Progress in the Use of Antisense Oligonucleotides for Vaccine Improvement

 ,
,  , ,
, ,  ,
,
Abstract
1. Introduction
1.1. Earlier Uses of Oligonucleotides in Vaccines
1.2. Birth of ASOs
2. Molecular Basis of ASOs
2.1. Pharmacokinetics
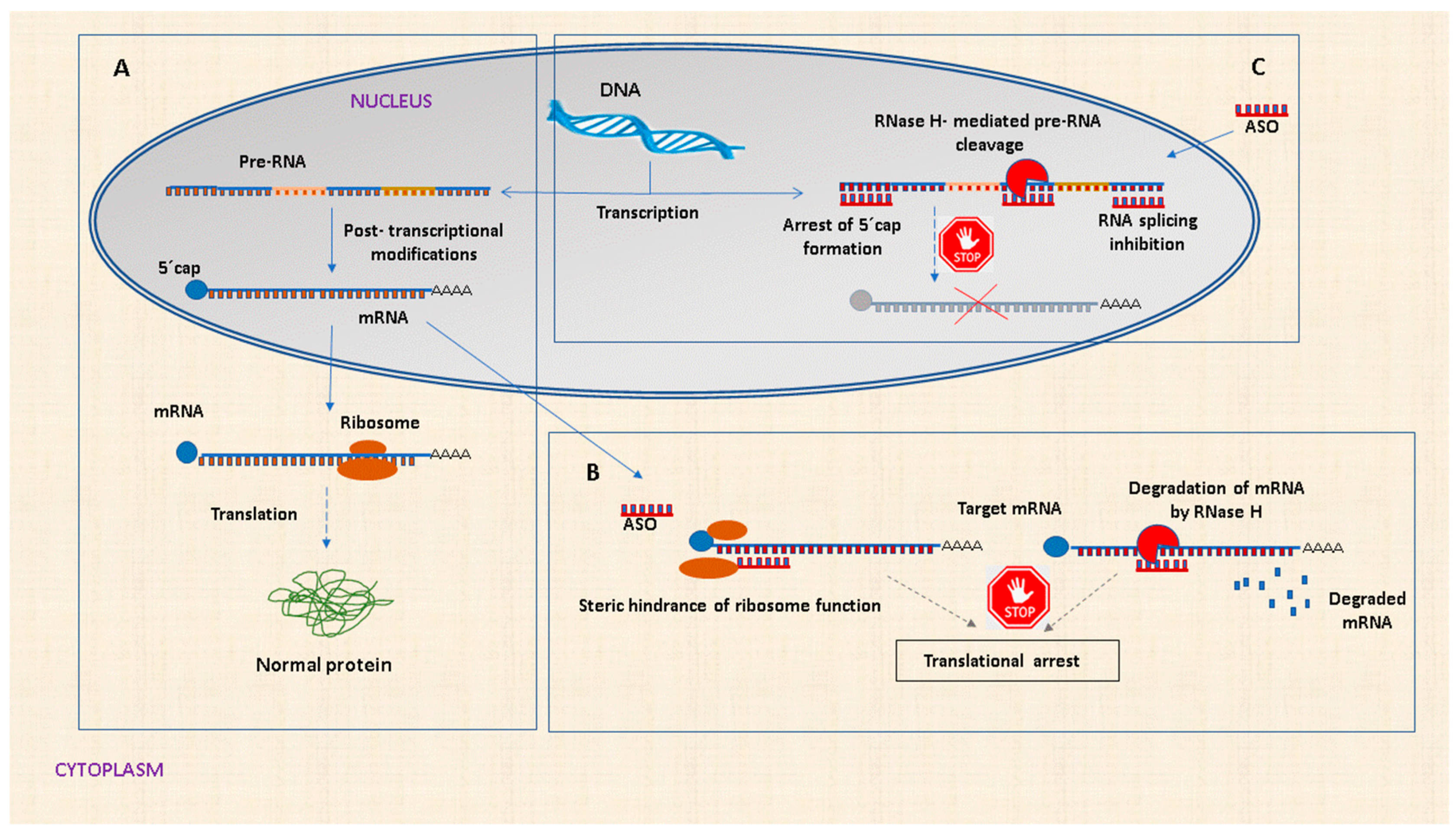
2.2. Mechanism of Action of ASOs
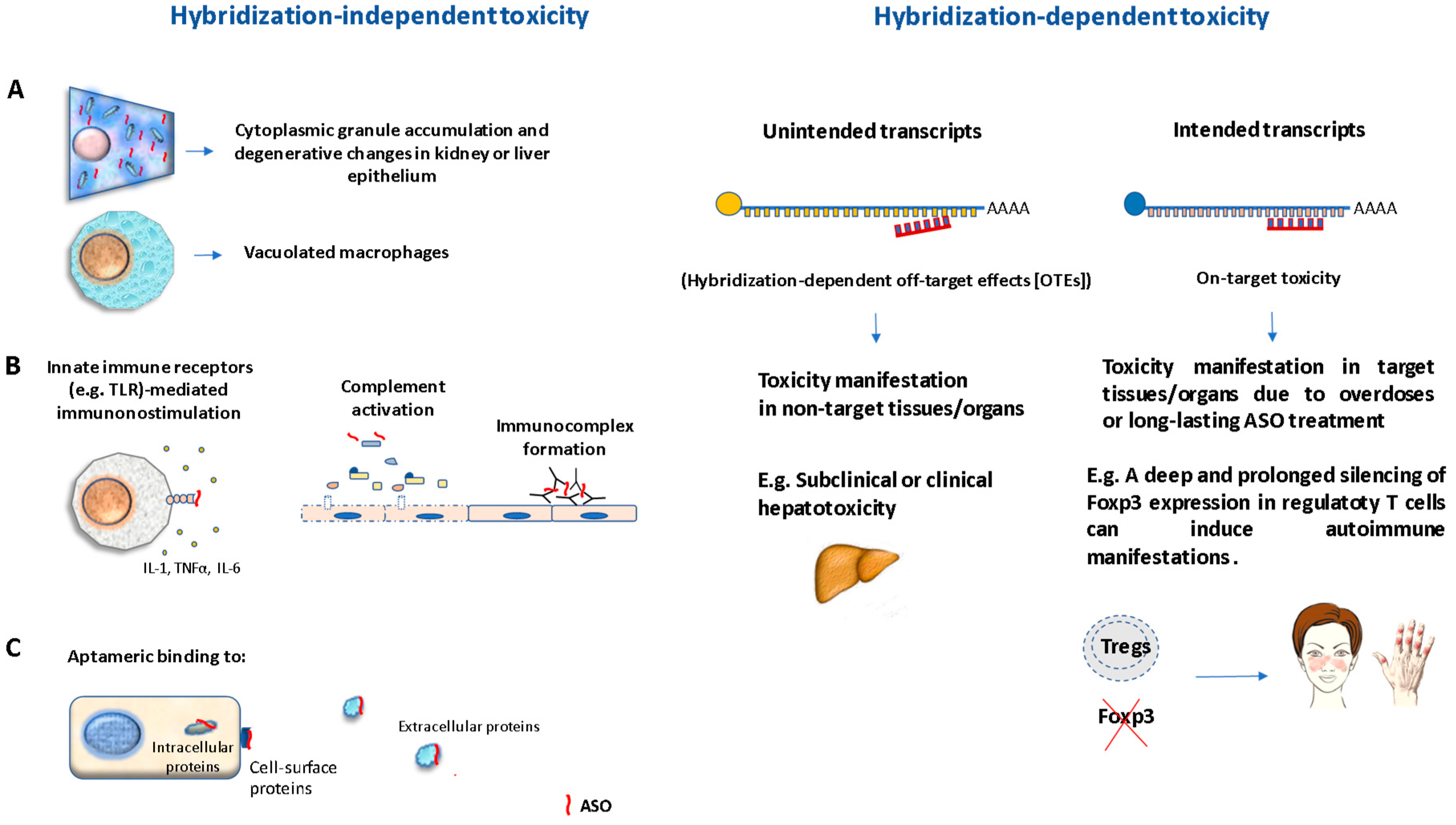
2.3. Toxicology of ASOs
2.4. Strategies to Improve ASOs Cell Targeting and Overcome Toxicity
3. ASOs in Vaccines
3.1. Antigen Modification
3.2. Targeting Host Immune Mechanisms
ASOs as Vaccine Adjuvants in Subunit Vaccines
4. Challenges and Opportunities for ASOs Application in Vaccinology
- Discovery of new suitable genes to improve vaccine protective immunogenicity against specific infectious or tumoral disease using ASOs.
- Development of bioinformatic tools and in vitro systems for ASOs screening to vaccine application.
- Discovery of delivery systems that can promote effective ASOs cellular uptake in the immune system.
- Studies of stability and antigen-ASOs compatibility in vaccine formulations.
- Immunotoxicity studies to discover potential consequences of immune overstimulation.
- Studies of efficacy/safety in different genetic contexts.
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Rappuoli, R.; Mandl, C.W.; Black, S.; De Gregorio, E. Vaccines for the twenty-first society. Nature Rev. Immunol. 2011, 11, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Finco, O.; Rappuoli, R. Designing vaccines for the twenty-first century society. Front. Immunol. 2014, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Afrough, B.; Dowall, S.; Hewson, R. Emerging viruses and current strategies for vaccine intervention. Clin. Exp. Immunol. 2019, 196, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; Jasny, E.; Schmidt, K.E.; Petsch, B. New Vaccine Technologies to Combat Outbreak Situations. Front. Immunol. 2018, 9, 1963. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Grandi, G.; Rappuoli, R. Structural vaccinology starts to deliver. Nat. Rev. Microbiol. 2012, 10, 807–813. [Google Scholar] [CrossRef]
- Myhr, A.I. DNA Vaccines: Regulatory Considerations and Safety Aspects. Curr. Issues Mol. Biol. 2017, 22, 79–88. [Google Scholar] [CrossRef]
- Ghaffarifar, F. Plasmid DNA vaccines: Where are we now? Drugs Today (Barc) 2018, 54, 315–333. [Google Scholar] [CrossRef]
- Geall, A.J.; Mandl, C.W.; Ulmer, J.B. RNA: The new revolution in nucleic acid vaccines. Semin. Immunol. 2013, 25, 152–159. [Google Scholar] [CrossRef]
- Kramps, T.; Elbers, K. Introduction to RNA Vaccines. Methods Mol. Biol. 2017, 1499, 1–11. [Google Scholar]
- Bode, C.; Zhao, G.; Steinhagen, F.; Kinjo, T.; Klinman, D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 2011, 10, 499–511. [Google Scholar] [CrossRef]
- Scheiermann, J.; Klinman, D.M. Clinical evaluation of CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases and cancer. Vaccine 2014, 32, 6377–6389. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Bacon, T.A.; Wickstrom, E. Oligodeoxynucleoside phosphorothioate stability in subcellular extracts, culture media, sera and cerebrospinal fluid. J. Biochem. Biophys. Methods 1990, 20, 259–267. [Google Scholar] [CrossRef]
- Hyjek, M.; Figiel, M.; Nowotny, M. RNases H: Structure and mechanism. DNA Repair (Amst). 2019, 84, 102672. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef]
- Coley, W.B. The treatment of malignant tumors by repeated inoculations of Erysipelas, with a report of ten original cases. Am. J. Med. Sci. 1893, 105, 487–511. [Google Scholar] [CrossRef]
- Taliaferro, W.H.; Jaroslow, B.N. The restoration of hemolysin formation in x-rayed rabbits by nucleic acid derivatives and antagonists of nucleic acid synthesis. J. Infect. Dis. 1960, 107, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Reist, E.J.; Benitez, A.; Goodman, L. The synthesis of some 5′-thiopentofuranosylpyrimidines. J. Org. Chem. 1964, 29, 554–558. [Google Scholar] [CrossRef]
- Codington, J.F.; Doerr, I.L.; Fox, J.J. Nucleosides. XVIII. Synthesis of 2’-fluorothymidine, 2’-fluorodeoxyuridine, and other 2’-halogeno-2¢-deoxy nucleosides. J. Org. Chem. 1964, 29, 558–564. [Google Scholar] [CrossRef]
- Eckstein, F. Nucleoside phosphorothioates. J. Am. Chem. Soc. 1966, 88, 4292–4294. [Google Scholar] [CrossRef]
- Bobst, A.M.; Rottman, F.; Cerutti, P.A. Effect of the methylation of the 2’-hydroxyl groups in polyadenylic acid on its structure in weakly acidic and neutral solutions and on its capability to form ordered complexes with polyuridylic acid. J. Mol. Biol. 1969, 46, 221–234. [Google Scholar] [CrossRef]
- Braun, W.; Nakano, M. Influence of oligodeoxyribonucleotides on early events in antibody formation. Proc. Soc. Exp. Biol. Med. 1965, 119, 701–707. [Google Scholar] [CrossRef]
- Braun, W.; Nakano, M. Antibody formation: Stimulation by polyadenylic and polycytidylic acids. Science 1967, 157, 819–821. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, A.D.; Baron, S.; Talal, N. The pathogenesis of autoimmunity in New Zealand mice, I. Induction of antinucleic acid antibodies by polyinosinic-polycytidylic acid. Proc. Natl. Acad. Sci. USA 1969, 63, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Field, A.K.; Tytell, A.A.; Lampson, G.P.; Hilleman, M.R. Inducers of interferon and host resistance. II. Multistranded synthetic polynucleotide complexes. Proc. Natl. Acad. Sci. USA 1967, 58, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Tokunaga, T.; Yamamoto, H.; Shimada, S.; Abe, H.; Fukuda, T.; Fujisawa, Y.; Furutani, Y.; Yano, O.; Kataoka, T.; Sudo, T.; et al. Antitumor activity of deoxyribonucleic acid fraction from Mycobacterium bovis BCG. I. Isolation, physicochemical characterization, and antitumor activity. J. Natl. Cancer Inst. 1984, 72, 955–962. [Google Scholar]
- Yamamoto, S.; Yamamoto, T.; Shimada, S.; Kuramoto, E.; Yano, O.; Kataoka, T.; Tokunaga, T. DNA from bacteria, but not from vertebrates, induces interferons, activates natural killer cells and inhibits tumor growth. Microbiol. Immunol. 1992, 36, 983–997. [Google Scholar] [CrossRef]
- Kuramoto, E.; Yano, O.; Kimura, Y.; Baba, M.; Makino, T.; Yamamoto, S.; Yamamoto, T.; Kataoka, T.; Tokunaga, T. Oligonucleotide sequences required for natural killer cell activation. Jpn. J. Cancer Res. 1992, 83, 1128–1131. [Google Scholar] [CrossRef]
- Krieg, A.M.; Yi, A.K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546–549. [Google Scholar] [CrossRef]
- Vollmer, J.; Krieg, A.M. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv. Drug Deliv. Rev. 2009, 61, 195–204. [Google Scholar] [CrossRef]
- Campbell, J.D. Development of the CpG adjuvant 1018: A case study. Methods Mol. Biol. 2017, 1494, 15–27. [Google Scholar] [PubMed]
- Krieg, A.M. CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 2002, 20, 709–760. [Google Scholar] [CrossRef] [PubMed]
- Sacher, T.; Knolle, P.; Nichterlein, T.; Arnold, B.; Hämmerling, G.J.; Limmer, A. CpG-ODN-induced inflammation is sufficient to cause T-cell-mediated autoaggression against hepatocytes. Eur. J. Immunol. 2002, 32, 3628–3637. [Google Scholar] [CrossRef]
- Tadema, H.; Abdulahad, W.H.; Lepse, N.; Stegeman, C.A.; Kallenberg, C.G.; Heeringa, P. Bacterial DNA motifs trigger ANCA production in ANCA-associated vasculitis in remission. Rheumatology (Oxford) 2011, 50, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Guerrier, T.; Youinou, P.; Pers, J.O.; Jamin, C. TLR9 drives the development of transitional B cells towards the marginal zone pathway and promotes autoimmunity. J. Autoimmun. 2012, 39, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef]
- Donis-Keller, H. Site specific enzymatic cleavage of RNA. Nucleic Acids Res. 1979, 7, 179–192. [Google Scholar] [CrossRef]
- Simons, R.W.; Kleckner, N. Translational control of IS10 transposition. Cell 1983, 34, 683–691. [Google Scholar] [CrossRef]
- Izant, J.G.; Weintraub, H. Inhibition of thymidine kinase gene expression by anti-sense RNA: A molecular approach to genetic analysis. Cell 1984, 36, 1007–1015. [Google Scholar] [CrossRef]
- Harland, R.; Weintraub, H. Translation of mRNA injected into Xenopus oocytes is specifically inhibited by antisense RNA. J. Cell Biol. 1985, 101, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Melton, D.A. Injected anti-sense RNAs specifically block messenger RNA translation in vivo. Proc. Natl. Acad. Sci. USA 1985, 82, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Matsukura, M.; Zon, G.; Shinozuka, K.; Robert-Guroff, M.; Shimada, T.; Stein, C.A.; Mitsuya, H.; Wong-Staal, F.; Cohen, J.S.; Broder, S. Regulation of viral expression of human immunodeficiency virus in vitro by an antisense phosphorothioate oligodeoxynucleotide against rev (art/trs) in chronically infected cells. Proc. Natl. Acad. Sci. USA 1989, 86, 4244–4248. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.D.; Biernat, J.; McManus, J.; Köster, H. Polymer support oligonucleotide synthesis XVIII: Use of beta-cyanoethyl-N,N-dialkylamino-/N-morpholino phosphoramidite of deoxynucleosides for the synthesis of DNA fragments simplifying deprotection and isolation of the final product. Nucleic Acids Res. 1984, 12, 4539–4557. [Google Scholar] [CrossRef] [PubMed]
- Usman, N.; Ogilvie, K.K.; Jiang, M.Y.; Cedergren, R.J. The automated chemical synthesis of long oligoribuncleotides using 2’-O-silylated ribonucleoside 3’-O-phosphoramidites on a controlled-pore glass support: Synthesis of a 43-nucleotide sequence similar to the 3’-half molecule of an Escherichia coli formylmethionine tRNA. J. Am. Chem. Soc. 1987, 109, 7845–7854. [Google Scholar]
- Walder, J.A.; Walder, R.Y. Nucleic acid hybridization and amplification method for detection of specific sequences in which a complementary labeled nucleic acid probe is cleaved. U.S. Patent 5,403,711, 4 April 1995. [Google Scholar]
- Dias, N.; Stein, C.A. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Roehr, B. Fomivirsen approved for CMV retinitis. J. Int. Assoc. Physicians AIDS Care 1998, 4, 14–16. [Google Scholar]
- Wong, E.; Goldberg, T. Mipomersen (kynamro): A novel antisense oligonucleotide inhibitor for the management of homozygous familial hypercholesterolemia. P. T. 2014, 39, 119–122. [Google Scholar]
- Aartsma-Rus, A. FDA Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid Ther. 2017, 27, 67–69. [Google Scholar] [CrossRef]
- Yu, E.Y.; Ellard, S.L.; Hotte, S.J.; Gingerich, J.R.; Joshua, A.M.; Gleave, M.E.; Chi, K.N. A randomized phase 2 study of a HSP27 targeting antisense, apatorsen with prednisone versus prednisone alone, in patients with metastatic castration resistant prostate cancer. Invest. New Drugs 2018, 36, 278–287. [Google Scholar] [CrossRef]
- Spigel, D.R.; Shipley, D.L.; Waterhouse, D.M.; Jones, S.F.; Ward, P.J.; Shih, K.C.; Hemphill, B.; McCleod, M.; Whorf, R.C.; Page, R.D.; et al. A Randomized, Double-Blinded, Phase II Trial of Carboplatin and Pemetrexed with or without Apatorsen (OGX-427) in Patients with Previously Untreated Stage IV Non-Squamous-Non-Small-Cell Lung Cancer: The SPRUCE Trial. Oncologist 2019, 24, e1409–e1416. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Zhang, Y.J. Antisense Phosphorodiamidate Morpholino Oligomers as Novel Antiviral Compounds. Front Microbiol. 2018, 9, 750. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef]
- Rüger, J.; Ioannou, S.; Castanotto, D.; Stein, C.A. Oligonucleotides to the (Gene) Rescue: FDA Approvals 2017-2019. Trends Pharmacol. Sci. 2020, 41, 27–41. [Google Scholar] [CrossRef]
- Yu, R.Z.; Grundy, J.S.; Geary, R.S. Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin. Drug Metab. Toxicol. 2013, 9, 169–182. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef]
- Gaus, H.J.; Gupta, R.; Chappell, A.E.; Østergaard, M.E.; Swayze, E.E.; Seth, P.P. Characterization of the interactions of chemically-modified therapeutic nucleic acids with plasma proteins using a fluorescence polarization assay. Nucleic Acids Res. 2019, 47, 1110–1122. [Google Scholar] [CrossRef]
- Moulton, J.D. Guide for morpholino users: Toward therapeutics. J. Drug Discov. Dev. Deliv. 2016, 3, 1023. [Google Scholar]
- Dirin, M.; Winkler, J. Influence of diverse chemical modifications on the ADME characteristics and toxicology of antisense oligonucleotides. Expert Opin. Biol. Ther. 2013, 13, 875–888. [Google Scholar] [CrossRef]
- Yu, R.Z.; Kim, T.W.; Hong, A.; Watanabe, T.A.; Gaus, H.J.; Geary, R.S. Cross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotide, ISIS 301012, targeting human apolipoprotein B-100. Drug Metab. Dispos. 2007, 35, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.M.; Tanowitz, M.; Donner, A.J.; Prakash, T.P.; Swayze, E.E.; Harris, E.N.; Seth, P.P. Receptor-Mediated Uptake of Phosphorothioate Antisense Oligonucleotides in Different Cell Types of the Liver. Nucleic Acid Ther. 2018, 28, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. Intracellular Trafficking and Endosomal Release of Oligonucleotides: What We Know and What We Don’t. Nucleic Acid Ther. 2018, 28, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Stecker, K.; Bennett, C.F. Cellular distribution of phosphorothioate oligodeoxynucleotides in normal rodent tissues. Lab. Invest. 1997, 77, 379–388. [Google Scholar] [PubMed]
- Bailey, J.K.; Shen, W.; Liang, X.H.; Crooke, S.T. Nucleic acid binding proteins affect the subcellular distribution of phosphorothioate antisense oligonucleotides. Nucleic Acids Res. 2017, 45, 10649–10671. [Google Scholar] [CrossRef]
- Crooke, S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017, 27, 70–77. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Gagnon, K.T.; Corey, D.R. Guidelines for experiments using antisense oligonucleotides and double-stranded RNAs. Nucleic Acid Ther. 2019, 29, 116–122. [Google Scholar] [CrossRef]
- Hagedorn, P.H.; Pontoppidan, M.; Bisgaard, T.S.; Berrera, M.; Dieckmann, A.; Ebeling, M.; Møller, M.R.; Hudlebusch, H.; Jensen, M.L.; Hansen, H.F.; et al. Identifying and avoiding off-target effects of RNase H-dependent antisense oligonucleotides in mice. Nucleic Acids Res. 2018, 46, 5366–5380. [Google Scholar] [CrossRef]
- Yoshida, T.; Naito, Y.; Sasaki, K.; Uchida, E.; Sato, Y.; Naito, M.; Kawanishi, T.; Obika, S.; Inoue, T. Estimated number of off-target candidate sites for antisense oligonucleotides in human mRNA sequences. Genes Cells 2018, 23, 448–455. [Google Scholar] [CrossRef]
- Frazier, K.S. Antisense oligonucleotide therapies: The promise and the challenges from a toxicologic pathologist’s perspective. Toxicol. Pathol. 2015, 43, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Temsamani, J.; Iadarola, P.L.; Jiang, Z.; Agrawal, S. Effect of different chemically modified oligodeoxynucleotides on immune stimulation. Biochem. Pharmacol. 1996, 51, 173–182. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; McNeil, S.E. Immunological and hematological toxicities challenging clinical translation of nucleic acid-based therapeutics. Expert Opin. Biol. Ther. 2015, 15, 1023–1048. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A. Controversies in the cellular pharmacology of oligodeoxynucleotides. Ciba Found. Symp. 1997, 209, 79–89. [Google Scholar] [CrossRef]
- Swayze, E.E.; Siwkowski, A.M.; Wancewicz, E.V.; Migawa, M.T.; Wyrzykiewicz, T.K.; Hung, G.; Monia, B.P.; Bennett, C.F. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 2007, 35, 687–700. [Google Scholar] [CrossRef]
- Burel, S.A.; Hart, C.E.; Cauntay, P.; Hsiao, J.; Machemer, T.; Katz, M.; Watt, A.; Bui, H.H.; Younis, H.; Sabripour, M.; et al. Hepatotoxicity of high affinity gapmer antisense oligonucleotides is mediated by RNase H1 dependent promiscuous reduction of very long pre-mRNA transcripts. Nucleic Acids Res. 2016, 44, 2093–2109. [Google Scholar] [CrossRef]
- Chi, X.; Gatti, P.; Papoian, T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef]
- Grijalvo, S.; Aviñó, A.; Eritja, R. Oligonucleotide delivery: A patent review (2010 - 2013). Expert Opin. Ther. Pat. 2014, 24, 801–819. [Google Scholar] [CrossRef]
- Grijalvo, S.; Puras, G.; Zárate, J.; Sainz-Ramos, M.; Qtaish, N.A.L.; López, T.; Mashal, M.; Attia, N.; Díaz, D.; Pons, R.; et al. Cationic Niosomes as Non-Viral Vehicles for Nucleic Acids: Challenges and Opportunities in Gene Delivery. Pharmaceutics 2019, 11, 50. [Google Scholar] [CrossRef]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; McNeil, S.E. Strategy for selecting nanotechnology carriers to overcome immunological and hematological toxicities challenging clinical translation of nucleic acid-based therapeutics. Expert Opin. Drug Deliv. 2015, 12, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Epa, W.R.; Rong, P.; Bartlett, P.F.; Coulson, E.J.; Barrett, G.L. Enhanced downregulation of the p75 nerve growth factor receptor by cholesteryl and bis-cholesteryl antisense oligonucleotides. Antisense Nucleic Acid Drug Dev. 1998, 8, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Allen, N.; Prakash, T.P.; Liang, X.H.; Crooke, S.T. Lipid Conjugates Enhance Endosomal Release of Antisense Oligonucleotides Into Cells. Nucleic Acid Ther. 2019, 29, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Ellipilli, S.; Murthy, R.; Ganesh, K.N. Perfluoroalkylchain conjugation as a new tactic for enhancing cell permeability of peptide nucleic acids (PNAs) via reducing the nanoparticle size. Chem. Commun. 2016, 52, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Godeau, G.; Arnion, H.; Brun, C.; Staedel, C.; Barthélémy, P. Fluorocarbon oligonucleotide conjugates for nucleic acids delivery. Med. Chem. Commun. 2010, 1, 76–78. [Google Scholar] [CrossRef]
- Tanowitz, M.; Hettrick, L.; Revenko, A.; Kinberger, G.A.; Prakash, T.P.; Seth, P.P. Asialoglycoprotein receptor 1 mediates productive uptake of N-acetylgalactosamine-conjugated and unconjugated phosphorothioate antisense oligonucleotides into liver hepatocytes. Nucleic Acids Res. 2017, 45, 12388–12400. [Google Scholar] [CrossRef]
- Derossi, D.; Chassaing, G.; Prochiantz, A. Trojan peptides: The penetrating system for intracellular delivery. Trends Cell Biol. 1998, 8, 84–87. [Google Scholar] [CrossRef]
- Vives, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef]
- Juliano, R.L.; Akhtar, S. Liposomes as a drug delivery system for antisense oligonucleotides. Antisense Res. Dev. 1992, 2, 165–176. [Google Scholar] [CrossRef]
- Fraley, R.; Straubinger, R.M.; Rule, G.; Springer, E.L.; Papahadjopoulos, D. Liposome-mediated delivery of deoxyribonucleic acid to cells: Enhanced efficiency of delivery related to lipid composition and incubation conditions. Biochemistry 1981, 20, 6978–6987. [Google Scholar] [CrossRef]
- Farhood, H.; Serbina, N.; Huang, L. The role of dioleoyl phosphatidylethanolamine in cationic liposome mediated gene transfer. Biochim. Biophys. Acta 1995, 1235, 289–295. [Google Scholar] [CrossRef]
- Felgner, J.H.; Kumar, R.; Sridhar, C.N.; Wheeler, C.J.; Tsai, Y.J.; Border, R.; Ramsey, P.; Martin, M.; Felgner, P.L. Enhanced gene delivery and mechanism studies with a novel series of cationic lipid formulations. J. Biol. Chem. 1994, 269, 2550–2561. [Google Scholar]
- Ma, D.D.; Wei, A.Q. Enhanced delivery of synthetic oligonucleotides to human leukaemic cells by liposomes and immunoliposomes. Leuk. Res. 1996, 20, 925–930. [Google Scholar] [CrossRef]
- Fattal, E.; Couvreur, P.; Dubernet, C. “Smart” delivery of antisense oligonucleotides by Clement resistant ovarian cancer cells (NCI/ADR-RES). Int. J. Pharm. 2012, 431, 222–229. [Google Scholar]
- Cheng, X.; Yu, D.; Cheng, G.; Yung, B.C.; Liu, Y.; Li, H.; Kang, C.; Fang, X.; Tian, S.; Zhou, X.; et al. T7 Peptide-Conjugated Lipid Nanoparticles for Dual Modulation of Bcl-2 and Akt-1 in Lung and Cervical Carcinomas. Mol. Pharm. 2018, 15, 4722–4732. [Google Scholar] [CrossRef]
- Chen, D.; Parayath, N.; Ganesh, S.; Wang, W.; Amiji, M. The role of apolipoprotein- and vitronectin-enriched protein corona on lipid nanoparticles for in vivo targeted delivery and transfection of oligonucleotides in murine tumor models. Nanoscale 2019, 11, 18806–18824. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012, 51, 8529–8533. [Google Scholar] [CrossRef]
- Zheng, B.; Yang, S.; Tian, Q.; Xie, Y.; Zhang, S.; Lee, R.J. Delivery of Antisense oligonucleotide LOR-2501 Using Transferrin-conjugated Polyethylenimine-based Lipid Nanoparticle. Anticancer Res. 2019, 39, 1785–1793. [Google Scholar] [CrossRef]
- Yang, S.; Wang, D.; Sun, Y.; Zheng, B. Delivery of Antisense Oligonucleotide Using Polyethylenimine-Based Lipid Nanoparticle Modified with Cell Penetrating Peptide. Drug Deliv. 2019, 26, 965–974. [Google Scholar] [CrossRef]
- Li, H.; Liu, Y.; Chen, L.; Liu, Q.; Qi, S.; Cheng, X.; Lee, Y.B.; Ahn, C.H.; Kim, D.J.; Lee, R.J. Folate receptor-targeted lipid-albumin nanoparticles (F-LAN) for therapeutic delivery of an Akt1 antisense oligonucleotide. J. Drug Target. 2018, 26, 466–473. [Google Scholar] [CrossRef]
- Prakash, T.P.; Graham, M.J.; Yu, J.; Carty, R.; Low, A.; Chappell, A.; Schmidt, K.; Zhao, C.; Aghajan, M.; Murray, H.F.; et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014, 42, 8796–8807. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, R.; Eloy, J.O.; Marchetti, J.M.; Lopez, R.F.; Lee, R.J. Targeted lipid nanoparticles for antisense oligonucleotide delivery. Curr. Pharm. Biotechnol. 2014, 15, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Clarenc, J.P.; Degols, G.; Leonetti, J.P.; Milhaud, P.; Lebleu, B. Delivery of antisense oligonucleotides by poly(L-lysine) conjugation and liposome encapsulation. Anticancer Drug Des. 1993, 8, 81–94. [Google Scholar]
- Stewart, A.J.; Pichon, C.; Meunier, L.; Midoux, P.; Monsigny, M.; Roche, A.C. Enhanced biological activity of antisense oligonucleotides complexed with glycosylated poly-L-lysine. Mol. Pharmacol. 1996, 50, 1487–1494. [Google Scholar]
- Chavany, C.; Le Doan, T.; Couvreur, P.; Puisieux, F.; Helene, C. Polyalkylcyanoacrylate nanoparticles as polymeric carriers for antisense oligonucleotides. Pharm. Res. 1992, 9, 441–449. [Google Scholar] [CrossRef]
- Zobel, H.P.; Kreuter, J.; Werner, D.; Noe, C.R.; Kumel, G.; Zimmer, A. Cationic polyhexylcyanoacrylate nanoparticles as carriers for antisense oligonucleotides. Antisense Nucleic Acid Drug Dev. 1997, 7, 483–493. [Google Scholar] [CrossRef]
- Schwab, G.; Chavany, C.; Duroux, I.; Goubin, G.; Lebeau, J.; Helene, C.; Saison-Behmoaras, T. Antisense oligonucleotides adsorbed to polyalkylcyanoacrylate nanoparticles specifically inhibit mutated Ha-ras-mediated cell proliferation and tumorigenicity in nude mice. Proc. Natl. Acad. Sci. USA 1994, 91, 10460–10464. [Google Scholar] [CrossRef]
- Boussif, O.; Lezoualch, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef]
- Dheur, S.; Dias, N.; van Aerschot, A.; Herdewijn, P.; Bettinger, T.; Remy, J.S.; Helene, C.; Saison-Behmoaras, E.T. Polyethylenimine but not cationic lipid improves antisense activity of 3′-capped phosphodiester oligonucleotides. Antisense Nucleic Acid Drug Dev. 1999, 9, 515–525. [Google Scholar] [CrossRef]
- Bielinska, A.; Kukowska-Latallo, J.F.; Johnson, J.; Tomalia, D.A.; Baker, J.R., Jr. Regulation of in vitro gene expression using antisense oligonucleotides or antisense expression plasmids transfected using starburst PAMAM dendrimers. Nucleic Acids Res. 1996, 24, 2176–2182. [Google Scholar] [CrossRef]
- Huang, Y.Z.; Han, G.; Wang, H.; Liang, W.Q. Cationic niosomes as gene carriers: Preparation and cellular uptake in vitro. Pharmazie 2005, 60, 473–474. [Google Scholar] [PubMed]
- Bartelds, R.; Nematollahi, M.H.; Pols, T.; Stuart, M.C.; Pardakhty, A.; Asadikaram, G.; Poolman, B. Niosomes, an alternative for liposomal delivery. PLoS ONE 2018, 13, e0194179. [Google Scholar] [CrossRef] [PubMed]
- Goudsmit, J.; Geelen, J.; Keulen, W.; Notermans, D.; Kuiken, C.; Ramautarsing, C.; Smit, L.; Koole, L.; van Genderen, M.; Buck, H.; et al. Characterization of the African HIV-1 isolate CBL-4 (RUT) by partial sequence analysis and virus neutralization with peptide antibody and antisense phosphate-methylated DNA. AIDS 1990, 4, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Buck, H.M.; Koole, L.H.; van Genderen, M.H.; Smit, L.; Geelen, J.L.; Jurriaans, S.; Goudsmit, J. Phosphate-methylated DNA aimed at HIV-1 RNA loops and integrated DNA inhibits viral infectivity. Science 1990, 248, 208–212. [Google Scholar] [CrossRef]
- Moody, H.M.; Quaedflieg, P.J.L.M.; Koole, L.H.; van Genderen, M.H.P.; Buck, H.M.; Smit, L.; Jurriaans, S.; Geelen, J.L.M.C.; Goudsmit, J. Inhibition of HIV-1 Infectivity by Phosphate-Methylated DNA: Retraction. Science 1990, 250, 125–126. [Google Scholar] [CrossRef]
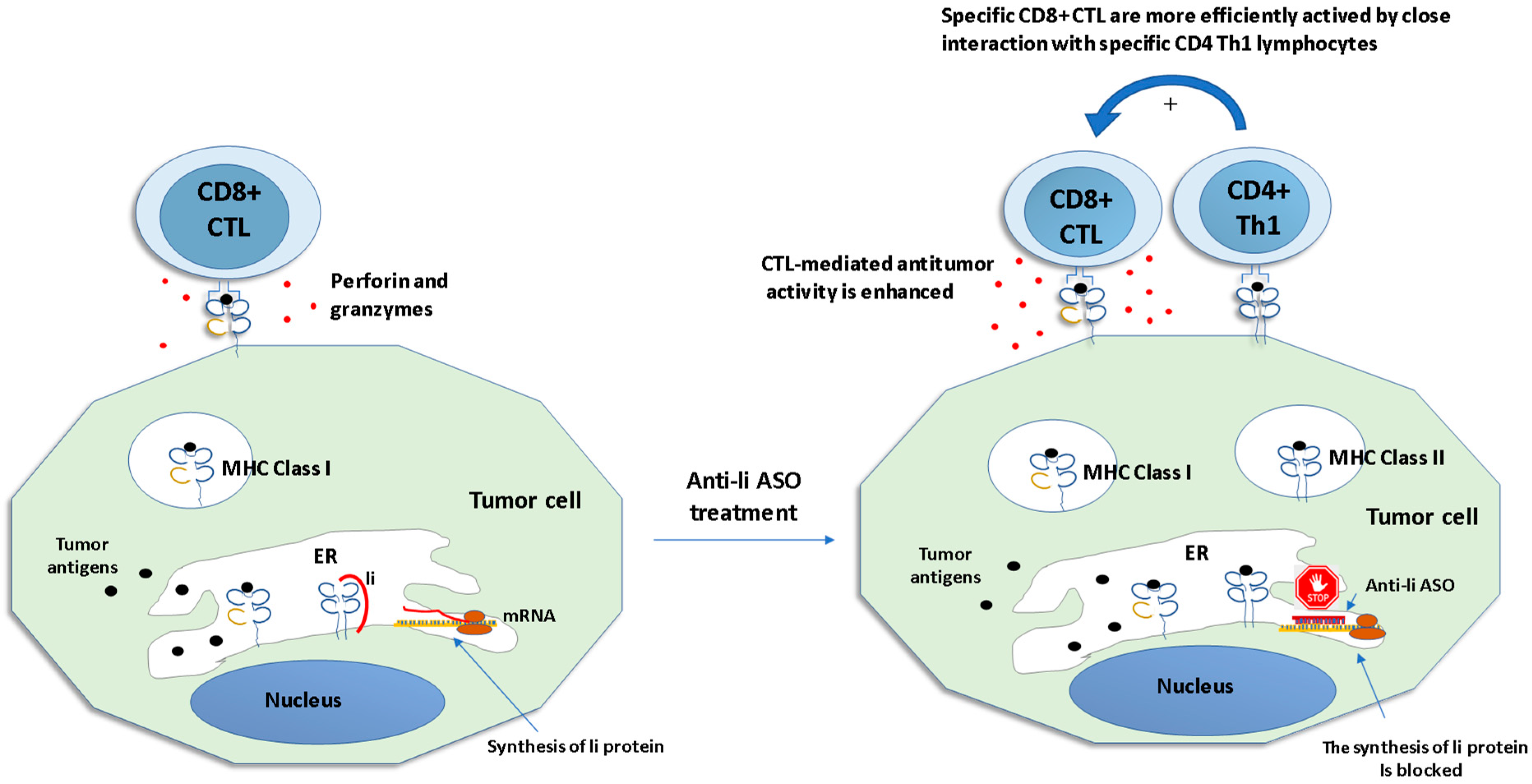
- Humphreys, R.E.; Hillman, G.G.; von Hofe, E.; Xu, M. Forcing tumor cells to present their own tumor antigens to the immune system: A necessary design for an efficient tumor immunotherapy. Cell. Mol. Immunol. 2004, 1, 180–185. [Google Scholar]
- Baskar, S.; Azarenko, V.; Garcia Marshall, E.; Hughes, E.; Ostrand-Rosenberg, S. MHC class II-transfected tumor cells induce long-term tumor-specific immunity in autologous mice. Cell Immunol. 1994, 155, 123–133. [Google Scholar] [CrossRef]
- Clements, V.K.; Baskar, S.; Armstrong, T.D.; Ostrand-Rosenberg, S. Invariant chain alters the malignant phenotype of MHC class II+ tumor cells. J. Immunol. 1992, 149, 2391–2396. [Google Scholar]
- Qiu, G.; Goodchild, J.; Humphreys, R.E.; Xu, M. Cancer immunotherapy by antisense suppression of Ii protein in MHC-class-II-positive tumorcells. Cancer Immunol. Immunother. 1999, 48, 499–506. [Google Scholar] [CrossRef]
- Lu, X.; Kallinteris, N.L.; Li, J.; Wu, S.; Li, Y.; Jiang, Z.; Hillman, G.G.; Gulfo, J.V.; Humphreys, R.E.; Xu, M. Tumor immunotherapy by converting tumor cells to MHC class II-positive, Ii protein-negative phenotype. Cancer Immunol. Immunother. 2003, 52, 592–598. [Google Scholar] [CrossRef]
- Hillman, G.G.; Kallinteris, N.L.; Li, J.; Wang, Y.; Lu, X.; Li, Y.; Wu, S.; Wright, J.L.; Slos, P.; Gulfo, J.V.; et al. Generating MHC Class II+/Ii- phenotype after adenoviral delivery of both an expressible gene for MHC Class II inducer and an antisense Ii-RNA construct in tumor cells. Gene Ther. 2003, 10, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, M.; Hollowell, C.M.; Guinan, P. Differentiated prostatic antigen expression in LNCaP cells following treatment with bispecific antisense oligonucleotides directed against BCL-2 and EGFR. Med. Oncol. 2012, 29, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, M.; Hollowell, C.M.P.; Guinan, P. Inhibition of BCL2 by antisense oligonucleotides is followed by a compensatory suppression of caspase-3 in LNCaP cells. Eur. J. Clin. Med. Oncol. 2011, 3, 1–6. [Google Scholar]
- Rubenstein, M.; Hollowell, C.M.P.; Guinan, P. Additional compensatory mechanisms altering antisense oligonucleotide suppression of BCL2: Effects upon AKT1 and STAT3. In Vivo 2014, 28, 867–870. [Google Scholar]
- Rubenstein, M.; Hollowell, C.M.P.; Guinan, P. In LNCaP cells enhanced expression of the androgen receptor compensates for BCL2 suppression by antisense oligonucleotides. Ther. Adv. Urology 2011, 3, 73–79. [Google Scholar]
- Rubenstein, M.; Hollowell, C.M.P.; Guinan, P. In LNCaP cells enhanced expression of both androgen receptor and co-stimulatory protein p300 compensate for antisense oligonucleotide suppression of BCL2. Ther. Adv. Urology 2011, 3, 243–250. [Google Scholar]
- Rubenstein, M.; Hollowell, C.M.P.; Guinan, P. Increased expression of the androgen receptor with p300 and IL-6 coactivators compensate for oligonucleotide suppression of BCL2. No increased CREBBP or IL-4 expression. Ther. Adv. Urology 2013, 5, 85–93. [Google Scholar] [CrossRef]
- Rubenstein, M.; Hollowell, C.M.P.; Guinan, P. Following inhibition of BCL2 by antisense oligonucleotides compensatory suppression of apoptosis involves the direct signal transduction pathway of LNCaP cell. Online J. Apoptosis 2015, 4, 1–10. [Google Scholar] [CrossRef][Green Version]
- Rubenstein, M.; Hollowell, C.M.; Guinan, P. Suppression of BCL2 by Antisense Oligonucleotides and Compensation by Non-Targeted Genes May Enhance Tumor Proliferation. In Vivo 2015, 29, 687–693. [Google Scholar]
- Tsamandas, A.C.; Kardamakis, D.; Ravazoula, P.; Zolota, V.; Salakou, S.; Tepetes, K.; Kalogeropoulou, C.; Tsota, I.; Kourelis, T.; Makatsoris, T.; et al. The potential role of TGFbeta1, TGFbeta2 and TGFbeta3 protein expression in colorectal carcinomas. Correlation with classic histopathologic factors and patient survival. Strahlenther. Onkol. 2004, 180, 201–208. [Google Scholar] [CrossRef]
- Dallas, S.L.; Zhao, S.; Cramer, S.D.; Chen, Z.; Peehl, D.M.; Bonewald, L.F. Preferential production of latent transforming growth factor beta-2 by primary prostatic epithelial cells and its activation by prostate-specific antigen. J. Cell Physiol. 2005, 202, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Polak, M.E.; Borthwick, N.J.; Gabriel, F.G.; Johnson, P.; Higgins, B.; Hurren, J.; McCormick, D.; Jager, M.J.; Cree, I.A. Mechanisms of local immunosuppression in cutaneous melanoma. Br. J. Cancer 2007, 96, 1879–1887. [Google Scholar] [CrossRef] [PubMed]
- Vagenas, K.; Spyropoulos, C.; Gavala, V.; Tsamandas, A.C. TGFbeta1, TGFbeta2, and TGFbeta3 protein expression in gastric carcinomas: Correlation with prognostics factors and patient survival. J. Surg. Res. 2007, 139, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.S.; Jen, J. TGF-beta signaling and the role of inhibitory Smads in non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 417–419. [Google Scholar] [CrossRef]
- Schlingensiepen, K.H.; Jaschinski, F.; Lang, S.A.; Moser, C.; Geissler, E.K.; Schlitt, H.J.; Kielmanowicz, M.; Schneider, A. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011, 102, 1193–1200. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Tzai, T.S.; Shiau, A.L.; Liu, L.L.; Wu, C.L. Immunization with TGF-beta antisense oligonucleotide-modified autologous tumor vaccine enhances the antitumor immunity ofMBT-2 tumor-bearing mice through upregulation of MHC clas I and Fas expressions. Anticancer Res. 2000, 20, 1557–1562. [Google Scholar]
- Schneider, T.; Becker, A.; Ringe, K.; Reinhold, A.; Firsching, R.; Sabel, B.A. Brain tumor therapy by combined vaccination and antisense oligonucleotide delivery with nanoparticles. J. Neuroimmunol. 2008, 195, 21–27. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Dillman, R.O.; Schwarzenberger, P.O.; Senzer, N.; Cunningham, C.; Cutler, J.; Tong, A.; Kumar, P.; Pappen, B.; Hamilton, C.; et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 4721–4730. [Google Scholar] [CrossRef]
- Giaccone, G.; Bazhenova, L.A.; Nemunaitis, J.; Tan, M.; Juhász, E.; Ramlau, R.; van den Heuvel, M.M.; Lal, R.; Kloecker, G.H.; Eaton, K.D.; et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine as maintenance therapy for non-small cell lung cancer. Eur. J. Cancer 2015, 51, 2321–2329. [Google Scholar] [CrossRef]
- Schlingensiepen, K.H.; Fischer-Blass, B.; Schmaus, S.; Ludwig, S. Antisense therapeutics for tumor treatment: The TGF-beta2 inhibitor AP 12009 in clinical development against malignant tumors. Recent Results Cancer Res. 2008, 177, 137–150. [Google Scholar] [PubMed]
- Vallières, L. Trabedersen, a TGFbeta2-specific antisense oligonucleotide for the treatment of malignant gliomas and other tumors overexpressing TGFbeta2. IDrugs 2009, 12, 445–453. [Google Scholar] [PubMed]
- D’Cruz, O.J.; Qazi, S.; Hwang, L.; Ng, K.; Trieu, V. Impact of targeting transforming growth factor β-2 with antisense OT-101 on the cytokine and chemokine profile in patients with advanced pancreatic cancer. Onco Targets Ther. 2018, 11, 2779–2796. [Google Scholar] [CrossRef] [PubMed]
- Bogdahn, U.; Hau, P.; Stockhammer, G.; Venkataramana, N.K.; Mahapatra, A.K.; Suri, A.; Balasubramaniam, A.; Nair, S.; Oliushine, V.; Parfenov, V.; et al. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: Results of randomized and controlled phase IIb study. Neuro Oncol. 2011, 13, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Papachristodoulou, A.; Silginer, M.; Weller, M.; Schneider, H.; Hasenbach, K.; Janicot, M.; Roth, P. Therapeutic targeting of TGF-β ligands in glioblastoma using novel antisense oligonucleotides reduces the growth of experimental gliomas. Clin. Cancer Res. 2019, 25, 7189–7201. [Google Scholar] [CrossRef] [PubMed]
- Schillaci, R.; Salatino, M.; Cassataro, J.; Proietti, C.J.; Giambartolomei, G.H.; Rivas, M.A.; Carnevale, R.P.; Charreau, E.H.; Elizalde, P.V. Immunization with murine breast cancer cells treated with antisense oligodeoxynucleotides to type I insulin-like growth factor receptor induced an antitumoral effect mediated by a CD8+ response involving Fas/Fas ligand cytotoxic pathway. J. Immunol. 2006, 176, 3426–3437. [Google Scholar] [CrossRef]
- Miguel, A.; Sendra, L.; Noé, V.; Ciudad, C.J.; Dasí, F.; Hervas, D.; Herrero, M.J.; Aliño, S.F. Silencing of Foxp3 enhances the antitumor efficacy of GM-CSF genetically modified tumor cell vaccine against B16 melanoma. Onco Targets Ther. 2017, 10, 503–514. [Google Scholar] [CrossRef]
- Moyle, P.M.; Toth, I. Modern subunit vaccines: Development, components, and research opportunities. Chem. Med. Chem. 2013, 8, 360–376. [Google Scholar] [CrossRef]
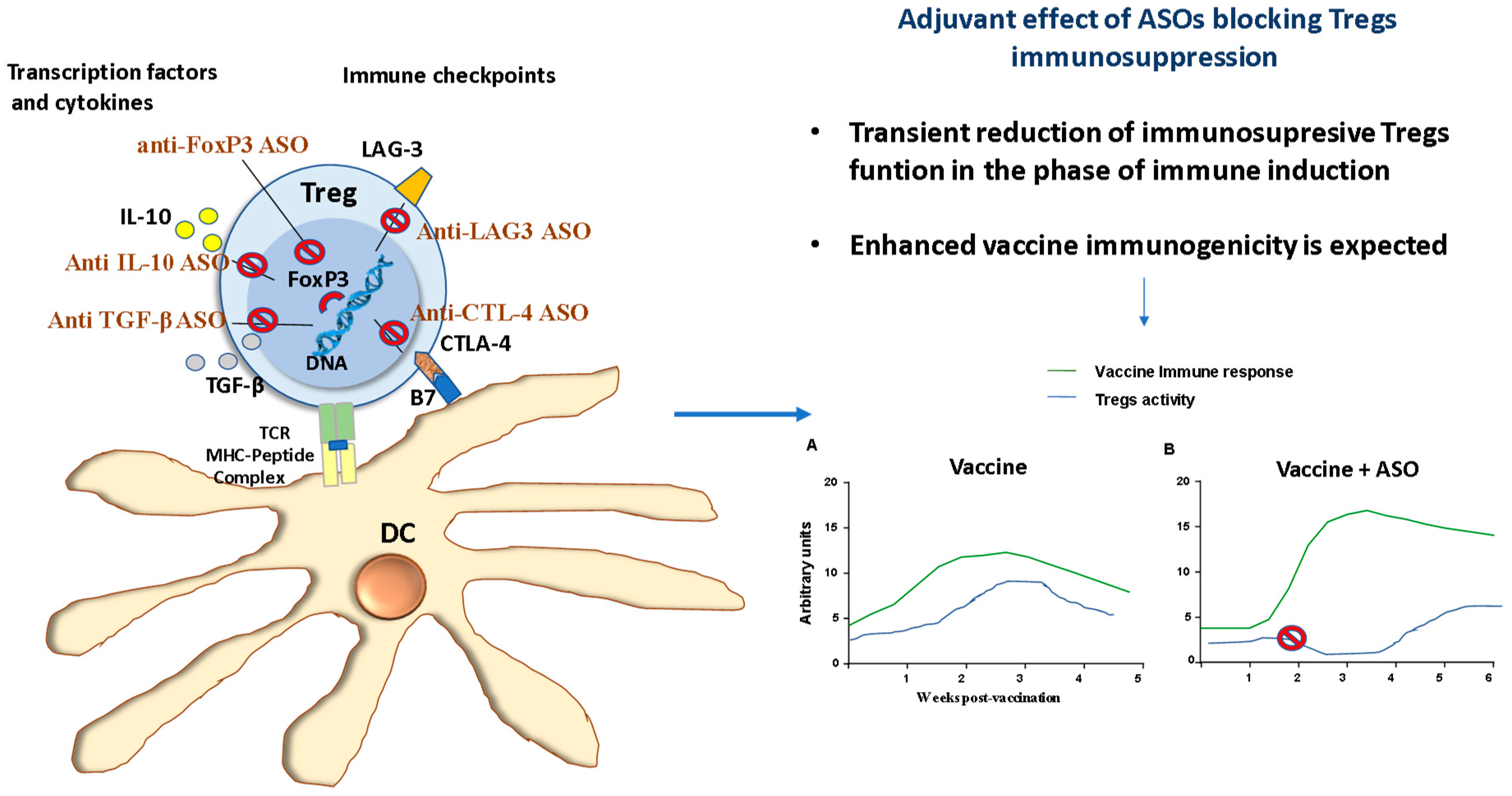
- Batista-Duharte, A.; Téllez-Martínez, D.; Fuentes, D.L.P.; Carlos, I.Z. Molecular adjuvants that modulate regulatory T cell function in vaccination: A critical appraisal. Pharmacol. Res. 2018, 129, 237–250. [Google Scholar] [CrossRef]
- Ripple, M.J.; You, D.; Honnegowda, S.; Giaimo, J.D.; Sewell, A.B.; Becnel, D.M.; Cormier, S.A. Immunomodulation with IL-4Rα antisense oligonucleotide prevents respiratory syncytial virus-mediated pulmonary disease. J. Immunol. 2010, 185, 4804–4811. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, N.; Lu, Y.; Huang, Z.; Zang, Y.; Chen, J.; Zhang, J.; Ding, Z. Phosphorothioated antisense oligodeoxynucleotide suppressing interleukin-10 is a safe and potent vaccine adjuvant. Vaccine 2019, 37, 4081–4088. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, L.; Zhao, P.; Yao, Y.; Lu, F.; Tu, L.; Liu, J.; Li, Z.; Yu, Y.; Wang, L. Adjuvanticity of a CTLA-4 3’ UTR complementary oligonucleotide for emulsion formulated recombinant subunit and inactivated vaccines. Vaccine 2017, 35, 2379–2389. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Song, Y.; Cui, C.; Lan, Y.; Li, X.; Liu, Y.; Lu, F.; Zhang, Y.; Yu, Y.; Wang, L. A LAG3-interfering oligonucleotide acts as an adjuvant to enhance the antibody responses induced by recombinant protein vaccines and inactivated influenza virus vaccines. Appl. Microbiol. Biotechnol. 2019, 103, 6543–6557. [Google Scholar] [CrossRef] [PubMed]
- Akl, M.R.; Ayoub, N.M. Tumor cell transformation using antisense oligonucleotide. Methods Mol. Biol. 2014, 1139, 259–268. [Google Scholar]
- Gajewski, T.F.; Meng, Y.; Blank, C.; Brown, I.; Kacha, A.; Kline, J.; Harlin, H. Immune resistance orchestrated by the tumor microenvironment. Immunol. Rev. 2006, 213, 131–145. [Google Scholar] [CrossRef]
- Pedersen, L.; Hagedorn, P.H.; Koch, T. Identifying Suitable Target Regions and Analyzing Off-Target Effects of Therapeutic Oligonucleotides. Methods Mol. Biol. 2019, 2036, 261–282. [Google Scholar]
- Batista-Duharte, A.; Martínez, D.T.; Carlos, I.Z. Efficacy and safety of immunological adjuvants. Where is the cut-off? Biomed. Pharmacother. 2018, 105, 616–624. [Google Scholar] [CrossRef]
- Schoch, K.M.; Miller, T.M. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Modifications | Characteristics | Mechanisms | Clinical Use | Limitations |
|---|---|---|---|---|
| First Generation | ||||
| Phosphorothioate (PTO), Methylphosphonate (MPO) | Either a sulfur atom (PTO), or a methyl group (MPO) substitutes the non-bridging oxygen atoms in the phosphodiester bond. | First generation ASOs promote degradation of target mRNA by RNase H enzyme. They also confer higher solubility, resistance to nuclease degradation, antisense activity and longer plasma half-life as compared with phosphodiester oligonucleotides. | PTO is the most widely used modification of ASOs. Fomivirsen, is a PTO-modified ASO, used as local treatment of cytomegalovirus (CMV) retinitis in patients with acquired immunodeficiency syndrome (AIDS) [48]. | High affinity for various cellular proteins and components of the innate immune system, such as Toll-like receptors (TLRs), with proinflammatory effects. Commonly reported side effects following systemic administration of PTO ASOs include fever, activated partial thromboplastin time prolongation, thrombocytopenia, and leukopenia. |
| Second Generation | ||||
| ASOs with 2’-O-alkyl modifications of the ribose. Chimeric ‘gapmer’ ASOs | 2’-O-Methyl (2’-OMe) and 2’-O-Methoxyethyl (2’-MOE) are the most widely studied. Chimeric ‘gapmer’ ASOs consist in a central ‘gap’ region containing 10 DNA or PTO DNA monomers, flanked on both 5’ and 3’extremities by alkyl modified nucleotides such as 2′-OM or 2’-MOE. | The PTO DNA induces RNase H cleavage while 2′-OME or 2′-MOE on both sides (5′- and 3′-directions) confers nuclease-resistance, and they can exert activity by a steric interference of translation process. They are safer than PTO-modified ASOs and exhibit enhanced affinity towards the complementary RNA with better tissue uptake and longer in vivo half-life. | Mipomersen is used as an adjunct therapy for homozygous familial hypercholesterolemia [49]. Nusinersen was approved for spinal muscular atrophy treatment [50]. Apatorsen is a HSP27 targeting ASO that is being studied in phase II clinical trials in patients with metastatic castration resistant prostate cancer [51] and Untreated Stage IV Non-Squamous-Non-Small-Cell Lung Cancer [52]. | A subset of 2´-MOE-modified ASOs induced pro-inflammatory cytokines and type I interferons (IFN-α/β) and interaction with innate immune receptors such as TLR9, melanoma-differentiation associated-5 (MDA-5) and IFN-β promoter stimulator-1 (IPS-1). |
| Third Generation | ||||
| Peptide nucleic acid (PNA) | PNA is a synthetic DNA in which the deoxyribose phosphate backbone is replaced by polyamide linkages. | PNA block the protein expression, by steric hindrance, forming sequence-specific duplex with the targeted mRNA. They are biologically stable and have good hybridization properties. | The potential of PNA as drugs in gene therapy has been hampered by the poor intrinsic uptake of PNA by living cells. Current strategies for improving PNA delivery into the cytosolic space and nucleus include microinjection, electroporation, co-transfection with DNA, or conjugation to lipophilic moieties, nanoparticles, cell-penetrating peptides (CPPs), oligo-aspartic acid, or nuclear localization signal (NLS) peptides to enhance cellular internalization | PNA do not activate the RNase H to cleave the target hybridized RNA. PNA have low solubility and cellular uptake. |
| Phosphoramidate morpholino oligomer (PMO) | PMOs are neutral ASOs. The pentose sugar is substituted by a morpholino ring and the inter-nucleotide linkages are phosphoramidate bonds in place of phosphodiester bonds. | The mechanism of PMO is the translational arrest mediated by steric interference of ribosomal assembly. PMO show fewer nonspecific properties and lesser toxicity than PTO. | Eteplirsen was approved for Duchenne muscular dystrophy (DMD) treatment [53]. Other potential applications include the treatment of viral infections, antibiotic-resistant bacterial infections, and cancers [54]. | PMOs exhibit reduced cellular uptake. Conjugation with peptides such as arginine-rich peptide (ARP) can enhance its cellular uptake and antisense efficacy. |
| Locked nucleic acid (LNA) | LNAs are chemically modified nucleotides with a ribose containing a methylene bridge between the 2′-oxygen and the 4′-carbon of the ribose. | LNA modifications improve the affinity of ASO hybridization towards mRNA target, by increase of the DNA/RNA heteroduplexes thermal stability. LNAs avoid nuclease degradation. | Diverse LNAs are currently in clinical trials by several biotechnology firms. | LNA does not activate RNase. LNA nucleotides can be incorporated at the ends of RNA and DNA sequences to form chimeric oligonucleotides resulting in restoration of RNase H-mediated cleavage of mRNA. |
| ASOs | mAb | |
|---|---|---|
| Molecular Weight | ~6 to10 kDa | ~150 kDa |
| Structure | Relatively simple structure. Usually 13–20 mer with chemical modifications | Glycoproteins with complex structure |
| Thermal Stability | Highly stable. Lyophilization and freezing does not modify its biological activity | Low stability. Cold chain through the storage, handling, and transportation is necessary |
| Cellular Bioavailability | ASOs can penetrate the cells and act on intracellular targets | They are unable to penetrate the cells |
| Immunogenicity | ASOs are not properly immunogenic | Highly immunogenic by xenogeneic differences, e.g., between mice and humans |
| Specificity | Highly specific but off target interaction can be observed | Highly specific but cross-reactivity can be observed |
| Toxicity | Relatively low toxicity | Different grades of toxicity have been described |
| Development and Manufacturing | ASOs are obtained synthetically.The use of vehicles can add complexity to the manufacture process | The production for pharmacological proposes requires high level of technological complexity |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batista-Duharte, A.; Sendra, L.; Herrero, M.J.; Téllez-Martínez, D.; Carlos, I.Z.; Aliño, S.F. Progress in the Use of Antisense Oligonucleotides for Vaccine Improvement. Biomolecules 2020, 10, 316. https://doi.org/10.3390/biom10020316
Batista-Duharte A, Sendra L, Herrero MJ, Téllez-Martínez D, Carlos IZ, Aliño SF. Progress in the Use of Antisense Oligonucleotides for Vaccine Improvement. Biomolecules. 2020; 10(2):316. https://doi.org/10.3390/biom10020316
Chicago/Turabian StyleBatista-Duharte, Alexander, Luis Sendra, Maria José Herrero, Damiana Téllez-Martínez, Iracilda Zeppone Carlos, and Salvador Francisco Aliño. 2020. "Progress in the Use of Antisense Oligonucleotides for Vaccine Improvement" Biomolecules 10, no. 2: 316. https://doi.org/10.3390/biom10020316
APA StyleBatista-Duharte, A., Sendra, L., Herrero, M. J., Téllez-Martínez, D., Carlos, I. Z., & Aliño, S. F. (2020). Progress in the Use of Antisense Oligonucleotides for Vaccine Improvement. Biomolecules, 10(2), 316. https://doi.org/10.3390/biom10020316