Pathological Crosstalk between Metastatic Breast Cancer Cells and the Bone Microenvironment


Abstract
1. Introduction
2. The Cellular Composition of the Bone Microenvironment
3. Physiological and Pathological Bone Remodeling
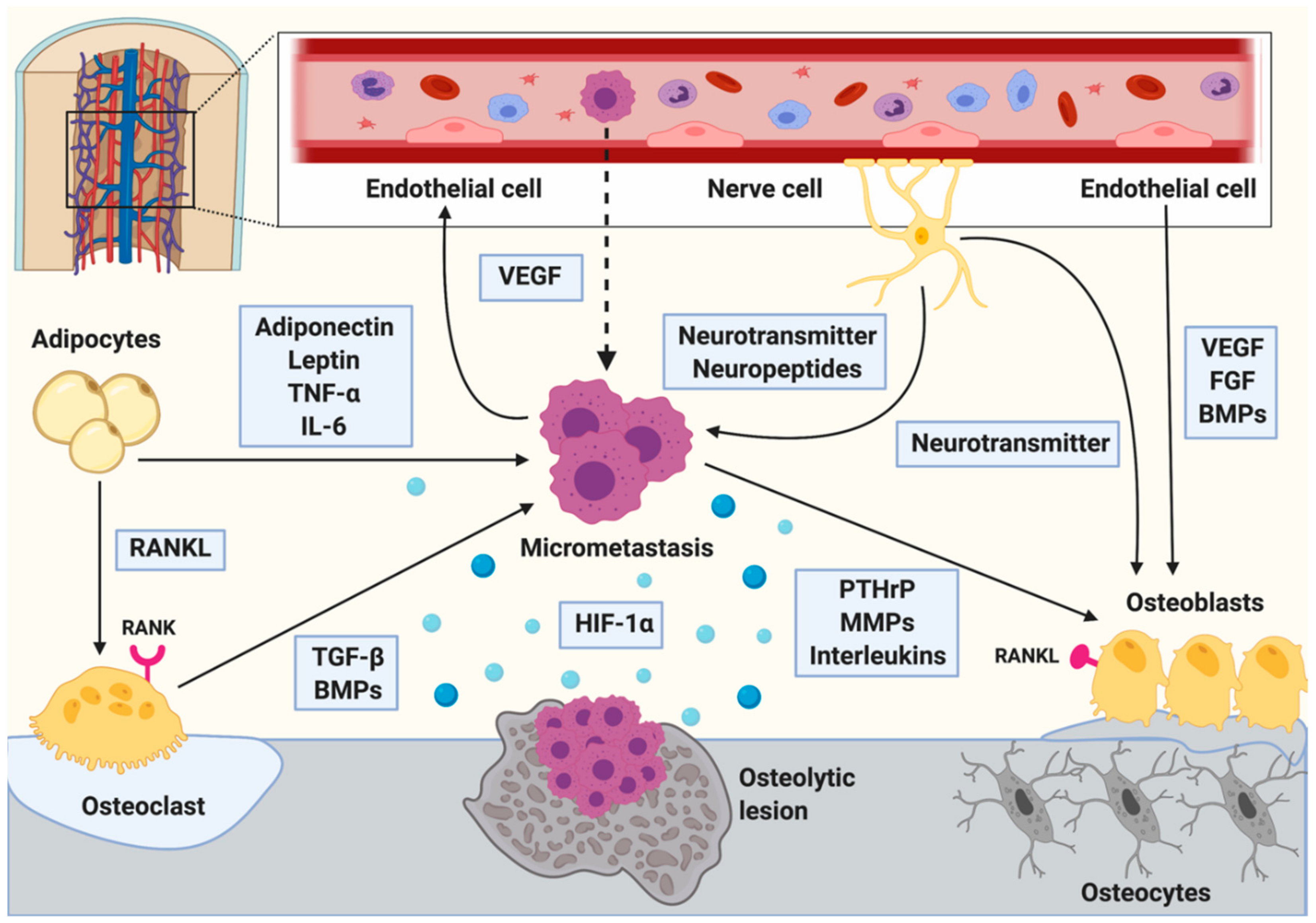
4. Bone Cells Regulating Breast Cancer Bone Metastasis
4.1. The Role of Osteoclasts in Breast Cancer Bone Metastasis
4.2. The Role of Osteoblasts in Breast Cancer Bone Metastasis
4.2.1. Osteoblasts during the Early Stages of Bone Metastasis
4.2.2. Osteoblasts during Later Stages of Bone Metastases
4.2.3. The Effect of Cancer Cell–Osteoblast Interactions on Bone Matrix and Bone Quality
4.3. The Role of Osteocytes in Breast Cancer Bone Metastasis
5. Adipocytes as Regulators of Bone Metastases
6. Bone Marrow Vasculature and Hypoxia as Regulators of Metastatic Bone Disease
6.1. The Role of Bone Marrow Vasculature in Bone Metastases
6.2. The Effect of Hypoxia in Metastatic Bone Environment
7. The Role of Nerve Cells in Bone Metastases
8. The Bone Marrow Microenvironment as a Target to Treat Breast Cancer Bone Metastases
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep Nature of Metastatic Inefficiency : Dormancy of Solitary Cells after Successful Extravasation and Limited Survival of Early Micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Wang, N.; Docherty, F.; Brown, H.K.; Reeves, K.; Fowles, A.; Lawson, M.; Ottewell, P.D.; Holen, I.; Croucher, P.I.; Eaton, C.L. Mitotic quiescence, but not unique “stemness,” marks the phenotype of bone metastasis-initiating cells in prostate cancer. FASEB J. 2015, 29, 3141–3150. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 571–573. [Google Scholar] [CrossRef]
- Jordan, C.T.; Lemischka, I.R. Clonal and systemic analysis of long-term hematopoiesis in the mouse. Genes Development 1990, 4, 220–232. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Rutkovskiy, A.; Stensløkken, K.O.; Vaage, I.J. Osteoblast Differentiation at a Glance. Med. Sci. Monit. Basic Res. 2016, 22, 95–106. [Google Scholar] [CrossRef]
- Nakamura, H. Morphology, Function, and Differentiation of Bone Cells. J. Hard Tissue Biol. 2007, 16, 15–22. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Bellido, T. Osteocytes and Skeletal Pathophysiology. Curr. Mol. Biol. Rep. 2015, 1, 157–167. [Google Scholar] [CrossRef]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef]
- Akune, T.; Ohba, S.; Kamekura, S.; Yamaguchi, M.; Chung, U.-I.; Kubota, N.; Terauchi, Y.; Harada, Y.; Azuma, Y.; Nakamura, K.; et al. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J. Clin. Invest. 2004, 113, 846–855. [Google Scholar] [CrossRef]
- Abdallah, B.M. Marrow adipocytes inhibit the differentiation of mesenchymal stem cells into osteoblasts via suppressing BMP-signaling. J. Biomed. Sci. 2017, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Hozumi, A.; Osaki, M.; Fukushima, T.; Sakamoto, K.; Yonekura, A.; Tomita, M.; Furukawa, K.; Shindo, H.; Baba, H. Primary human bone marrow adipocytes support TNF-α-induced osteoclast differentiation and function through RANKL expression. Cytokine 2011, 56, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.C.; Adams, R.H. Biology of bone: The vasculature of the skeletal system. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Sivaraj, K.K.; Adams, R.H. Blood vessel formation and function in bone. Development 2016, 143, 2706–2715. [Google Scholar] [CrossRef]
- Johnson, R.W.; Sowder, M.E.; Giaccia, A.J. Hypoxia and Bone Metastatic Disease. Curr. Osteoporos. Rep. 2017, 15, 231–238. [Google Scholar] [CrossRef]
- Campbell, J.P.; Karolak, M.R.; Ma, Y.; Perrien, D.S.; Masood-Campbell, S.K.; Penner, N.L.; Munoz, S.A.; Zijlstra, A.; Yang, X.; Sterling, J.A.; et al. Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PLoS Biol. 2012, 10, 13. [Google Scholar] [CrossRef]
- Elefteriou, F. Role of sympathetic nerves in the establishment of metastatic breast cancer cells in bone. J. Bone Oncol. 2016, 5, 132–134. [Google Scholar] [CrossRef]
- Elefteriou, F.; Campbell, P. Involvement of Sympathetic Nerves in Bone Metastasis, 2nd ed.; Academic Press: Cambridge, MA USA, 2015; pp. 591–597. ISBN 9780124167285. [Google Scholar]
- Baron, R.; Hesse, E. Update on bone anabolics in osteoporosis treatment: Rationale, current status, and perspectives. J. Clin. Endocrinol. Metab. 2012, 97, 311–325. [Google Scholar] [CrossRef]
- Rucci, N. Molecular biology of bone remodelling. Clin. Cases Miner. Bone Metab. 2008, 5, 49–56. [Google Scholar]
- Negishi-Koga, T.; Shinohara, M.; Komatsu, N.; Bito, H.; Kodama, T.; Friedel, R.H.; Takayanagi, H. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med. 2011, 17, 1473–1480. [Google Scholar] [CrossRef]
- Paiva, K.B.S.; Granjeiro, J.M. Matrix Metalloproteinases in Bone Resorption, Remodeling, and Repair. Prog. Mol. Biol. Transl. Sci. 2017, 148, 203–303. [Google Scholar]
- Käkönen, S.M.; Mundy, G.R. Mechanisms of osteolytic bone metastases in breast carcinoma. Cancer 2003, 97, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Sosnoski, D.M.; Mastro, A.M. Breast cancer metastasis to the bone: Mechanisms of bone loss. Breast Cancer Res. 2010, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Advances in bone biology: The osteoclast. Endocr. Rev. 1996, 17, 308–332. [Google Scholar] [PubMed]
- Suda, T.; Nakamura, I.; Jimi, E.; Takahashi, N. Regulation of osteoclast function. J. Bone Min. Res. 1997, 12, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Cell biology of the osteoclast. Exp. Hematol. 1999, 27, 1229–1241. [Google Scholar] [CrossRef]
- McCoy, E.M.; Hong, H.; Pruitt, H.C.; Feng, X. IL-11 produced by breast cancer cells augments osteoclastogenesis by sustaining the pool of osteoclast progenitor cells. BMC Cancer 2013, 13, 16. [Google Scholar] [CrossRef]
- Thomas, R.J.; Guise, T.A.; Yin, J.J.; Elliott, J.; Horwood, N.J.; Martin, T.J.; Gillespie, M.T. Breast Cancer Cells Interact with Osteoblasts to Support Osteoclast Formation. Endocrinology 1999, 140, 4451–4458. [Google Scholar] [CrossRef]
- Grimaud, E.; Soubigou, L.; Couillaud, S.; Coipeau, P.; Moreau, A.; Passuti, N.; Gouin, F.; Redini, F.; Heymann, D. Receptor Activator of Nuclear Factor κB Ligand (RANKL)/Osteoprotegerin (OPG) Ratio Is Increased in Severe Osteolysis. Am. J. Pathol. 2003, 163, 2021–2031. [Google Scholar] [CrossRef]
- Mundy, G.R. Mechanisms of bone metastasis. Cancer 1997, 80, 1546–1556. [Google Scholar] [CrossRef]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.-T.; Holen, I.; Dear, T.N.; Hunter, K.; Brown, H.K. Modifying the osteoblastic niche with zoledronic acid in vivo-potential implications for breast cancer bone metastasis. Bone 2014, 66, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Kolb, A.D.; Shupp, A.B.; Mukhopadhyay, D.; Marini, F.C.; Bussard, K.M. Osteoblasts are “educated” by crosstalk with metastatic breast cancer cells in the bone tumor microenvironment. Breast Cancer Res 2019, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.K.; Ottewell, P.D.; Evans, C.A.; Holen, I. Location matters: Osteoblast and osteoclast distribution is modified by the presence and proximity to breast cancer cells in vivo. Clin. Exp. Metastasis 2012, 29, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- CALVI, L.M. Osteoblastic Activation in the Hematopoietic Stem Cell Niche. Ann. N. Y. Acad. Sci. 2006, 1068, 477–488. [Google Scholar] [CrossRef]
- Jung, Y.; Wang, J.; Schneider, A.; Sun, Y.X.; Koh-Paige, A.J.; Osman, N.I.; McCauley, L.K.; Taichman, R.S. Regulation of SDF-1 (CXCL12) production by osteoblasts; a possible mechanism for stem cell homing. Bone 2006, 38, 497–508. [Google Scholar] [CrossRef]
- Ponomaryov, T.; Peled, A.; Petit, I.; Taichman, R.S.; Habler, L.; Sandbank, J.; Arenzana-Seisdedos, F.; Magerus, A.; Caruz, A.; Fujii, N.; et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J. Clin. Invest. 2000, 106, 1331–1339. [Google Scholar] [CrossRef]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Kim Lyerly, H.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340ra73. [Google Scholar] [CrossRef]
- Devignes, C.S.; Aslan, Y.; Brenot, A.; Devillers, A.; Schepers, K.; Fabre, S.; Chou, J.; Casbon, A.J.; Werb, Z.; Provot, S. HIF signaling in osteoblast-lineage cells promotes systemic breast cancer growth and metastasis in mice. Proc. Natl. Acad. Sci. USA 2018, 115, E992–E1001. [Google Scholar] [CrossRef] [PubMed]
- Allocca, G.; Hughes, R.; Wang, N.; Brown, H.K.; Ottewell, P.D.; Brown, N.J.; Holen, I. The bone metastasis niche in breast cancer-potential overlap with the haematopoietic stem cell niche. J. Bone Oncol. 2019, 17, 100244. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Venzon, D.J.; Mastro, A.M. Osteoblasts are a major source of inflammatory cytokines in the tumor microenvironment of bone metastatic breast cancer. J. Cell Biochem. 2010, 111, 1138–1148. [Google Scholar] [CrossRef]
- Vallet, S.; Bashari, M.H.; Fan, F.J.; Malvestiti, S.; Schneeweiss, A.; Wuchter, P.; Jäger, D.; Podar, K. Pre-Osteoblasts Stimulate Migration of Breast Cancer Cells via the HGF/MET Pathway. PLoS One 2016, 11, e0150507. [Google Scholar] [CrossRef] [PubMed]
- Menon, L.G.; Picinich, S.; Koneru, R.; Gao, H.; Lin, S.Y.; Koneru, M.; Mayer-Kuckuk, P.; Glod, J.; Banerjee, D. Differential Gene Expression Associated with Migration of Mesenchymal Stem Cells to Conditioned Medium from Tumor Cells or Bone Marrow Cells. Stem Cells 2007, 25, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Bodenstine, T.M.; Beck, B.H.; Cao, X.; Cook, L.M.; Ismail, A.; Powers, S.J.; Powers, J.K.; Mastro, A.M.; Welch, D.R. Pre-osteoblastic MC3T3-E1 cells promote breast cancer growth in bone in a murine xenograft model. Chin. J. Cancer 2011, 30, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Kinder, M.; Chislock, E.; Bussard, K.M.; Shuman, L.; Mastro, A.M. Metastatic breast cancer induces an osteoblast inflammatory response. Exp. Cell Res. 2008, 314, 173–183. [Google Scholar] [CrossRef]
- Abana, C.O.; Bingham, B.S.; Cho, J.H.; Graves, A.J.; Koyama, T.; Pilarski, R.T.; Chakravarthy, A.B.; Xia, F. IL-6 variant is associated with metastasis in breast cancer patients. PLoS One 2017, 12, e0181725. [Google Scholar] [CrossRef]
- Dutta, P.; Sarkissyan, M.; Paico, K.; Wu, Y.; Vadgama, J. V MCP-1 is overexpressed in triple-negative breast cancers and drives cancer invasiveness and metastasis. Breast Cancer Res. Treat 2018, 170, 477–486. [Google Scholar] [CrossRef]
- Phadke, P.A.; Mercer, R.R.; Harms, J.F.; Jia, Y.; Frost, A.R.; Jewell, J.L.; Bussard, K.M.; Nelson, S.; Moore, C.; Kappes, J.C.; et al. Kinetics of metastatic breast cancer cell trafficking in bone. Clin. Cancer Res. 2006, 12, 1431–1440. [Google Scholar] [CrossRef]
- Mastro, A.M.; Gay, C.V.; Welch, D.R.; Donahue, H.J.; Jewell, J.; Mercer, R.; DiGirolamo, D.; Chislock, E.M.; Guttridge, K. Breast cancer cells induce osteoblast apoptosis: A possible contributor to bone degradation. J. Cell Biochem. 2004, 91, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.R.; Miyasaka, C.; Mastro, A.M. Metastatic breast cancer cells suppress osteoblast adhesion and differentiation. Clin. Exp. Metastasis 2004, 21. [Google Scholar] [CrossRef] [PubMed]
- Sekita, A.; Matsugaki, A.; Nakano, T. Disruption of collagen/apatite alignment impairs bone mechanical function in osteoblastic metastasis induced by prostate cancer. Bone 2017, 97, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Matsugaki, A.; Sekita, A.; Nakano, T. Alteration of osteoblast arrangement via direct attack by cancer cells: New insights into bone metastasis. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Matsugaki, A.; Harada, T.; Kimura, Y.; Sekita, A.; Nakano, T. Dynamic Collision Behavior Between Osteoblasts and Tumor Cells Regulates the Disordered Arrangement of Collagen Fiber/Apatite Crystals in Metastasized Bone. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Wise-Milestone, L.; Akens, M.K.; Rosol, T.J.; Hojjat, S.P.; Grynpas, M.D.; Whyne, C.M. Evaluating the effects of mixed osteolytic/osteoblastic metastasis on vertebral bone quality in a new rat model. J. Orthop. Res. 2012, 30, 817–823. [Google Scholar] [CrossRef]
- Ma, Y.V.; Xu, L.; Mei, X.; Middleton, K.; You, L. Mechanically stimulated osteocytes reduce the bone-metastatic potential of breast cancer cells in vitro by signaling through endothelial cells. J. Cell Biochem. 2018. [Google Scholar] [CrossRef]
- Giuliani, N.; Ferretti, M.; Bolzoni, M.; Storti, P.; Lazzaretti, M.; Dalla Palma, B.; Bonomini, S.; Martella, E.; Agnelli, L.; Neri, A.; et al. Increased osteocyte death in multiple myeloma patients: Role in myeloma-induced osteoclast formation. Leukemia 2012, 26, 1391–1401. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Hiasa, M.; Chirgwin, J.M.; Carlesso, N.; Yoneda, T.; Mohammad, K.S.; Plotkin, L.I.; Roodman, G.D.; et al. Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma. Cancer Res. 2016, 76, 1089. [Google Scholar] [CrossRef]
- Toscani, D.; Palumbo, C.; Dalla Palma, B.; Ferretti, M.; Bolzoni, M.; Marchica, V.; Sena, P.; Martella, E.; Mancini, C.; Ferri, V.; et al. The Proteasome Inhibitor Bortezomib Maintains Osteocyte Viability in Multiple Myeloma Patients by Reducing Both Apoptosis and Autophagy: A New Function for Proteasome Inhibitors. J. Bone Min. Res. 2016, 31, 815–827. [Google Scholar] [CrossRef]
- Guise, T.A.; Kozlow, W.M.; Heras-Herzig, A.; Padalecki, S.S.; Yin, J.J.; Chirgwin, J.M. Molecular mechanisms of breast cancer metastases to bone. Clin. Breast Cancer 2005, 5, S46–S53. [Google Scholar] [CrossRef]
- Cui, Y.X.; Evans, B.A.; Jiang, W.G. New Roles of Osteocytes in Proliferation, Migration and Invasion of Breast and Prostate Cancer Cells. Anticancer Res. 2016, 36, 1193–1201. [Google Scholar]
- Myers, T.J.; Longobardi, L.; Willcockson, H.; Temple, J.D.; Tagliafierro, L.; Ye, P.; Li, T.; Esposito, A.; Moats-Staats, B.M.; Spagnoli, A. BMP2 Regulation of CXCL12 Cellular, Temporal, and Spatial Expression is Essential During Fracture Repair. J. Bone Min. Res. 2015, 30, 2014–2027. [Google Scholar] [CrossRef]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar]
- Wang, N.; Docherty, F.E.; Brown, H.K.; Reeves, K.J.; Fowles, A.C.; Ottewell, P.D.; Dear, T.N.; Holen, I.; Croucher, P.I.; Eaton, C.L. Prostate cancer cells preferentially home to osteoblast-rich areas in the early stages of bone metastasis: Evidence from in vivo models. J. Bone Min. Res 2014, 29, 2688–2696. [Google Scholar] [CrossRef]
- Herroon, M.K.; Rajagurubandara, E.; Hardaway, A.L.; Powell, K.; Turchick, A.; Feldmann, D.; Podgorski, I. Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget 2013, 4, 2108–2123. [Google Scholar] [CrossRef]
- Morris, E.V.; Edwards, C.M. The role of bone marrow adipocytes in bone metastasis. J. Bone Oncol. 2016, 5, 121–123. [Google Scholar] [CrossRef]
- Luo, G.; He, Y.; Yu, X. Bone Marrow Adipocyte: An intimate partner with tumor cells in bone metastasis. Front. Endocrinol. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Herroon, M.K.; Rajagurubandara, E.; Diedrich, J.D.; Heath, E.I.; Podgorski, I. Adipocyte-Activated oxidative and ER stress pathways promote tumor survival in bone via upregulation of Heme Oxygenase 1 and Survivin. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Diedrich, J.D.; Rajagurubandara, E.; Herroon, M.K.; Mahapatra, G.; Hüttemann, M.; Podgorski, I. Bone marrow adipocytes promote the warburg phenotype in metastatic prostate tumors via HIF-1α activation. Oncotarget 2016, 7, 64854–64877. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- Caers, J.; Deleu, S.; Belaid, Z.; De Raeve, H.; Van Valckenborgh, E.; De Bruyne, E.; DeFresne, M.P.; Van Riet, I.; Van Camp, B.; Vanderkerken, K. Neighboring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia 2007, 21, 1580–1584. [Google Scholar] [CrossRef] [PubMed]
- Jöhrer, K.; Ploner, C.; Thangavadivel, S.; Wuggenig, P.; Greil, R. Adipocyte-derived players in hematologic tumors: Useful novel targets? Expert Opin. Biol. Ther. 2015, 15, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.-T.; Phuong, T.N.T.; Tien, N.L.B.; Tran, D.-K.; Nguyen, T.-T.; Thanh, V.V.; Quang, T.L.; Minh, L.B.; Pham, V.H.; Ngoc, V.T.N.; et al. The Effects of Adipocytes on the Regulation of Breast Cancer in the Tumor Microenvironment: An Update. Cells 2019, 8, 857. [Google Scholar] [CrossRef]
- Jia, Z.; Liu, Y.; Cui, S. Adiponectin induces breast cancer cell migration and growth factor expression. Cell Biochem. Biophys. 2014, 70, 1239–1245. [Google Scholar] [CrossRef]
- Libby, E.F.; Frost, A.R.; Demark-Wahnefried, W.; Hurst, D.R. Linking adiponectin and autophagy in the regulation of breast cancer metastasis. J. Mol. Med. 2014, 92, 1015–1023. [Google Scholar] [CrossRef][Green Version]
- Snoussi, K.; Strosberg, A.D.; Bouaouina, N.; Ahmed, S.B.; Helal, A.N.; Chouchane, L. Leptin and leptin receptor polymorphisms are associated with increased risk and poor prognosis of breast carcinoma. BMC Cancer 2006, 6, 1–10. [Google Scholar] [CrossRef]
- Templeton, Z.S.; Lie, W.-R.; Wang, W.; Rosenberg-Hasson, Y.; Alluri, R.V.; Tamaresis, J.S.; Bachmann, M.H.; Lee, K.; Maloney, W.J.; Contag, C.H.; et al. Breast Cancer Cell Colonization of the Human Bone Marrow Adipose Tissue Niche. Neoplasia 2015, 17, 849–861. [Google Scholar] [CrossRef]
- Gonzalez-Perez, R.R.; Xu, Y.; Guo, S.; Watters, A.; Zhou, W.; Leibovich, S.J. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFκB/HIF-1α activation. Cell. Signal. 2010, 22, 1350–1362. [Google Scholar] [CrossRef]
- Maes, C.; Goossens, S.; Bartunkova, S.; Drogat, B.; Coenegrachts, L.; Stockmans, I.; Moermans, K.; Nyabi, O.; Haigh, K.; Naessens, M.; et al. Increased skeletal VEGF enhances Β-catenin activity and results in excessively ossified bones. EMBO J. 2010, 29, 424–441. [Google Scholar] [CrossRef]
- Filipowska, J.; Tomaszewski, K.A.; Niedźwiedzki, Ł.; Walocha, J.A.; Niedźwiedzki, T. The role of vasculature in bone development, regeneration and proper systemic functioning. Angiogenesis 2017, 20, 291–302. [Google Scholar] [PubMed]
- Nyangoga, H.; Mercier, P.; Libouban, H.; Baslé, M.F.; Chappard, D. Three-dimensional characterization of the vascular bed in bone metastasis of the rat by microcomputed tomography (MicroCT). PLoS One 2011, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Shomento, S.H.; Wan, C.; Cao, X.; Faugere, M.C.; Bouxsein, M.L.; Clemens, T.L.; Riddle, R.C. Hypoxia-inducible factors 1α and 2α exert both distinct and overlapping functions in long bone development. J. Cell. Biochem. 2010, 109, 196–204. [Google Scholar] [CrossRef]
- Wang, Y.; Wan, C.; Deng, L.; Liu, X.; Cao, X.; Gilbert, S.R.; Bouxsein, M.L.; Faugere, M.C.; Guldberg, R.E.; Gerstenfeld, L.C.; et al. The hypoxia-inducible factor α pathway couples angiogenesis to osteogenesis during skeletal development. J. Clin. Invest. 2007, 117, 1616–1626. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Höckel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.R.; Manova-Todorova, K.; Massagué, J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef]
- Shyu, K.G.; Hsu, F.L.; Wang, M.J.; Wang, B.W.; Lin, S. Hypoxia-inducible factor 1α regulates lung adenocarcinoma cell invasion. Exp. Cell Res. 2007, 313, 1181–1191. [Google Scholar] [CrossRef]
- Sullivan, R.; Graham, C.H. Hypoxia-driven selection of the metastatic phenotype. Cancer Metastasis Rev. 2007, 26, 319–331. [Google Scholar] [CrossRef]
- Hiraga, T.; Kizaka-Kondoh, S.; Hirota, K.; Hiraoka, M.; Yoneda, T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007, 67, 4157–4163. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.K.; Mohammad, K.S.; Fournier, P.G.J.; McKenna, C.R.; Davis, H.W.; Niewolna, M.; Peng, X.H.; Chirgwin, J.M.; Guise, T.A. Hypoxia and TGF-β drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yan, C.H.; Yuan, M.; Wei, Y.; Hu, G.; Kang, Y. In vivo dynamics and distinct functions of hypoxia in primary tumor growth and organotropic metastasis of breast cancer. Cancer Res. 2010, 70, 3905–3914. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef]
- Okada, Y.; Eibl, G.; Duffy, J.P.; Reber, H.A.; Hines, O.J. Glial cell-derived neurotrophic factor upregulates the expression and activation of matrix metalloproteinase-9 in human pancreatic cancer. Surgery 2003, 134, 293–299. [Google Scholar] [CrossRef]
- Okada, Y.; Eibl, G.; Guha, S.; Duffy, J.P.; Reber, H.A.; Hines, O.J. Nerve growth factor stimulates MMP-2 expression and activity and increases invasion by human pancreatic cancer cells. Clin. Exp. Metastasis 2004, 21, 285–292. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.-S.; Ye, S.-L.; Luo, P.; Wang, B.-L. Blockage of tropomyosin receptor kinase a (TrkA) enhances chemo-sensitivity in breast cancer cells and inhibits metastasis in vivo. Int. J. Clin. Exp. Med. 2015, 8, 634–641. [Google Scholar]
- Gattelli, A.; Nalvarte, I.; Boulay, A.; Roloff, T.C.; Schreiber, M.; Carragher, N.; Macleod, K.K.; Schlederer, M.; Lienhard, S.; Kenner, L.; et al. Ret inhibition decreases growth and metastatic potential of estrogen receptor positive breast cancer cells. EMBO Mol. Med. 2013, 5, 1335–1350. [Google Scholar] [CrossRef]
- Kuol, N.; Stojanovska, L.; Apostolopoulos, V.; Nurgali, K. Role of the nervous system in cancer metastasis. J. Exp. Clin. Cancer Res. 2018, 37, 1–12. [Google Scholar] [CrossRef]
- Madden, K.S.; Szpunar, M.J.; Brown, E.B. β-Adrenergic receptors (β-AR) regulate VEGF and IL-6 production by divergent pathways in high β-AR-expressing breast cancer cell lines. Breast Cancer Res. Treat. 2011, 130, 747–758. [Google Scholar] [CrossRef]
- Medeiros, P.J.; Jackson, D.N. Neuropeptide y Y5-receptor activation on breast cancer cells acts as a paracrine system that stimulates VEGF expression and secretion to promote angiogenesis. Peptides 2013, 48, 106–113. [Google Scholar] [PubMed]
- Li, S.; Sun, Y.; Gao, D. Role of the nervous system in cancer metastasis (Review). Oncol. Lett. 2013, 5, 1101–1111. [Google Scholar] [PubMed]
- Sloan, E.K.; Priceman, S.J.; Cox, B.F.; Yu, S.; Pimentel, M.A.; Tangkanangnukul, V.; Arevalo, J.M.G.; Morizono, K.; Karanikolas, B.D.W.; Wu, L.; et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010, 70, 7042–7052. [Google Scholar] [CrossRef] [PubMed]
- Szpunar, M.J.; Burke, K.A.; Dawes, R.P.; Brown, E.B.; Madden, K.S. The antidepressant desipramine and α2-adrenergic receptor activation promote breast tumor progression in association with altered collagen structure. Cancer Prev. Res. 2013, 6, 1262–1272. [Google Scholar]
- Gumireddy, K.; Li, A.; Kossenkov, A.V.; Sakurai, M.; Yan, J.; Li, Y.; Xu, H.; Wang, J.; Zhang, P.J.; Zhang, L.; et al. The mRNA-edited form of GABRA3 suppresses GABRA3-mediated Akt activation and breast cancer metastasis. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, D.B. Mechanism of action, pharmacokinetic and pharmacodynamic profile, and clinical applications of nitrogen-containing bisphosphonates. J. Dent. Res. 2007, 86, 1022–1033. [Google Scholar] [CrossRef]
- Hanley, D.A.; Adachi, J.D.; Bell, A.; Brown, V. Denosumab: Mechanism of action and clinical outcomes. Int. J. Clin. Pract. 2012, 66, 1139–1146. [Google Scholar]
- Le Gall, C.; Bellahcène, A.; Bonnelye, E.; Gasser, J.A.; Castronovo, V.; Green, J.; Zimmermann, J.; Clézardin, P. A cathepsin K inhibitor reduces breast cancer induced osteolysis and skeletal tumor burden. Cancer Res. 2007, 67, 9894–9902. [Google Scholar] [CrossRef]
- Duong, L.T.; Wesolowski, G.A.; Leung, P.; Oballa, R.; Pickarski, M. Efficacy of a cathepsin K inhibitor in a preclinical model for prevention and treatment of breast cancer bone metastasis. Mol. Cancer Ther. 2014, 13, 2898–2909. [Google Scholar]
- Irelli, A.; Sirufo, M.M.; Scipioni, T.; De Pietro, F.; Pancotti, A.; Ginaldi, L.; De Martinis, M. mTOR Links Tumor Immunity and Bone Metabolism: What are the Clinical Implications? Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Sousa, S.; Clézardin, P. Bone-Targeted Therapies in Cancer-Induced Bone Disease. Calcif. Tissue Int. 2018, 102, 227–250. [Google Scholar] [CrossRef] [PubMed]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab Treatment in Postmenopausal Women with Osteoporosis. N. Engl. J. Med. 2016, 375, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Cosman, F. The evolving role of anabolic therapy in the treatment of osteoporosis. Curr. Opin. Rheumatol. 2019, 31, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Hesse, E.; Schröder, S.; Brandt, D.; Pamperin, J.; Saito, H.; Taipaleenmäki, H. Sclerostin inhibition alleviates breast cancer-induced bone metastases and muscle weakness. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Ukita, M.; Yamaguchi, T.; Ohata, N.; Tamura, M. Sclerostin Enhances Adipocyte Differentiation in 3T3-L1 Cells. J. Cell. Biochem. 2016, 117, 1419–1428. [Google Scholar] [CrossRef]
- Kim, S.P.; Frey, J.L.; Li, Z.; Kushwaha, P.; Zoch, M.L.; Tomlinson, R.E.; Da, H.; Aja, S.; Noh, H.L.; Kim, J.K.; et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc. Natl. Acad. Sci. USA 2017, 114, E11238–E11247. [Google Scholar] [CrossRef]
- De Paula, F.J.A.; Rosen, C.J. Marrow Adipocytes: Origin, Structure, and Function. 2020, 1–24. [Google Scholar] [CrossRef]
- Fairfield, H.; Falank, C.; Harris, E.; Demambro, V.; McDonald, M.; Pettitt, J.A.; Mohanty, S.T.; Croucher, P.; Kramer, I.; Kneissel, M.; et al. The skeletal cell-derived molecule sclerostin drives bone marrow adipogenesis. J. Cell. Physiol. 2018, 233, 1156–1167. [Google Scholar] [CrossRef]
- Zhou, J.Z.; Riquelme, M.A.; Gu, S.; Kar, R.; Gao, X.; Sun, L.; Jiang, J.X. Osteocytic connexin hemichannels suppress breast cancer growth and bone metastasis. Oncogene 2016, 35, 5597–5607. [Google Scholar] [CrossRef]
- Rodan, G.A.; Fleisch, H.A. Bisphosphonates: Mechanisms of action. J. Clin. Invest. 1996, 97, 2692–2696. [Google Scholar] [CrossRef]
- Clezardin, P. Mechanisms of action of bisphosphonates in oncology: A scientific concept evolving from antiresorptive to anticancer activities. BoneKEy Rep. 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Luckman, S.P.; Hughes, D.E.; Coxon, F.P.; Graham, R.; Russell, G.; Rogers, M.J. Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J. Bone Min. Res. 1998, 13, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Daubiné, F.; Le Gall, C.; Gasser, J.; Green, J.; Clézardin, P. Antitumor effects of clinical dosing regimens of bisphosphonates in experimental breast cancer bone metastasis. J. Natl. Cancer Inst. 2007, 99, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Rico, M.; Baglioni, M.; Bondarenko, M.; Laluce, N.C.; Rozados, V.; André, N.; Carré, M.; Scharovsky, O.G.; Márquez, M.M. Metformin and propranolol combination prevents cancer progression and metastasis in different breast cancer models. Oncotarget 2017, 8, 2874–2889. [Google Scholar] [CrossRef] [PubMed]
- Montoya, A.; Varela-Ramirez, A.; Dickerson, E.; Pasquier, E.; Torabi, A.; Aguilera, R.; Nahleh, Z.; Bryan, B. The beta adrenergic receptor antagonist propranolol alters mitogenic and apoptotic signaling in late stage breast cancer. Biomed. J. 2019, 42, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Cardwell, C.R.; Pottegård, A.; Vaes, E.; Garmo, H.; Murray, L.J.; Brown, C.; Vissers, P.A.J.; O’Rorke, M.; Visvanathan, K.; Cronin-Fenton, D.; et al. Propranolol and survival from breast cancer: A pooled analysis of European breast cancer cohorts. Breast Cancer Res. 2016, 18. [Google Scholar] [CrossRef]
- Kingsley, L.A.; Fournier, P.G.J.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol. Cancer Ther. 2007, 6, 2609–2617. [Google Scholar] [CrossRef]


{kind=link}
| Cell/Molecule | Key Findings | Reference |
|---|---|---|
| Bone Cells | ||
| Osteoclasts | Osteoclasts are not involved in tumor cell homing, but drive osteolysis | [34,35] |
| Osteoblasts | Interaction between osteoblasts and breast cancer cells via CXCL12/CXCR4 is important for tumor cell homing | [41] |
| Breast cancer cells modify osteoblast migration | [46] | |
| Co-injection of breast cancer cells and osteoblasts promotes tumor growth | [47] | |
| IL-6, IL-8, MCP-1, MIP-2 and VEGF are increased in osteoblasts in the presence of breast cancer cells | [44,48] | |
| OPNhigh and aSMAlow osteoblasts decrease cancer cell proliferation and may regulate dormancy | [35] | |
| Cancer cells alter osteoblast arrangement and collagen organization | [54,55,56] | |
| Osteocytes | Osteocytes secrete RANKL, MMPs, TNFα, sclerostin which influence cancer cell proliferation and migration | [24,62] |
| Osteocyte conditioned medium increased proliferation of human prostate and breast cancer cells | [63] | |
| Vasculature | ||
| Type H/type L capillaries | Expression of CD31/endomucin distinguishes vessels in metaphysis and diaphysis | [13] |
| Endothelial cells | Breast cancer cells localize in metaphysis and around the vasculature | [43,44] |
| Endothelial cells regulate cancer cell dormancy via thrombospondin-1 | [84] | |
| Hypoxia | ||
| HIF signaling | HIF1α overexpression stimulates bone metastasis, HIF1α knockdown shows reverse effects | [92,93] |
| HIF signaling stimulates cancer cell dissemination to bone via CXCR4/CXCL12 | [42,95] | |
| LIFR/STAT3 | Loss of LIFR/STAT3 regulates dormancy escape of breast cancer cells | [95] |
| Nerve Cells | ||
| β2AR | Stimulates RANKL production and metastasis | [17] |
| GDNF, NGF | Increase invasiveness of pancreatic cancer cells via MMP2 and MMP9 | [97,98] |
| Neurotransmittors, Neuropeptides, βAR, | Regulation of VEGF and angiogenesis | [19,101,102,103] |
| α2-AR | Stromal cells expressing α2-AR promote breast cancer cell metastasis | [105] |
| Gabra3 | Overexpression stimulates metastasis via AKT signaling | [106] |
| Adipocytes | ||
| Lipids | Lipids act as energy source for cancer cells, affect their metabolism and increase their invasiveness | [67,72,73,74] |
| Adipokines | Adiponectin has anti-tumoral effects via mTOR and NF-KB signaling | [75] |
| Leptin promotes metastasis via regulation of VEGF | [80] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarrer, J.; Haider, M.-T.; Smit, D.J.; Taipaleenmäki, H. Pathological Crosstalk between Metastatic Breast Cancer Cells and the Bone Microenvironment. Biomolecules 2020, 10, 337. https://doi.org/10.3390/biom10020337
Zarrer J, Haider M-T, Smit DJ, Taipaleenmäki H. Pathological Crosstalk between Metastatic Breast Cancer Cells and the Bone Microenvironment. Biomolecules. 2020; 10(2):337. https://doi.org/10.3390/biom10020337
Chicago/Turabian StyleZarrer, Jennifer, Marie-Therese Haider, Daniel J. Smit, and Hanna Taipaleenmäki. 2020. "Pathological Crosstalk between Metastatic Breast Cancer Cells and the Bone Microenvironment" Biomolecules 10, no. 2: 337. https://doi.org/10.3390/biom10020337
APA StyleZarrer, J., Haider, M.-T., Smit, D. J., & Taipaleenmäki, H. (2020). Pathological Crosstalk between Metastatic Breast Cancer Cells and the Bone Microenvironment. Biomolecules, 10(2), 337. https://doi.org/10.3390/biom10020337