Notch Signaling in Skeletal Development, Homeostasis and Pathogenesis


Abstract
1. Introduction
2. Notch Signaling in Human Skeletal Diseases
3. Notch Signaling in Chondrogenesis
4. Notch Signaling and Osteoarthritis
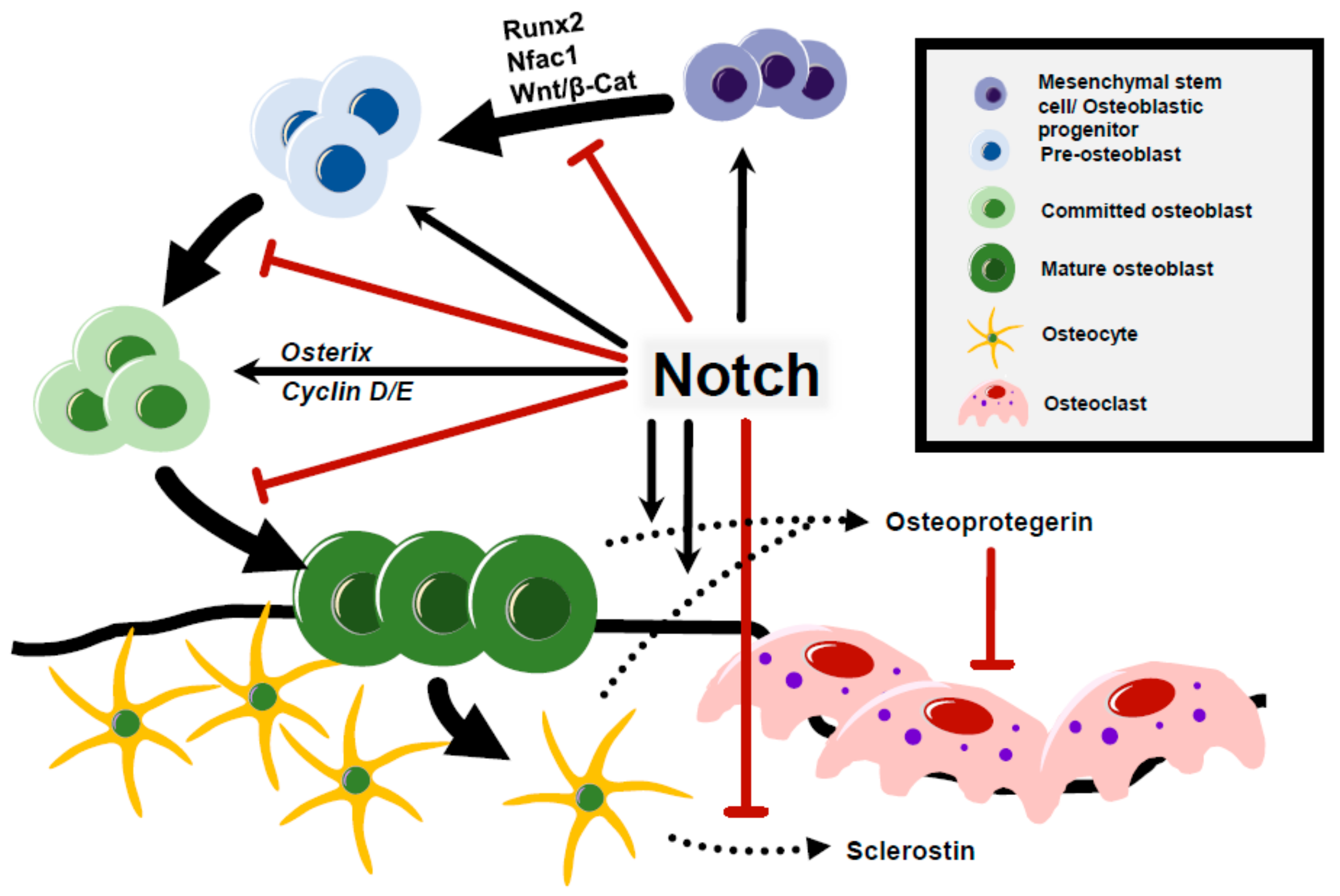
5. Notch Regulation of Osteoblast Differentiation and Osteocyte Function
6. Notch Signaling in Osteoclastogenesis and Bone Resorption
7. Notch Function in Skeletal Stem Cells and Fracture Healing
8. Pathological Function of Notch Signaling in Cancer
9. Concluding Remarks
Funding
Conflicts of Interest
References
- Stittrich, A.B.; Lehman, A.; Boidan, D.L.; Ashworth, J.; Zong, Z.; Li, H.; Lam, P.; Khromykh, A.; Iyer, R.K.; Vockley, J.G.; et al. Mutations in NOTCH1 cause Adams-Oliver syndrome. Am. J. Hum. Genet. 2014, 95, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Hassed, S.J.; Wiley, G.B.; Wang, S.; Lee, J.Y.; Li, S.; Xu, W.; Zhao, Z.J.; Mulvihill, J.J.; Robertson, J.; Warner, J.; et al. RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am. J. Hum. Genet. 2012, 91, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Meester, J.A.; Southgate, L.; Stittrich, A.B.; Vensellar, H.; Beekmans, S.J.; den Hollnader, N.; Bijlsma, E.K.; Helderman-van den Enden, A.; Verheij, J.B.; Glusman, G.; et al. Heterozygous Loss-of-Function Mutations in DLL4 Cause Adams-Oliver Syndrome. Am. J. Hum. Genet. 2015, 97, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Emerick, K.M.; Rand, E.B.; Goldmuntz, E.; Krantz, I.D.; Spinner, N.B.; Piccoli, D.A. Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis. Hepatology 1999, 29, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.C.; Laney, A.O.; Smith, R.; Gerfen, J.; Morrissette, J.J.; Woyciechowski, S.; Garbarini, J.; Loomes, K.M.; Krantz, I.D.; Urban, Z.; et al. Jagged1 (JAG1) mutations in patients with tetralogy of Fallot or pulmonic stenosis. Hum. Mutat. 2010, 31, 594–601. [Google Scholar] [CrossRef]
- Crosnier, C.; Driancourt, C.; Raynaud, N.; Dhorne-Pollet, S.; Pollet, N.; Bernard, O.; Hadchouel, M.; Meunier-Rotival, M. Mutations in JAGGED1 gene are predominantly sporadic in Alagille syndrome. Gastroenterology 1999, 116, 1141–1148. [Google Scholar] [CrossRef]
- Morrissette, J.D.; Colliton, R.P.; Spinner, N.B. Defective intracellular transport and processing of JAG1 missense mutations in Alagille syndrome. Hum. Mol. Genet. 2001, 10, 405–413. [Google Scholar] [CrossRef]
- Boyer-Di Ponio, J.; Wright-Crosnier, C.; Groyer-Picard, M.T.; Driancourt, C.; Beau, I.; Hadchouel, M.; Meunier-Rotival, M. Biological function of mutant forms of JAGGED1 proteins in Alagille syndrome: Inhibitory effect on Notch signaling. Hum. Mol. Genet. 2007, 16, 2683–2692. [Google Scholar] [CrossRef]
- Kamath, B.M.; Bauer, R.C.; Loomes, K.M.; Chao, G.; Gerfen, J.; Hutchinson, A.; Hardikar, W.; Hirschfield, G.; Jara, P.; Krantz, I.D.; et al. NOTCH2 mutations in Alagille syndrome. J. Med. Genet. 2012, 49, 138–144. [Google Scholar] [CrossRef]
- McDaniell, R.; Warthen, D.M.; Sanchez-Lara, P.A.; Pai, A.; Krantz, I.D.; Piccoli, D.A.; Spinner, N.B. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am. J. Hum. Genet. 2006, 79, 169–173. [Google Scholar] [CrossRef]
- Xue, Y.; Gao, X.; Lindsell, C.E.; Norton, C.R.; Chang, B.; Hicks, C.; Gendron-Maguire, M.; Rand, E.B.; Weinmaster, G.; Gridley, T. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum. Mol. Genet. 1999, 8, 723–730. [Google Scholar] [CrossRef] [PubMed]
- McCright, B.; Lozier, J.; Gridley, T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 2002, 129, 1075–1082. [Google Scholar] [PubMed]
- Rimoin, D.L.; Fletcher, B.D.; McKusick, V.A. Spondylocostal dysplasia. A dominantly inherited form of short-trunked dwarfism. Am. J. Med. 1968, 45, 948–953. [Google Scholar] [CrossRef]
- Turnpenny, P.D.; Whittock, N.; Duncan, J.; Dunwoodie, S.; Kusumi, K.; Ellard, S. Novel mutations in DLL3, a somitogenesis gene encoding a ligand for the Notch signalling pathway, cause a consistent pattern of abnormal vertebral segmentation in spondylocostal dysostosis. J. Med. Genet. 2003, 40, 333–339. [Google Scholar] [CrossRef]
- Whittock, N.V.; Sparrow, D.B.; Wouters, M.A.; Sillence, D.; Ellard, S.; Dunwoodie, S.L.; Turnpenny, P.D. Mutated MESP2 causes spondylocostal dysostosis in humans. Am. J. Hum. Genet. 2004, 74, 1249–1254. [Google Scholar] [CrossRef]
- Sparrow, D.B.; Guillén-Navarro, E.; Fatkin, D.; Dunwoodie, S.L. Mutation of Hairy-and-Enhancer-of-Split-7 in humans causes spondylocostal dysostosis. Hum. Mol Genet. 2008, 17, 3761–3766. [Google Scholar] [CrossRef]
- Sparrow, D.B.; Chapman, G.; Wouters, M.A.; Whittock, N.V.; Ellard, S.; Fatkin, D.; Turnpenny, P.D.; Kusumi, K.; Sillence, D.; Dunwoodie, S.L. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am. J. Hum. Genet. 2006, 78, 28–37. [Google Scholar] [CrossRef]
- Bessho, Y.; Sakata, R.; Komatsu, S.; Shiota, K.; Yamada, S.; Kageyama, R. Dynamic expression and essential functions of Hes7 in somite segmentation. Genes. Dev. 2001, 15, 2642–2647. [Google Scholar] [CrossRef]
- Okubo, Y.; Sugawara, T.; Abe-Koduka, N.; Kanno, J.; Kimura, A.; Saga, Y. Lfng regulates the synchronized oscillation of the mouse segmentation clock via trans-repression of Notch signalling. Nat. Commun. 2013, 3, 1141. [Google Scholar] [CrossRef]
- Sparrow, D.B.; Sillence, D.; Wouters, M.A.; Turnpenny, P.D.; Dunwoodie, S.L. Two novel missense mutations in HAIRY-AND-ENHANCER-OF-SPLIT-7 in a family with spondylocostal dysostosis. Eur. J. Hum. Genet. 2010, 18, 674–679. [Google Scholar] [CrossRef]
- Han, M.S.; Ko, J.M.; Cho, T.J.; Park, W.Y.; Cheong, H.I. A novel NOTCH2 mutation identified in a Korean family with Hajdu-Cheney syndrome showing phenotypic diversity. Ann. Clin. Lab. Sci. 2015, 45, 110–114. [Google Scholar] [PubMed]
- Majewski, J.; Schwartzentruber, J.A.; Caqueret, A.; Patry, L.; Marcadier, J.; Fryns, J.P.; Boycott, K.M.; Ste-Marie, L.G.; McKiernan, F.E.; Marik, I.; et al. Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum. Mutat. 2011, 32, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Schilling, L.; Yee, S.P.; Lee, S.K.; Zanotti, S. Hajdu Cheney Mouse Mutants Exhibit Osteopenia, Increased Osteoclastogenesis, and Bone Resorption. J. Biol. Chem. 2016, 291, 1538–1551. [Google Scholar] [CrossRef] [PubMed]
- Lehman, R.A.; Stears, J.C.; Wesenberg, R.L.; Nusbaum, E.D. Familial osteosclerosis with abnormalities of the nervous system and meninges. J. Pediatr. 1977, 90, 49–54. [Google Scholar] [CrossRef]
- Gripp, K.W.; Scott, C.I., Jr.; Hughes, H.E.; Wallerstein, R.; Nicholson, L.; States, L.; Bason, L.D.; Kaplan, P.; Zderic, S.A.; Duhaime, A.C.; et al. Lateral meningocele syndrome: Three new patients and review of the literature. Am. J. Med. Genet. 1997, 70, 229–239. [Google Scholar] [CrossRef]
- Gripp, K.W.; Robbins, K.M.; Sobreira, N.L.; Witmer, P.D.; Bird, L.M.; Avela, K.; Makitie, O.; Alves, D.; Hogue, J.S.; Zackai, E.H.; et al. Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome. Am. J. Med. Genet. A 2015, 167, 271–281. [Google Scholar] [CrossRef]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef]
- Wuelling, M.; Vortkamp, A. Transcriptional networks controlling chondrocyte proliferation and differentiation during endochondral ossification. Pediatr. Nephrol. 2010, 25, 625–631. [Google Scholar] [CrossRef]
- Miraoui, H.; Marie, P.J. Fibroblast growth factor receptor signaling crosstalk in skeletogenesis. Sci. Signal. 2010, 3, 9. [Google Scholar] [CrossRef]
- Plaas, A.; Velasco, J.; Gorski, D.J.; Cole, A.; Christopherson, K.; Sandy, J.D. The relationship between fibrogenic TGFbeta1 signaling in the joint and cartilage degradation in post-injury osteoarthritis. Osteoarthr. Cartil. 2011, 19, 1081–1090. [Google Scholar] [CrossRef]
- Baldridge, D.; Shchelochkov, O.; Kelley, B.; Lee, B. Signaling pathways in human skeletal dysplasias. Annu. Rev. Genom. Hum. Genet. 2010, 11, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Tezuka, Y.; Matsuno, K.; Miyatani, S.; Morimura, N.; Yasuda, M.; Fujimaki, R.; Kuroda, K.; Hiraki, Y.; Hozumi, N.; et al. Suppression of differentiation and proliferation of early chondrogenic cells by Notch. J. Bone Miner. Metab. 2003, 21, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Jesse, A.M.; Kohn, A.; Gunnell, L.M.; Honjo, T.; Zuscik, M.J.; O’Keefe, R.J.; Hilton, M.J. RBPjkappa-dependent Notch signaling regulates mesenchymal progenitor cell proliferation and differentiation during skeletal development. Development 2010, 137, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, R.; Toyama, Y.; Hozumi, N.; Tezuka, K. Involvement of Notch signaling in initiation of prechondrogenic condensation and nodule formation in limb bud micromass cultures. J. Bone Miner. Metab. 2006, 24, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Grogan, S.P.; Olee, T.; Hiraoka, K.; Lotz, M.K. Repression of chondrogenesis through binding of notch signaling proteins HES-1 and HEY-1 to N-box domains in the COL2A1 enhancer site. Arthritis Rheum. 2008, 58, 2754–2763. [Google Scholar] [CrossRef]
- Mead, T.J.; Yutzey, K.E. Notch pathway regulation of chondrocyte differentiation and proliferation during appendicular and axial skeleton development. Proc. Natl. Acad. Sci. USA 2009, 106, 14420–14425. [Google Scholar] [CrossRef]
- Crowe, R.; Zikherman, J.; Niswander, L. Delta-1 negatively regulates the transition from prehypertrophic to hypertrophic chondrocytes during cartilage formation. Development 1999, 126, 987–998. [Google Scholar]
- Rutkowski, T.P.; Kohn, A.; Sharma, D.; Ren, Y.; Mirando, A.J.; Hilton, M.J. HES factors regulate specific aspects of chondrogenesis and chondrocyte hypertrophy during cartilage development. J. Cell Sci. 2016, 129, 2145–2155. [Google Scholar] [CrossRef]
- Karlsson, C.; Brantsing, C.; Kageyama, R.; Lindahl, A. HES1 and HES5 are dispensable for cartilage and endochondral bone formation. Cells Tissues Organs. 2010, 192, 17–27. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, Y.; Fu, Q.; Chang, M.; Wang, Y.; Shang, X.; Wan, C.; Marymont, J.V.; Dong, Y. Notch inhibits chondrogenic differentiation of mesenchymal progenitor cells by targeting Twist1. Mol. Cell Endocrinol. 2015, 403, 30–38. [Google Scholar] [CrossRef]
- Hilton, M.J.; Tu, X.; Wu, X.; Bai, S.; Zhao, H.; Kobayashi, T.; Kronenberg, H.M.; Teitelbaum, S.L.; Ross, F.P.; Kopan, R.; et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat. Med. 2008, 14, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Chen, J.; Lim, J.; Karner, C.M.; Lee, S.Y.; Heisig, J.; Wiese, C.; Surendran, K.; Kopan, R.; Gessler, M.; et al. Physiological notch signaling maintains bone homeostasis via RBPjk and Hey upstream of NFATc1. PLoS Genet. 2018, 8, e1002577. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Eberspaecher, H.; Lefebvre, V.; De Crombrugghe, B. Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev. Dyn. 1997, 209, 377–386. [Google Scholar] [CrossRef]
- Chen, S.; Tao, J.; Bae, Y.; Jiang, M.M.; Bertin, T.; Chen, Y.; Yang, T.; Lee, B. Notch gain of function inhibits chondrocyte differentiation via Rbpj-dependent suppression of Sox9. J. Bone Miner. Res. 2013, 28, 649–659. [Google Scholar] [CrossRef]
- Dahlqvist, C.; Blokzijl, A.; Chapman, G.; Falk, A.; Dannaeus, K.; Ibâñez, C.F.; Lendahl, U. Functional Notch signaling is required for BMP4-induced inhibition of myogenic differentiation. Development 2003, 130, 6089–6099. [Google Scholar] [CrossRef]
- Itoh, F.; Itoh, S.; Goumans, M.J.; Valdimarsdottir, G.; Iso, T.; Dotto, G.P.; Hamamori, Y.; Kedes, L.; Kato, M.; ten Dijke, P. Synergy and antagonism between Notch and BMP receptor signaling pathways in endothelial cells. EMBO J. 2004, 23, 541–551. [Google Scholar] [CrossRef]
- Blokzijl, A.; Dahlqvist, C.; Reissmann, E.; Falk, A.; Moliner, A.; Lendahl, U.; Ibáñez, C.F. Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J. Cell Biol. 2003, 163, 723–728. [Google Scholar] [CrossRef]
- Ross, D.A.; Kadesch, T. The notch intracellular domain can function as a coactivator for LEF-1. Mol. Cell Biol. 2001, 21, 7537–7544. [Google Scholar] [CrossRef]
- Axelrod, J.D.; Matsuno, K.; Artavanis-Tsakonas, S.; Perrimon, N. Interaction between Wingless and Notch signaling pathways mediated by dishevelled. Science 1996, 271, 1826–1832. [Google Scholar] [CrossRef]
- Hayward, P.; Brennan, K.; Sanders, P.; Balayo, T.; DasGupta, R.; Perrimon, N.; Arias, A.M. Notch modulates Wnt signalling by associating with Armadillo/beta-catenin and regulating its transcriptional activity. Development 2005, 132, 1819–1830. [Google Scholar] [CrossRef]
- Vacca, A.; Felli, M.P.; Palermo, R.; Di Mario, G.; Calce, A.; Di Giovine, M.; Frati, L.; Gulino, A.; Screpanti, I. Notch3 and pre-TCR interaction unveils distinct NF-kappaB pathways in T-cell development and leukemia. EMBO J. 2006, 25, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Vilimas, T.; Mascarenhas, J.; Palomero, T.; Mandal, M.; Buonamici, S.; Meng, F.; Thompson, B.; Spaulding, C.; Macaroun, S.; Alegre, M.L. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat. Med. 2007, 13, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shelly, L.; Miele, L.; Boykins, R.; Norcross, M.A.; Guan, E. Human Notch-1 inhibits NF-kappa B activity in the nucleus through a direct interaction involving a novel domain. J. Immunol. 2001, 167, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Kohn, A.; Dong, Y.; Mirando, A.J.; Jesse, A.M.; Honjo, T.; Zuscik, M.J.; O’Keefe, R.J.; Hilton, M.J. Cartilage-specific RBPjkappa-dependent and -independent Notch signals regulate cartilage and bone development. Development 2012, 139, 1198–1212. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Goldring, S.R. Osteoarthritis. J. Cell Physiol. 2007, 213, 626–634. [Google Scholar] [CrossRef]
- Goldring, M.B.; Marcu, K.B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 2009, 11, 224. [Google Scholar] [CrossRef]
- Pacifici, M.; Koyama, E.; Iwamoto, M. Mechanisms of synovial joint and articular cartilage formation: Recent advances, but many lingering mysteries. Birth Defects Res. C Embryo Today 2005, 75, 237–248. [Google Scholar] [CrossRef]
- Sandell, L.J. Etiology of osteoarthritis: Genetics and synovial joint development. Nat. Rev. Rheumatol. 2012, 8, 77–89. [Google Scholar] [CrossRef]
- Karlsson, C.; Jonsson, M.; Asp, J.; Brantsing, C.; Kageyama, R.; Lindahl, A. Notch and HES5 are regulated during human cartilage differentiation. Cell Tissue Res. 2007, 327, 539–551. [Google Scholar] [CrossRef]
- Ustunel, I.; Ozenci, A.M.; Sahin, Z.; Ozbey, O.; Acar, N.; Tanriover, G.; Celik-Ozenci, C.; Demir, R. The immunohistochemical localization of notch receptors and ligands in human articular cartilage, chondroprogenitor culture and ultrastructural characteristics of these progenitor cells. Acta Histochem. 2008, 110, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, C.; Brantsing, C.; Egell, S.; Lindahl, A. Notch1, Jagged1, and HES5 are abundantly expressed in osteoarthritis. Cells Tissues Organs. 2008, 188, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, J.; Mirando, A.J.; Wang, C.; Zuscik, M.J.; O’Keefe, R.J.; Hilton, M.J. A dual role for NOTCH signaling in joint cartilage maintenance and osteoarthritis. Sci. Signal 2015, 8, ra71. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, Y.; Saito, T.; Sugita, S.; Hikata, T.; Kobayashi, H.; Fukai, A.; Taniguchi, Y.; Hirata, M.; Akiyama, H.; Chung, U.I.; et al. Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proc. Natl. Acad. Sci. USA 2013, 110, 1875–1880. [Google Scholar] [CrossRef]
- Mirando, A.J.; Liu, Z.; Moore, T.; Lang, A.; Kohn, A.; Osinski, A.M.; O’Keefe, R.J.; Mooney, R.A.; Zuscik, M.J.; Hilton, M.J. RBP-Jkappa-dependent Notch signaling is required for murine articular cartilage and joint maintenance. Arthritis Rheum. 2013, 65, 2623–2633. [Google Scholar]
- Canalis, E.; Parker, K.; Feng, J.Q.; Zanotti, S. Osteoblast lineage-specific effects of notch activation in the skeleton. Endocrinology 2013, 154, 623–634. [Google Scholar] [CrossRef]
- Zanotti, S.; Smerdel-Ramoya, A.; Stadmeyer, L.; Durant, D.; Radtke, F.; Canalis, E. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology 2008, 149, 3890–3899. [Google Scholar] [CrossRef]
- Lawal, R.A.; Zhou, X.; Batey, K.; Hoffman, C.M.; Georger, M.A.; Radtke, F.; Hilton, M.J.; Xing, L.; Frisch, B.J.; Calvi, L.M. The Notch Ligand Jagged1 Regulates the Osteoblastic Lineage by Maintaining the Osteoprogenitor Pool. J. Bone Miner. Res. 2017, 32, 1320–1331. [Google Scholar] [CrossRef]
- Muguruma, Y.; Hozumi, K.; Warita, H.; Yahata, T.; Uno, T.; Ito, M.; Ando, K. Maintenance of Bone Homeostasis by DLL1-Mediated Notch Signaling. J. Cell Physiol. 2017, 232, 2569–2580. [Google Scholar] [CrossRef]
- Engin, F.; Yao, Z.; Yang, T.; Zhou, G.; Bertin, T.; Jiang, M.M.; Chen, Y.; Wang, L.; Zheng, H.; Sutton, R.E.; et al. Dimorphic effects of Notch signaling in bone homeostasis. Nat Med 2008, 14, 299–305. [Google Scholar] [CrossRef]
- Canalis, E.; Adams, D.J.; Boskey, A.; Parker, K.; Kranz, L.; Zanotti, S. Notch signaling in osteocytes differentially regulates cancellous and cortical bone remodeling. J. Biol. Chem. 2013, 288, 25614–25625. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Bridgewater, D.; Schilling, L.; Zanotti, S. Canonical Notch activation in osteocytes causes osteopetrosis. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E171–E182. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Chen, S.; Lee, B. Alteration of Notch signaling in skeletal development and disease. Ann. N. Y. Acad. Sci. 2010, 1192, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Engin, F.; Lee, B. NOTCHing the bone: Insights into multi-functionality. Bone 2010, 46, 274–280. [Google Scholar] [CrossRef]
- Barak, H.; Surendran, K.; Boyle, S.C. The role of Notch signaling in kidney development and disease. Adv. Exp. Med. Biol. 2017, 727, 99–113. [Google Scholar]
- Chen, S.; Lee, B.H.; Bae, Y. Notch signaling in skeletal stem cells. Calcif. Tissue Int. 2014, 94, 68–77. [Google Scholar] [CrossRef]
- Canalis, E. Notch in skeletal physiology and disease. Osteoporos. Int 2018, 29, 2611–2621. [Google Scholar] [CrossRef]
- Lee, S.Y.; Long, F. Notch signaling suppresses glucose metabolism in mesenchymal progenitors to restrict osteoblast differentiation. J. Clin. Invest. 2018, 128, 5573–5586. [Google Scholar] [CrossRef]
- Ziouti, F.; Ebert, R.; Rummler, M.; Krug, M.; Müller-Deubert, S.; Lüdemann, M.; Jakob, F.; Willie, B.M.; Jundt, F. NOTCH Signaling Is Activated through Mechanical Strain in Human Bone Marrow-Derived Mesenchymal Stromal Cells. Stem. Cells Int. 2019, 2019, 5150634. [Google Scholar] [CrossRef]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Lüthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Sekine, C.; Koyanagi, A.; Koyama, N.; Hozumi, K.; Chiba, S.; Yagita, H. Differential regulation of osteoclastogenesis by Notch2/Delta-like 1 and Notch1/Jagged1 axes. Arthritis Res. Ther. 2012, 14, R45. [Google Scholar] [CrossRef]
- Yamada, T.; Yamazaki, H.; Yamane, T.; Yoshino, M.; Okuyama, H.; Tsuneto, M.; Kurino, T.; Hayashi, S.; Sakano, S. Regulation of osteoclast development by Notch signaling directed to osteoclast precursors and through stromal cells. Blood 2003, 101, 2227–2234. [Google Scholar] [CrossRef]
- Fukushima, H.; Nakao, A.; Okamoto, F.; Shin, M.; Kajiya, H.; Sakano, S.; Bigas, A.; Jimi, E.; Okabe, K. The association of Notch2 and NF-kappaB accelerates RANKL-induced osteoclastogenesis. Mol. Cell Biol. 2008, 28, 6402–6412. [Google Scholar] [CrossRef]
- Bai, S.; Kopan, R.; Zou, W.; Hilton, M.J.; Ong, C.T.; Long, F.; Ross, F.P.; Teitelbaum, S.L. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J. Biol. Chem. 2008, 283, 6509–6518. [Google Scholar] [CrossRef]
- Ashley, J.W.; Ahn, J.; Hankenson, K.D. Notch signaling promotes osteoclast maturation and resorptive activity. J. Cell Biochem. 2015, 116, 2598–2609. [Google Scholar] [CrossRef]
- Zhao, B.; Grimes, S.N.; Li, S.; Hu, X.; Ivashkiv, L.B. TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. J. Exp. Med. 2012, 209, 319–334. [Google Scholar] [CrossRef]
- Zanotti, S.; Smerdel-Ramoya, A.; Canalis, E. HES1 (hairy and enhancer of split 1) is a determinant of bone mass. J. Biol. Chem. 2010, 286, 2648–2657. [Google Scholar] [CrossRef]
- Zanotti, S.; Yu, J.; Sanjay, A.; Shilling, L.; Schoenherr, C.; Economides, A.N.; Canalis, E. Sustained Notch2 signaling in osteoblasts, but not in osteoclasts, is linked to osteopenia in a mouse model of Hajdu-Cheney syndrome. J. Biol. Chem. 2017, 292, 12232–12244. [Google Scholar] [CrossRef]
- Dishowitz, M.I.; Terkhorn, S.P.; Bostic, S.A.; Hankenson, K.D. Notch signaling components are upregulated during both endochondral and intramembranous bone regeneration. J. Orthop. Res. 2013, 30, 296–303. [Google Scholar] [CrossRef]
- Dishowitz, M.I.; Mutyaba, P.L.; Takacs, J.D.; Barr, A.M.; Engiles, J.B.; Ahn, J.; Hankenson, K.D. Systemic inhibition of canonical Notch signaling results in sustained callus inflammation and alters multiple phases of fracture healing. PLoS ONE 2013, 8, e68726. [Google Scholar] [CrossRef]
- Wang, C.; Inzana, J.A.; Mirando, A.J.; Ren, Y.; Liu, Z.; Shen, J.; O’Keefe, R.J.; Awad, H.A.; Hilton, M.J. NOTCH signaling in skeletal progenitors is critical for fracture repair. J. Clin. Invest. 2016, 126, 1471–1481. [Google Scholar] [CrossRef]
- Matthews, B.G.; Grcevic, D.; Wang, L.; Hagiwara, Y.; Roguljic, H.; Joshi, P.; Shin, D.G.; Adams, D.J.; Kalajzic, I. Analysis of alphaSMA-labeled progenitor cell commitment identifies notch signaling as an important pathway in fracture healing. J. Bone Miner. Res. 2014, 29, 1283–1294. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, Y.; Xue, T.; Chen, L.; Shi, B.; Shu, B.; Xie, C.; Max Morandi, M.; Jaeblon, T.; Marymount, J.V.; et al. Notch activation enhances mesenchymal stem cell sheet osteogenic potential by inhibition of cellular senescence. Cell Death Dis. 2017, 8, e2595. [Google Scholar] [CrossRef]
- Youngstrom, D.W.; Senos, R.; Zondervan, R.L.; Brodeur, J.D.; Lints, A.R.; Young, D.R.; Mitchell, T.L.; Moore, M.E.; Myers, M.H.; Tseng, W.J.; et al. Intraoperative delivery of the Notch ligand Jagged-1 regenerates appendicular and craniofacial bone defects. NPJ Regen. Med. 2017, 2, 32. [Google Scholar] [CrossRef]
- Ramasamy, S.K.; Kusumbe, A.P.; Wang, L.; Adams, R.H. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 2014, 507, 376–380. [Google Scholar] [CrossRef]
- Benedito, R.; Hellstrom, M. Notch as a hub for signaling in angiogenesis. Exp Cell Res. 2013, 319, 1281–1288. [Google Scholar] [CrossRef]
- Hausman, M.R.; Schaffler, M.B.; Majeska, R.J. Prevention of fracture healing in rats by an inhibitor of angiogenesis. Bone 2001, 29, 560–564. [Google Scholar] [CrossRef]
- Engin, F.; Bertin, T.; Ma, O.; Jiang, M.M.; Wang, L.; Sutton, R.E.; Donehower, L.A.; Lee, B. Notch signaling contributes to the pathogenesis of human osteosarcomas. Hum. Mol. Genet. 2009, 18, 1464–1470. [Google Scholar] [CrossRef]
- Tao, J.; Jiang, M.M.; Jiang, L.; Salvo, J.S.; Zeng, H.C.; Dawson, B.; Bertin, T.K.; Rao, P.H.; Chen, R.; Donehower, L.A.; et al. Notch activation as a driver of osteogenic sarcoma. Cancer Cell 2014, 26, 390–401. [Google Scholar] [CrossRef]
- Hughes, D.P. How the NOTCH pathway contributes to the ability of osteosarcoma cells to metastasize. Cancer Treat. Res. 2009, 152, 479–496. [Google Scholar]
- Tanaka, M.; Setoguchi, T.; Hirotsu, M.; Gao, H.; Sasaki, H.; Matsunoshita, Y.; Komiya, S. Inhibition of Notch pathway prevents osteosarcoma growth by cell cycle regulation. Br. J. Cancer 2009, 100, 1957–1965. [Google Scholar] [CrossRef]
- Vermezovic, J.; Adamowicz, M.; Santarpia, L.; Rustighi, A.; Forcato, M.; Lucano, C.; Massimiliano, L.; Costanzo, V.; Bicciato, S.; Del Sal, G.; et al. Notch is a direct negative regulator of the DNA-damage response. Nat. Struct. Mol. Biol. 2015, 22, 417–424. [Google Scholar] [CrossRef]
- Adamowicz, M.; Vermezovic, J.; di Fagagna, F.D. NOTCH1 Inhibits Activation of ATM by Impairing the Formation of an ATM-FOXO3a-KAT5/Tip60 Complex. Cell Rep. 2016, 6, 2068–2076. [Google Scholar] [CrossRef]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2018, 27, 193–210. [Google Scholar] [CrossRef]
- Cappariello, A.; Loftus, A.; Muraca, M.; Maurizi, A.; Rucci, N.; Teti, A. Osteoblast-Derived Extracellular Vesicles Are Biological Tools for the Delivery of Active Molecules to Bone. J. Bone Miner. Res. 2018, 33, 517–533. [Google Scholar] [CrossRef]
- Cook, L.M.; Shay, G.; Araujo, A.; Lynch, C.C. Integrating new discoveries into the “vicious cycle” paradigm of prostate to bone metastases. Cancer Metastasis Rev. 2015, 33, 511–525. [Google Scholar] [CrossRef]
- Santagata, S.; Demichelis, F.; Riva, A.; Varambally, S.; Hofer, M.D.; Kutok, J.L.; Kim, R.; Tang, J.; Montie, J.E.; Chinnaiyan, A.M.; et al. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res. 2004, 64, 6854–6857. [Google Scholar] [CrossRef]
- Zayzafoon, M.; Abdulkadir, S.A.; McDonald, J.M. Notch signaling and ERK activation are important for the osteomimetic properties of prostate cancer bone metastatic cell lines. J. Biol. Chem. 2004, 279, 3662–3670. [Google Scholar] [CrossRef]
- Zheng, H.; Bae, Y.; Kasimir-Bauer, S.; Tang, R.; Chen, J.; Ren, G.; Yuan, M.; Esposito, M.; Li, W.; Wei, Y.; et al. Therapeutic Antibody Targeting Tumor- and Osteoblastic Niche-Derived Jagged1 Sensitizes Bone Metastasis to Chemotherapy. Cancer Cell 2017, 32, 731–747. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Mutation | Notch Effect | Symptoms |
|---|---|---|---|
| Adams Oliver Syndrome | NOTCH1 DLL4 RBPJK | Loss of function | Underdeveloped skull and absent or scarred skin (Aplasia cutis congenital), mild to severe limb defect (Terminal transverse limb defects), cardiovascular malformations/dysfunctions, brain anomalies, and less frequent renal, liver and eye anomalies |
| Alagille Syndrome | JAG1 NOTCH2 | Loss of function | Multisystem disorder with a wide spectrum of clinical variability; bile duct paucity, cholestasis, cardiac defect, butterfly vertebrae, craniosynostosis and characteristic facial features, low bone mass and increased fracture incidence |
| Spondylocostal Dysostosis | DLL3 MESP2 HES7 LFNG | Loss of function | Vertebral segmentation defects, rib abnormalities |
| Hajdu-Cheney Syndrome | NOTCH2 | Gain of function | Short stature, coarse and dysmorphic facies, bowing of long bones, and vertebral anomalies; focal bone destruction (acroosteolysis) and osteoporosis |
| Lateral Meningocele Syndrome | NOTCH3 | Gain of function | Facial anomalies, hypotonia, meningocele, short stature, scoliosis, Wormian bones, and thick calvariae |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zieba, J.T.; Chen, Y.-T.; Lee, B.H.; Bae, Y. Notch Signaling in Skeletal Development, Homeostasis and Pathogenesis. Biomolecules 2020, 10, 332. https://doi.org/10.3390/biom10020332
Zieba JT, Chen Y-T, Lee BH, Bae Y. Notch Signaling in Skeletal Development, Homeostasis and Pathogenesis. Biomolecules. 2020; 10(2):332. https://doi.org/10.3390/biom10020332
Chicago/Turabian StyleZieba, Jennifer T., Yi-Ting Chen, Brendan H. Lee, and Yangjin Bae. 2020. "Notch Signaling in Skeletal Development, Homeostasis and Pathogenesis" Biomolecules 10, no. 2: 332. https://doi.org/10.3390/biom10020332
APA StyleZieba, J. T., Chen, Y.-T., Lee, B. H., & Bae, Y. (2020). Notch Signaling in Skeletal Development, Homeostasis and Pathogenesis. Biomolecules, 10(2), 332. https://doi.org/10.3390/biom10020332