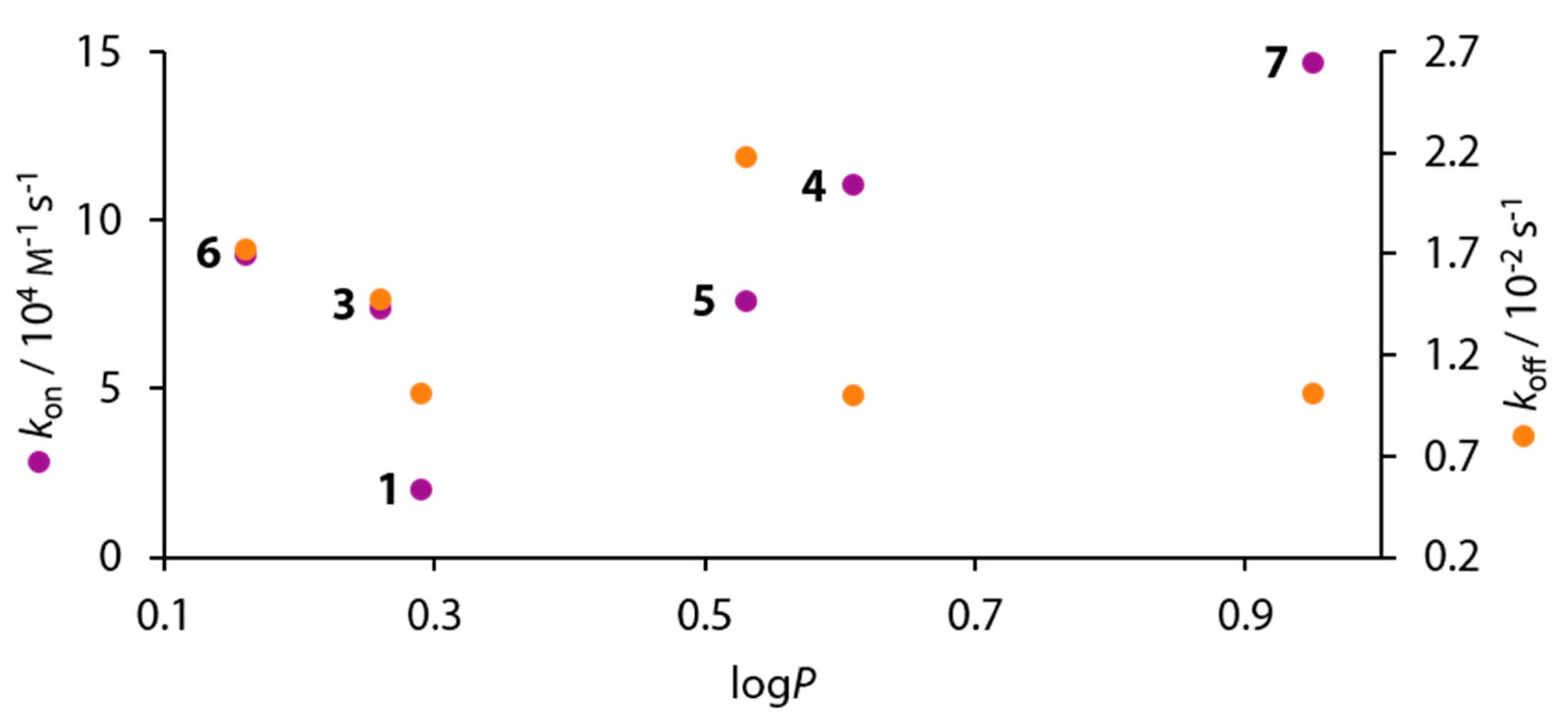
3.6. Kinetic Results
4. Discussion
4.1. Comparison of Thermodynamic Data with Earlier Studies
4.2. Comparison of Crystallographic with Thermodynamic Data
4.3. Comparison of Crystallographic with Kinetic Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Riess, J.G. Understanding the Fundamentals of Perfluorocarbons and Perfluorocarbon Emulsions Relevant to in Vivo Oxygen Delivery. Artif. Cells, Blood Substitutes. Immobil. Biotechnol. 2005, 33, 47–63. [Google Scholar] [CrossRef]
- Champagne, P.A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Monofluorination of Organic Compounds: 10 Years of Innovation. Chem. Rev. 2015, 115, 9073–9174. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Yudin, A.K. Macrocycles: Lessons from the distant past, recent developments, and future directions. Chem. Sci. 2015, 6, 30–49. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Kitteringham, N.R.; O’Neill, P.M. Metabolism of Fluorine-Containing Drugs. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–470. [Google Scholar] [CrossRef] [PubMed]
- Skora, L.; Jahnke, W. 19F-NMR-Based Dual-Site Reporter Assay for the Discovery and Distinction of Catalytic and Allosteric Kinase Inhibitors. ACS Med. Chem. Lett. 2017, 8, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Fried, J.; Sabo, E.F. 9α-Fluoro Derivatives of Cortisone and Hydrocortisone. J. Am. Chem. Soc. 1954, 76, 1455–1456. [Google Scholar] [CrossRef]
- Street, I.P.; Armstrong, C.R.; Withers, S.G. Hydrogen Bonding and Specificity: Fluorodeoxy Sugars as Probes of Hydrogen Bonding in the Glycogen Phosphorylase-Glucose Complex. Biochemistry 1986, 25, 6021–6027. [Google Scholar] [CrossRef]
- Holmgren, S.K.; Taylor, K.M.; Bretscher, L.E.; Raines, R.T. Code for collagen’s stability deciphered. Nature 1998, 392, 666–667. [Google Scholar] [CrossRef]
- Biffinger, J.C.; Kim, H.W.; Dimagno, S.G. The Polar Hydrophobicity of Fluorinated Compounds. ChemBioChem 2004, 5, 622–627. [Google Scholar] [CrossRef]
- Qiu, J.; Stevenson, S.H.; O’Beirne, M.J.; Silverman, R.B. 2,6-Difluorophenol as a Bioisostere of a Carboxylic Acid: Bioisosteric Analogues of γ-Aminobutyric Acid. J. Med. Chem. 1999, 42, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-Y.; Chang, J.S.; Doyon, J.B.; Baird, T.T., Jr.; Fierke, C.A.; Jain, A.; Christianson, D.W. Contribution of Fluorine to Protein–Ligand Affinity in the Binding of Fluoroaromatic Inhibitors to Carbonic Anhydrase II. J. Am. Chem. Soc. 2000, 122, 12125–12134. [Google Scholar] [CrossRef]
- Giroud, M.; Harder, M.; Kuhn, B.; Haap, W.; Trapp, N.; Schweizer, W.B.; Schirmeister, T.; Diederich, F. Fluorine Scan of Inhibitors of the Cysteine Protease Human Cathepsin L: Dipolar and Quadrupolar Effects in the π-Stacking of Fluorinated Phenyl Rings on Peptide Amide Bonds. ChemMedChem 2016, 11, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Schönemann, W.; Cramer, J.; Mühlethaler, T.; Fiege, B.; Silbermann, M.; Rabbani, S.; Dätwyler, P.; Zihlmann, P.; Jakob, R.P.; Sager, C.P.; et al. Improvement of Aglycone π-Stacking Yields Nanomolar to Sub-nanomolar FimH Antagonists. ChemMedChem 2019, 14, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.D.; Phillips, C.; Alex, A.; Flocco, M.; Bent, A.; Randall, A.; O’Brien, R.; Damian, L.; Jones, L.H. Thermodynamic Optimisation in Drug Discovery: A Case Study using Carbonic Anhydrase Inhibitors. ChemMedChem 2009, 4, 1985–1989. [Google Scholar] [CrossRef]
- Dudutienė, V.; Zubrienė, A.; Smirnov, A.; Gylytė, J.; Timm, D.; Manakova, E.; Gražulis, S.; Matulis, D. 4-Substituted-2,3,5,6-tetrafluorobenzenesulfonamides as inhibitors of carbonic anhydrases I, II, VII, XII, and XIII. Bioorg. Med. Chem. 2013, 21, 2093–2106. [Google Scholar] [CrossRef]
- Dudutienė, V.; Zubrienė, A.; Smirnov, A.; Timm, D.D.; Smirnovienė, J.; Kazokaitė, J.; Michailovienė, V.; Zakšauskas, A.; Manakova, E.; Gražulis, S.; et al. Functionalization of Fluorinated Benzenesulfonamides and Their Inhibitory Properties toward Carbonic Anhydrases. ChemMedChem 2015, 10, 662–687. [Google Scholar] [CrossRef]
- Krishnamurthy, V.M.; Bohall, B.R.; Kim, C.-Y.; Moustakas, D.T.; Christianson, D.W.; Whitesides, G.M. Thermodynamic Parameters for the Association of Fluorinated Benzenesulfonamides with Bovine Carbonic Anhydrase II. Chem. Asian J. 2007, 2, 94–105. [Google Scholar] [CrossRef]
- Olsen, J.A.; Banner, D.W.; Seiler, P.; Sander, U.O.; D’Arcy, A.; Stihle, M.; Müller, K.; Diederich, F. A Fluorine Scan of Thrombin Inhibitors to Map the Fluorophilicity/Fluorophobicity of an Enzyme Active Site: Evidence for C–F···C=O Interactions. Angew. Chem. Int. Ed. 2003, 42, 2507–2511. [Google Scholar] [CrossRef]
- Glöckner, S.; Ngo, K.; Sager, C.P.; Hüfner-Wulsdorf, T.; Heine, A.; Klebe, G. Conformational Changes in Alkyl Chains Determine the Thermodynamic and Kinetic Binding Profiles of Carbonic Anhydrase Inhibitors. ACS Chem. Biol. 2020. [Google Scholar] [CrossRef]
- vg v.1.6.1. Available online: https://pypi.org/project/vg/ (accessed on 5 December 2019).
- Van Der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy array: A structure for efficient numerical computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef]
- Delano, W.L. The PyMOL molecular graphics system 2002. Available online: http://www.pymol.org (accessed on 5 December 2019).
- Ribeiro, J.; Ríos-Vera, C.; Melo, F.; Schüller, A. Calculation of accurate interatomic contact surface areas for the quantitative analysis of non-bonded molecular interactions. Bioinformatics 2019, 35, 3499–3501. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, R.; Rechlin, C.; Heine, A.; Bottegoni, G.; Rocchia, W.; Schwarz, D.; Bomke, J.; Gerber, H.-D.; Klebe, G.; Cavalli, A. Kinetic and Structural Insights into the Mechanism of Binding of Sulfonamides to Human Carbonic Anhydrase by Computational and Experimental Studies. J. Med. Chem. 2016, 59, 4245–4256. [Google Scholar] [CrossRef] [PubMed]
- Cimmperman, P.; Baranauskienė, L.; Jachimovičiūtė; Jachno, J.; Torresan, J.; Michailovienė, V.; Matulienė, J.; Sereikaitė, J.; Bumelis, V.; Matulis, D. A Quantitative Model of Thermal Stabilization and Destabilization of Protein by Ligands. Biophys. J. 2008, 95, 3222–3231. [Google Scholar] [CrossRef]
- Piñeiro, Á.; Muñoz, E.; Sabín, J.; Costas, M.; Bastos, M.; Velázquez-Campoy, A.; Garrido, P.F.; Dumas, P.; Ennifar, E.; García-Río, L.; et al. AFFINImeter: A software to analyze molecular recognition processes from experimental data. Anal. Biochem. 2019, 577, 117–134. [Google Scholar] [CrossRef]
- Krimmer, S.G.; Klebe, G. Thermodynamics of protein-ligand interactions as a reference for computational analysis: How to assess accuracy, reliability and relevance of experimental data. J. Comput. Aided. Mol. Des. 2015, 29, 867–883. [Google Scholar] [CrossRef]
- Mizoue, L.S.; Tellinghuisen, J. The role of backlash in the “first injection anomaly” in isothermal titration calorimetry. Anal. Biochem. 2004, 326, 125–127. [Google Scholar] [CrossRef]
- Velazquez-Campoy, A.; Freire, E. Isothermal titration calorimetry to determine association constants for high-affinity ligands. Nat. Protoc. 2006, 1, 186–191. [Google Scholar] [CrossRef]
- Burnouf, D.; Ennifar, E.; Guedich, S.; Puffer, B.; Hoffmann, G.; Bec, G.; Disdier, F.; Baltzinger, M.; Dumas, P. kinITC: A New Method for Obtaining Joint Thermodynamic and Kinetic Data by Isothermal Titration Calorimetry. J. Am. Chem. Soc. 2012, 134, 559–565. [Google Scholar] [CrossRef]
- Dumas, P.; Ennifar, E.; Da Veiga, C.; Bec, G.; Palau, W.; Di Primo, C.; Piñeiro, A.; Sabín, J.; Muñoz, E.; Rial, J. Extending ITC to Kinetics with kinITC. Methods in Enzymology, 2016; 567, 157–180. [Google Scholar]
- Muñoz, E.; Piñeiro, A. AFFINImeter Software: From its Beginnings to Future Trends - A Literature review. J. Appl. Bioanal. 2018, 4, 124–139. [Google Scholar] [CrossRef]
- Mueller, U.; Förster, R.; Hellmig, M.; Huschmann, F.U.; Kastner, A.; Malecki, P.; Pühringer, S.; Röwer, M.; Sparta, K.; Steffien, M.; et al. The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: Current status and perspectives. Eur. Phys. J. Plus 2015, 130, 141–150. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Sparta, K.M.; Krug, M.; Heinemann, U.; Mueller, U.; Weiss, M.S. XDSAPP2.0. J. Appl. Crystallogr. 2016, 49, 1085–1092. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Avvaru, B.S.; Kim, C.U.; Sippel, K.H.; Gruner, S.M.; Agbandje-McKenna, M.; Silverman, D.N.; McKenna, R. A Short, Strong Hydrogen Bond in the Active Site of Human Carbonic Anhydrase II. Biochemistry 2010, 49, 249–251. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Tam, K.Y.; Takács-Novák, K. Multi-wavelength spectrophotometric determination of acid dissociation constants: A validation study. Anal. Chim. Acta 2001, 434, 157–167. [Google Scholar] [CrossRef]
- Box, K.; Bevan, C.; Comer, J.; Hill, A.; Allen, R.; Reynolds, D. High-Throughput Measurement of pKa Values in a Mixed-Buffer Linear pH Gradient System. Anal. Chem. 2003, 75, 883–892. [Google Scholar] [CrossRef]
- Schmid, M.; Nogueira, E.S.; Monnard, F.W.; Ward, T.R.; Meuwly, M. Arylsulfonamides as inhibitors for carbonic anhydrase: Prediction & validation. Chem. Sci. 2012, 3, 690–700. [Google Scholar]
- Krishnamurthy, V.M.; Kaufman, G.K.; Urbach, A.R.; Gitlin, I.; Gudiksen, K.L.; Weibel, D.B.; Whitesides, G.M. Carbonic Anhydrase as a Model for Biophysical and Physical-Organic Studies of Proteins and Protein-Ligand Binding. Chem. Rev. 2008, 108, 946–1051. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. Van der waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Baker, B.M.; Murphy, K.P. Evaluation of Linked Protonation Effects in Protein Binding Reactions Using Isothermal Titration Calorimetry. Biophys. J. 1996, 71, 2049–2055. [Google Scholar] [CrossRef]
- Brautigam, C.A.; Zhao, H.; Vargas, C.; Keller, S.; Schuck, P. Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat. Protoc. 2016, 11, 882–894. [Google Scholar] [CrossRef]
- Schmidtke, P.; Luque, F.J.; Murray, J.B.; Barril, X. Shielded Hydrogen Bonds as Structural Determinants of Binding Kinetics: Application in Drug Design. J. Am. Chem. Soc. 2011, 133, 18903–18910. [Google Scholar] [CrossRef]
- Zihlmann, P.; Silbermann, M.; Sharpe, T.; Jiang, X.; Mühletaler, T.; Jakob, R.P.; Rabbani, S.; Sager, C.P.; Frei, P.; Pang, L.; et al. KinITC—One Method Supports both Thermodynamic and Kinetic SARs as Exemplified on FimH Antagonists. Chem. Eur. J. 2018, 24, 13049–13057. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Fluorinated Positions | R | PDB Entry | Occupancyb | α / °c | Torsion Angle τ / °d | d / Å | RMSD Thr200e | Interface area F/protein meta / Å2f | Interface Area F/protein ortho / Å2g |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | - | H | 6GDCa | 1.0 | 0.9 | 49.2 | 3.5 | - | ||
| 2 | - | Me | 6GM9a | 1.0 | 2.4 | 50.9 | 3.5 | 0 | ||
| 3 | 2 | H | 6RIT | 1.0 | 5.0 | 55.7 | 3.6 | 0.183 | ||
| 4 | 3 | H | 6RQI | 1.0 | 2.9 | 49.1 | 3.6 | 0.072 | 127.4 | |
| 5 | 4 | F | 6RKN | 1.0 | 1.6 | 50.2 | 3.5 | 0.022 | ||
| 6 | 2,3 | H | 6RJJ | 1.0 | 40.1 | 0.5 | 4.0 | 0.086, 0.221 | 161.1 | 140.3 |
| 7 | 2,5 | H | 6RNP | 1.0 | 4.7 | 54.8 | 3.5 | 0.205 | 114.34 | |
| 8 | 2,6 | H | 6ROE | 0.66, 0.34 | 4.7 | 2.1, 56.9 | 3.6 | 0.061, 0.272 | 138.8 | |
| 9 | 3,5 | H | 6RRG | 0.69, 0.31 | 2.0 | 2.3, 54.7 | 3.6 | 0.07 | 157.3 | |
| 10 | 2,3,5,6 | H | 6RRI | 1.0 | 42.2 | 2.0 | 4.0 | 0.077 | 142.0 | 138.3 |
| 11 | 2,3,5,6 | Me | 6RS5 | 0.35, 0.65 | 5.3 | 0.1, 58.2 | 3.7 | 0.068, 0.298 | 157.4 | 137.0 |
| 12 | 2,3,5,6 | Et | 6RSZ | 1.0 | 42.0 | 1.2 | 4.0 | 0.065 | 165.6 | 139.9 |
| 13 | 2,3,5,6 | Pr | 6S9G | 1.0 | 41.2 | 2.1 | 4.0 | 0.097 | 159.9 | 138.9 |
| 14 | 2,3,4,5,6 | F | 6SD7 | 1.0 | 41.8 | 4.2 | 4.1 | 0.059 | 155.7 | 138.5 |
| 15 | - | CN | 6ROB | 1.0 | 2.4 | 48.8 | 3.6 | 0.039 | - | - |
| 16 | - | NO2 | 6RH4 | 1.0 | 3.2 | 49.2 | 3.6 | 0.038 | - | - |
| 17 | - | NH2 | 6RL9 | 1.0 | 2.2 | 50.1 | 3.6 | 0.015 | - | - |
| Compound | pKa |
|---|---|
| 1 | 9.96 ± 0.01 |
| 2 | 10.15 ± 0.01 |
| 3 | 9.38 ± 0.01 |
| 4 | 9.57 ± 0.01 |
| 5 | 9.84 ± 0.01 |
| 6 | 8.98 ± 0.01 |
| 7 | 8.99 ± 0.01 |
| 8 | 8.90 ± 0.01 |
| 9 | 9.18 ± 0.01 |
| 10 | 8.08 ± 0.01 |
| 11a | 8.20 ± 0.01 |
| 15 | 9.13 ± 0.01 |
| 16 | 9.06 ± 0.01 |
| 17 | 2.12 ± 0.01, 10.55 ± 0.01 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glöckner, S.; Ngo, K.; Wagner, B.; Heine, A.; Klebe, G. The Influence of Varying Fluorination Patterns on the Thermodynamics and Kinetics of Benzenesulfonamide Binding to Human Carbonic Anhydrase II. Biomolecules 2020, 10, 509. https://doi.org/10.3390/biom10040509
Glöckner S, Ngo K, Wagner B, Heine A, Klebe G. The Influence of Varying Fluorination Patterns on the Thermodynamics and Kinetics of Benzenesulfonamide Binding to Human Carbonic Anhydrase II. Biomolecules. 2020; 10(4):509. https://doi.org/10.3390/biom10040509
Chicago/Turabian StyleGlöckner, Steffen, Khang Ngo, Björn Wagner, Andreas Heine, and Gerhard Klebe. 2020. "The Influence of Varying Fluorination Patterns on the Thermodynamics and Kinetics of Benzenesulfonamide Binding to Human Carbonic Anhydrase II" Biomolecules 10, no. 4: 509. https://doi.org/10.3390/biom10040509
APA StyleGlöckner, S., Ngo, K., Wagner, B., Heine, A., & Klebe, G. (2020). The Influence of Varying Fluorination Patterns on the Thermodynamics and Kinetics of Benzenesulfonamide Binding to Human Carbonic Anhydrase II. Biomolecules, 10(4), 509. https://doi.org/10.3390/biom10040509