Biomarkers to Personalize the Treatment of Rheumatoid Arthritis: Focus on Autoantibodies and Pharmacogenetics

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Role of Autoantibodies in Predicting the Response to RA Treatment
3. Pharmacogenetic Biomarkers
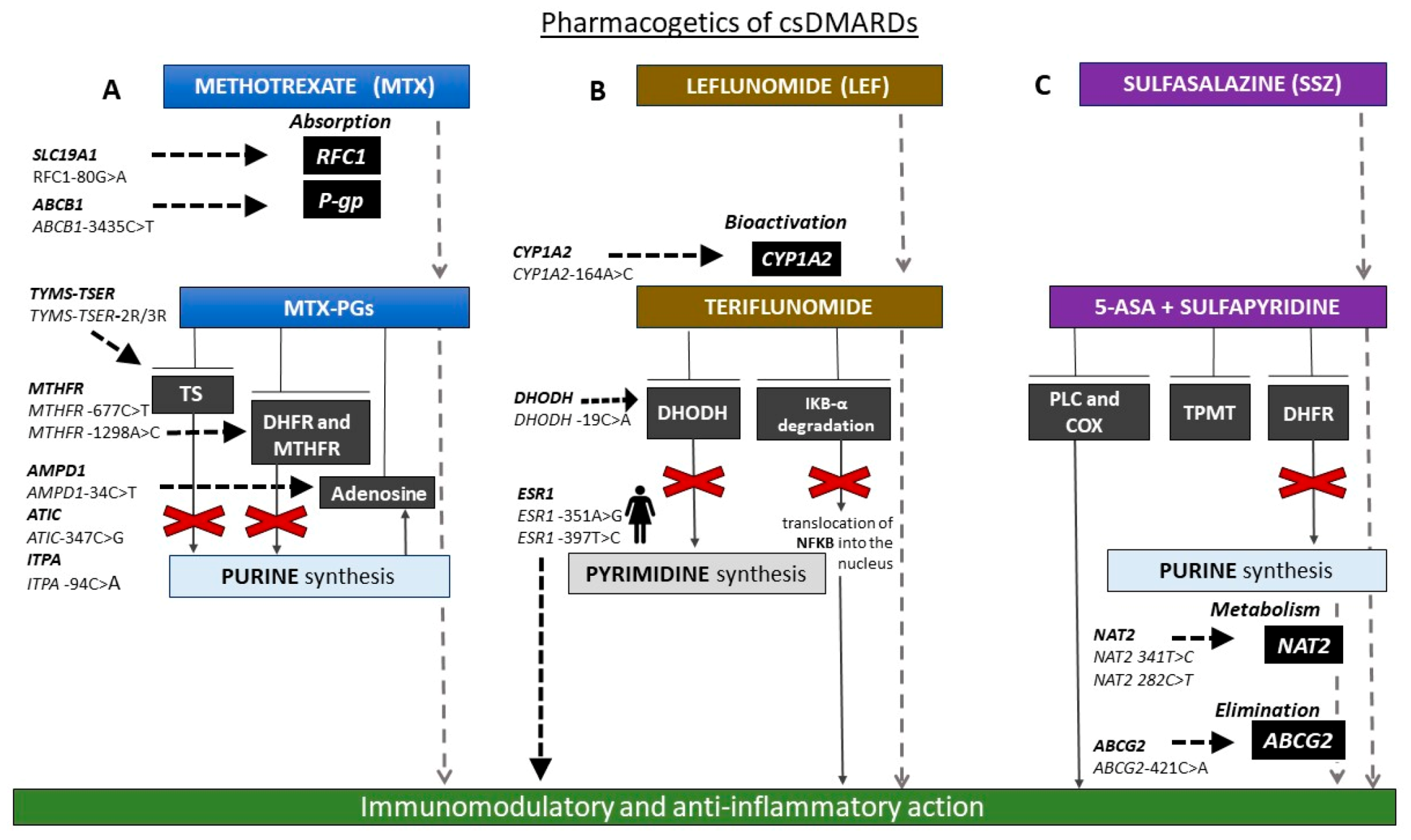
3.1. Pharmacogenetics of csDMARDs
3.2. Pharmacogenetics of Other csDMARDS
3.3. Pharmacogenetics of bDMARDs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Sokka, T.; Toloza, S.; Cutolo, M.; Kautiainen, H.; Makinen, H.; Gogus, F.; Skakic, V.; Badsha, H.; Peets, T.; Baranauskaite, A.; et al. QUEST-RA Group. Women, men, and rheumatoid arthritis: Analyses of disease activity, disease characteristics, and treatments in the QUEST-RA study. Arthritis Res. Ther. 2009, 11, R7. [Google Scholar] [CrossRef] [PubMed]
- Kvien, T.K.; Uhlig, T.; Ødegård, S.; Heiberg, M.S. Epidemiological aspects of rheumatoid arthritis: The sex ratio. Ann. N. Y. Acad. Sci. 2006, 69, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Kanda, N.; Tsuchida, T.; Tamaki, K. Estrogen enhancement of anti-double-stranded DNA antibody and immunoglobulin G production in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Arthritis Rheum. 1999, 42, 328–337. [Google Scholar] [CrossRef]
- Kramer, P.R.; Kramer, S.F.; Guan, G. 17 beta-estradiol regulates cytokine release through modulation of CD16 expression in monocytes and monocyte-derived macrophages. Arthritis Rheum. 2004, 50, 1967–1975. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Capellino, S.; Montagna, P.; Villaggio, B.; Sulli, A.; Seriolo, B.; Straub, R.H. New roles for estrogens in rheumatoid arthritis. Clin. Exp. Rheumatol. 2003, 21, 687–690. [Google Scholar]
- Smolen, J.S.; Landewé, R.; Bijlsma, J.W.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; Van Vollenhoven, R.F.; De Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020, 76, 685–699. [Google Scholar] [CrossRef]
- Cronstein, B.N. Low-dose methotrexate: A mainstay in the treatment of rheumatoid arthritis. Pharmacol. Rev. 2005, 57, 163–172. [Google Scholar] [CrossRef]
- Bedoui, Y.; Guillot, X.; Sélambarom, J.; Guiraud, P.; Giry, C.; Jaffar-Bandjee, M.C.; Ralandison, S.; Gasque, P. Methotrexate an Old Drug with New Tricks. Int. J. Mol. Sci. 2019, 20, 5023. [Google Scholar] [CrossRef]
- Italian Medicines Agency Home Page. Available online: https://www.aifa.gov.it/documents/20142/847374/Methotrexate_public_health_communication_IT.pdf/a8fb75db-4b62-2783-528b-6701ce5cfc66 (accessed on 15 November 2020).
- Holers, V.M.; Demoruelle, M.K.; Kuhn, K.A.; Buckner, J.H.; Robinson, W.H.; Okamoto, Y.; Norris, J.M.; Deane, K.D. Rheumatoid Arthritis and the Mucosal Origins Hypothesis: Protection Turns to Destruction. Nat. Rev. Rheumatol. 2018, 14, 542–557. [Google Scholar] [CrossRef]
- Trouw, L.A.; Rispens, T.; Toes, R.E.M. Beyond Citrullination: Other Post-Translational Protein Modifications in Rheumatoid Arthritis. Nat. Rev. Rheumatol. 2017, 13, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid Arthritis Classification Criteria: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Ann. Rheum. Dis. 2010, 69, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- van Boekel, M.A.M.; Vossenaar, E.R.; van den Hoogen, F.H.J.; van Venrooij, W.J. Autoantibody Systems in Rheumatoid Arthritis: Specificity, Sensitivity and Diagnostic Value. Arthritis Res. 2002, 4, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, G.A.; Visser, H.; de Jong, B.A.; van den Hoogen, F.H.; Hazes, J.M.; Breedveld, F.C.; van Venrooij, W.J. The Diagnostic Properties of Rheumatoid Arthritis Antibodies Recognizing a Cyclic Citrullinated Peptide. Arthritis Rheum. 2000, 43, 155–163. [Google Scholar] [CrossRef]
- Nishimura, K.; Sugiyama, D.; Kogata, Y.; Tsuji, G.; Nakazawa, T.; Kawano, S.; Saigo, K.; Morinobu, A.; Koshiba, M.; Kuntz, K.M.; et al. Meta-Analysis: Diagnostic Accuracy of Anti-Cyclic Citrullinated Peptide Antibody and Rheumatoid Factor for Rheumatoid Arthritis. Ann. Intern. Med. 2007, 146, 797–808. [Google Scholar] [CrossRef]
- Verpoort, K.N.; Jol-van der Zijde, C.M.; Papendrecht-van der Voort, E.A.M.; Ioan-Facsinay, A.; Drijfhout, J.W.; van Tol, M.J.D.; Breedveld, F.C.; Huizinga, T.W.J.; Toes, R.E.M. Isotype Distribution of Anti-Cyclic Citrullinated Peptide Antibodies in Undifferentiated Arthritis and Rheumatoid Arthritis Reflects an Ongoing Immune Response. Arthritis Rheum. 2006, 54, 3799–3808. [Google Scholar] [CrossRef]
- Reparon-Schuijt, C.C.; van Esch, W.J.; van Kooten, C.; Schellekens, G.A.; de Jong, B.A.; van Venrooij, W.J.; Breedveld, F.C.; Verweij, C.L. Secretion of Anti-Citrulline-Containing Peptide Antibody by B Lymphocytes in Rheumatoid Arthritis. Arthritis Rheum. 2001, 44, 41–47. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y. Peptidylarginine Deiminases in Citrullination, Gene Regulation, Health and Pathogenesis. Biochim. Biophys. Acta 2013, 1829, 1126–1135. [Google Scholar] [CrossRef]
- Wysocki, T.; Olesińska, M.; Paradowska-Gorycka, A. Current Understanding of an Emerging Role of HLA-DRB1 Gene in Rheumatoid Arthritis-From Research to Clinical Practice. Cells 2020, 9, 1127. [Google Scholar] [CrossRef]
- Gregersen, P.K. Genetics of Rheumatoid Arthritis: Confronting Complexity. Arthritis Res. 1999, 1, 37–44. [Google Scholar] [CrossRef][Green Version]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of Rheumatoid Arthritis Contributes to Biology and Drug Discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef] [PubMed]
- van der Woude, D.; Houwing-Duistermaat, J.J.; Toes, R.E.M.; Huizinga, T.W.J.; Thomson, W.; Worthington, J.; van der Helm-van Mil, A.H.M.; de Vries, R.R.P. Quantitative Heritability of Anti-Citrullinated Protein Antibody-Positive and Anti-Citrullinated Protein Antibody-Negative Rheumatoid Arthritis. Arthritis Rheum. 2009, 60, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Frisell, T.; Holmqvist, M.; Källberg, H.; Klareskog, L.; Alfredsson, L.; Askling, J. Familial Risks and Heritability of Rheumatoid Arthritis: Role of Rheumatoid Factor/Anti-Citrullinated Protein Antibody Status, Number and Type of Affected Relatives, Sex, and Age. Arthritis Rheum. 2013, 65, 2773–2782. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, J.R.; Nepom, B.S.; Haire, C.; Gersuk, V.H.; Gaur, L.; Moore, G.F.; Drymalski, W.; Palmer, W.; Eckhoff, P.J.; Klassen, L.W.; et al. HLA-DRB1 Typing in Rheumatoid Arthritis: Predicting Response to Specific Treatments. Ann. Rheum. Dis. 1998, 57, 209–213. [Google Scholar] [CrossRef]
- Zanelli, E.; Breedveld, F.C.; de Vries, R.R. HLA Class II Association with Rheumatoid Arthritis: Facts and Interpretations. Hum. Immunol. 2000, 61, 1254–1261. [Google Scholar] [CrossRef]
- Lard, L.R.; Boers, M.; Verhoeven, A.; Vos, K.; Visser, H.; Hazes, J.M.W.; Zwinderman, A.H.; Schreuder, G.M.T.; Breedveld, F.C.; De Vries, R.R.P.; et al. Early and Aggressive Treatment of Rheumatoid Arthritis Patients Affects the Association of HLA Class II Antigens with Progression of Joint Damage. Arthritis Rheum. 2002, 46, 899–905. [Google Scholar] [CrossRef]
- Van Snick, J.L.; Van Roost, E.; Markowetz, B.; Cambiaso, C.L.; Masson, P.L. Enhancement by IgM Rheumatoid Factor of in Vitro Ingestion by Macrophages and in Vivo Clearance of Aggregated IgG or Antigen-Antibody Complexes. Eur. J. Immunol. 1978, 8, 279–285. [Google Scholar] [CrossRef]
- Hogben, D.N.; Devey, M.E. Studies on Rheumatoid Factor: I. The Effect of Rheumatoid Factor on the Clearance of Preformed Immune Complexes in Mice. Clin. Exp. Immunol. 1986, 66, 648–653. [Google Scholar]
- Falkenburg, W.J.J.; van Schaardenburg, D.; Ooijevaar-de Heer, P.; Wolbink, G.; Rispens, T. IgG Subclass Specificity Discriminates Restricted IgM Rheumatoid Factor Responses From More Mature Anti-Citrullinated Protein Antibody-Associated or Isotype-Switched IgA Responses. Arthritis Rheumatol. 2015, 67, 3124–3134. [Google Scholar] [CrossRef]
- Ingegnoli, F.; Castelli, R.; Gualtierotti, R. Rheumatoid Factors: Clinical Applications. Dis. Mark. 2013, 35, 727–734. [Google Scholar] [CrossRef]
- Verheul, M.K.; Fearon, U.; Trouw, L.A.; Veale, D.J. Biomarkers for Rheumatoid and Psoriatic Arthritis. Clin. Immunol. 2015, 161, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Bojesen, S.E.; Schnohr, P.; Nordestgaard, B.G. Elevated Rheumatoid Factor and Long Term Risk of Rheumatoid Arthritis: A Prospective Cohort Study. BMJ 2012, 345, e5244. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, T.; Arinbjarnarson, S.; Thorsteinsson, J.; Steinsson, K.; Geirsson, A.J.; Jónsson, H.; Valdimarsson, H. Raised IgA Rheumatoid Factor (RF) but Not IgM RF or IgG RF Is Associated with Extra-Articular Manifestations in Rheumatoid Arthritis. Scand. J. Rheumatol. 1995, 24, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, Y.; Suzuki, A.; Sawada, T.; Ohsaka, M.; Inoue, T.; Yamada, R.; Yamamoto, K. Citrullinated Fibrinogen Detected as a Soluble Citrullinated Autoantigen in Rheumatoid Arthritis Synovial Fluids. Ann. Rheum. Dis. 2006, 65, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, H.; Koller, T.; Engström, A.; Nandakumar, K.S.; Turnay, J.; Kraetsch, H.G.; Kalden, J.R.; Holmdahl, R. Epitope-Specific Recognition of Type II Collagen by Rheumatoid Arthritis Antibodies Is Shared with Recognition by Antibodies That Are Arthritogenic in Collagen-Induced Arthritis in the Mouse. Arthritis Rheum. 2002, 46, 2339–2348. [Google Scholar] [CrossRef] [PubMed]
- Fialová, L. Avidity of Selected Autoantibodies—Usefulness of Their Determination for Clinical Purposes. Epidemiol. Mikrobiol. Imunol. Cas. Spol. Pro Epidemiol. Mikrobiol. Ces. Lek. Spol. J.E. Purkyne 2016, 65, 155–163. [Google Scholar]
- Nielen, M.M.J.; van Schaardenburg, D.; Reesink, H.W.; van de Stadt, R.J.; van der Horst-Bruinsma, I.E.; de Koning, M.H.M.T.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A.C. Specific Autoantibodies Precede the Symptoms of Rheumatoid Arthritis: A Study of Serial Measurements in Blood Donors. Arthritis Rheum. 2004, 50, 380–386. [Google Scholar] [CrossRef]
- Gan, R.W.; Trouw, L.A.; Shi, J.; Toes, R.E.M.; Huizinga, T.W.J.; Demoruelle, M.K.; Kolfenbach, J.R.; Zerbe, G.O.; Deane, K.D.; Edison, J.D.; et al. Anti-Carbamylated Protein Antibodies Are Present Prior to Rheumatoid Arthritis and Are Associated with Its Future Diagnosis. J. Rheumatol. 2015, 42, 572–579. [Google Scholar] [CrossRef]
- Shi, J.; Knevel, R.; Suwannalai, P.; van der Linden, M.P.; Janssen, G.M.C.; van Veelen, P.A.; Levarht, N.E.W.; van der Helm-van Mil, A.H.M.; Cerami, A.; Huizinga, T.W.J.; et al. Autoantibodies Recognizing Carbamylated Proteins Are Present in Sera of Patients with Rheumatoid Arthritis and Predict Joint Damage. Proc. Natl. Acad. Sci. USA 2011, 108, 17372–17377. [Google Scholar] [CrossRef]
- Kinslow, J.D.; Blum, L.K.; Deane, K.D.; Demoruelle, M.K.; Okamoto, Y.; Parish, M.C.; Kongpachith, S.; Lahey, L.J.; Norris, J.M.; Robinson, W.H.; et al. Elevated IgA Plasmablast Levels in Subjects at Risk of Developing Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 2372–2383. [Google Scholar] [CrossRef]
- van de Stadt, L.A.; Witte, B.I.; Bos, W.H.; van Schaardenburg, D.A. Prediction Rule for the Development of Arthritis in Seropositive Arthralgia Patients. Ann. Rheum. Dis. 2013, 72, 1920–1926. [Google Scholar] [CrossRef] [PubMed]
- Verheul, M.K.; Böhringer, S.; van Delft, M.A.M.; Jones, J.D.; Rigby, W.F.C.; Gan, R.W.; Holers, V.M.; Edison, J.D.; Deane, K.D.; Janssen, K.M.J.; et al. Triple Positivity for Anti-Citrullinated Protein Autoantibodies, Rheumatoid Factor, and Anti-Carbamylated Protein Antibodies Conferring High Specificity for Rheumatoid Arthritis: Implications for Very Early Identification of At-Risk Individuals. Arthritis Rheumatol. 2018, 70, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Husby, G.; Gran, J.T.; Johannessen, A. Epidemiological and Genetic Aspects of IgM Rheumatoid Factors. Scand. J. Rheumatol. Suppl. 1988, 75, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Reynisdottir, G.; Karimi, R.; Joshua, V.; Olsen, H.; Hensvold, A.H.; Harju, A.; Engström, M.; Grunewald, J.; Nyren, S.; Eklund, A.; et al. Structural Changes and Antibody Enrichment in the Lungs Are Early Features of Anti-Citrullinated Protein Antibody-Positive Rheumatoid Arthritis. Arthritis Rheumatol. 2014, 66, 31–39. [Google Scholar] [CrossRef]
- Roosnek, E.; Lanzavecchia, A. Efficient and Selective Presentation of Antigen-Antibody Complexes by Rheumatoid Factor B Cells. J. Exp. Med. 1991, 173, 487–489. [Google Scholar] [CrossRef]
- Daha, N.A.; Banda, N.K.; Roos, A.; Beurskens, F.J.; Bakker, J.M.; Daha, M.R.; Trouw, L.A. Complement Activation by (Auto-) Antibodies. Mol. Immunol. 2011, 48, 1656–1665. [Google Scholar] [CrossRef]
- Trouw, L.A.; Haisma, E.M.; Levarht, E.W.N.; van der Woude, D.; Ioan-Facsinay, A.; Daha, M.R.; Huizinga, T.W.J.; Toes, R.E. Anti-Cyclic Citrullinated Peptide Antibodies from Rheumatoid Arthritis Patients Activate Complement via Both the Classical and Alternative Pathways. Arthritis Rheum. 2009, 60, 1923–1931. [Google Scholar] [CrossRef]
- Aleyd, E.; Al, M.; Tuk, C.W.; van der Laken, C.J.; van Egmond, M. IgA Complexes in Plasma and Synovial Fluid of Patients with Rheumatoid Arthritis Induce Neutrophil Extracellular Traps via FcαRI. J. Immunol. 2016, 197, 4552–4559. [Google Scholar] [CrossRef]
- Paoliello-Paschoalato, A.B.; Marchi, L.F.; de Andrade, M.F.; Kabeya, L.M.; Donadi, E.A.; Lucisano-Valim, Y.M. Fcγ and Complement Receptors and Complement Proteins in Neutrophil Activation in Rheumatoid Arthritis: Contribution to Pathogenesis and Progression and Modulation by Natural Products. Evid. Based Complement. Altern. Med. 2015, 2015, 429878. [Google Scholar] [CrossRef]
- Nell, V.P.K.; Machold, K.P.; Stamm, T.A.; Eberl, G.; Heinzl, H.; Uffmann, M.; Smolen, J.S.; Steiner, G. Autoantibody Profiling as Early Diagnostic and Prognostic Tool for Rheumatoid Arthritis. Ann. Rheum. Dis. 2005, 64, 1731–1736. [Google Scholar] [CrossRef]
- de Moel, E.C.; Derksen, V.F.A.M.; Stoeken, G.; Trouw, L.A.; Bang, H.; Goekoop, R.J.; Speyer, I.; Huizinga, T.W.J.; Allaart, C.F.; Toes, R.E.M.; et al. Baseline Autoantibody Profile in Rheumatoid Arthritis Is Associated with Early Treatment Response but Not Long-Term Outcomes. Arthritis Res. Ther. 2018, 20, 33. [Google Scholar] [CrossRef] [PubMed]
- Martin-Mola, E.; Balsa, A.; García-Vicuna, R.; Gómez-Reino, J.; González-Gay, M.A.; Sanmartí, R.; Loza, E. Anti-Citrullinated Peptide Antibodies and Their Value for Predicting Responses to Biologic Agents: A Review. Rheumatol. Int. 2016, 36, 1043–1063. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.J. Tumor Necrosis Factor Inhibitors: Clinical Implications of Their Different Immunogenicity Profiles. Semin. Arthritis Rheum. 2005, 34, 19–22. [Google Scholar] [CrossRef] [PubMed]
- van Schouwenburg, P.A.; Rispens, T.; Wolbink, G.J. Immunogenicity of Anti-TNF Biologic Therapies for Rheumatoid Arthritis. Nat. Rev. Rheumatol. 2013, 9, 164–172. [Google Scholar] [CrossRef]
- Bartelds, G.M.; Krieckaert, C.L.M.; Nurmohamed, M.T.; van Schouwenburg, P.A.; Lems, W.F.; Twisk, J.W.R.; Dijkmans, B.A.C.; Aarden, L.; Wolbink, G.J. Development of Antidrug Antibodies against Adalimumab and Association with Disease Activity and Treatment Failure during Long-Term Follow-Up. JAMA 2011, 305, 1460–1468. [Google Scholar] [CrossRef]
- Pascual-Salcedo, D.; Plasencia, C.; Ramiro, S.; Nuño, L.; Bonilla, G.; Nagore, D.; Ruiz Del Agua, A.; Martínez, A.; Aarden, L.; Martín-Mola, E.; et al. Influence of Immunogenicity on the Efficacy of Long-Term Treatment with Infliximab in Rheumatoid Arthritis. Rheumatology 2011, 50, 1445–1452. [Google Scholar] [CrossRef]
- Hambardzumyan, K.; Hermanrud, C.; Marits, P.; Vivar, N.; Ernestam, S.; Wallman, J.K.; van Vollenhoven, R.F.; Fogdell-Hahn, A.; Saevarsdottir, S. Association of Female Sex and Positive Rheumatoid Factor with Low Serum Infliximab and Anti-Drug Antibodies, Related to Treatment Failure in Early Rheumatoid Arthritis: Results from the SWEFOT Trial Population. Scand. J. Rheumatol. 2019, 48, 362–366. [Google Scholar] [CrossRef]
- Ngo, S.T.; Steyn, F.J.; McCombe, P.A. Gender Differences in Autoimmune Disease. Front. Neuroendocrinol. 2014, 35, 347–369. [Google Scholar] [CrossRef]
- Quistrebert, J.; Hässler, S.; Bachelet, D.; Mbogning, C.; Musters, A.; Tak, P.P.; Wijbrandts, C.A.; Herenius, M.; Bergstra, S.A.; Akdemir, G.; et al. Incidence and Risk Factors for Adalimumab and Infliximab Anti-Drug Antibodies in Rheumatoid Arthritis: A European Retrospective Multicohort Analysis. Semin. Arthritis Rheum. 2019, 48, 967–975. [Google Scholar] [CrossRef]
- Bendtzen, K.; Geborek, P.; Svenson, M.; Larsson, L.; Kapetanovic, M.C.; Saxne, T. Individualized Monitoring of Drug Bioavailability and Immunogenicity in Rheumatoid Arthritis Patients Treated with the Tumor Necrosis Factor Alpha Inhibitor Infliximab. Arthritis Rheum. 2006, 54, 3782–3789. [Google Scholar] [CrossRef]
- Dore, R.K.; Mathews, S.; Schechtman, J.; Surbeck, W.; Mandel, D.; Patel, A.; Zhou, L.; Peloso, P. The Immunogenicity, Safety, and Efficacy of Etanercept Liquid Administered Once Weekly in Patients with Rheumatoid Arthritis. Clin. Exp. Rheumatol. 2007, 25, 40–46. [Google Scholar] [PubMed]
- Garcês, S.; Demengeot, J.; Benito-Garcia, E. The Immunogenicity of Anti-TNF Therapy in Immune-Mediated Inflammatory Diseases: A Systematic Review of the Literature with a Meta-Analysis. Ann. Rheum. Dis. 2013, 72, 1947–1955. [Google Scholar] [CrossRef] [PubMed]
- Moots, R.J.; Xavier, R.M.; Mok, C.C.; Rahman, M.U.; Tsai, W.-C.; Al-Maini, M.H.; Pavelka, K.; Mahgoub, E.; Kotak, S.; Korth-Bradley, J.; et al. The Impact of Anti-Drug Antibodies on Drug Concentrations and Clinical Outcomes in Rheumatoid Arthritis Patients Treated with Adalimumab, Etanercept, or Infliximab: Results from a Multinational, Real-World Clinical Practice, Non-Interventional Study. PLoS ONE 2017, 12, e0175207. [Google Scholar] [CrossRef]
- Pecoraro, V.; De Santis, E.; Melegari, A.; Trenti, T. The Impact of Immunogenicity of TNFα Inhibitors in Autoimmune Inflammatory Disease. A Systematic Review and Meta-Analysis. Autoimmun. Rev. 2017, 16, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.; Abdullah, A.; Frleta, M.; MacDonald, J.; McGucken, A. The Potential Value of Blood Monitoring of Biologic Drugs Used in the Treatment of Rheumatoid Arthritis. Ther. Adv. Musculoskelet. Dis. 2020, 12, 1759720X20904850. [Google Scholar] [CrossRef] [PubMed]
- Garcês, S.; Antunes, M.; Benito-Garcia, E.; da Silva, J.C.; Aarden, L.; Demengeot, J. A Preliminary Algorithm Introducing Immunogenicity Assessment in the Management of Patients with RA Receiving Tumour Necrosis Factor Inhibitor Therapies. Ann. Rheum. Dis. 2014, 73, 1138–1143. [Google Scholar] [CrossRef]
- Mehta, P.; Manson, J.J. What Is the Clinical Relevance of TNF Inhibitor Immunogenicity in the Management of Patients With Rheumatoid Arthritis? Front. Immunol. 2020, 11, 589. [Google Scholar] [CrossRef]
- Singh, J.A.; Saag, K.G.; Bridges, S.L.J.; Akl, E.A.; Bannuru, R.R.; Sullivan, M.C.; Vaysbrot, E.; McNaughton, C.; Osani, M.; Shmerling, R.H.; et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res. 2016, 68, 1–25. [Google Scholar] [CrossRef]
- Friedman, B.; Cronstein, B. Methotrexate Mechanism in Treatment of Rheumatoid Arthritis. Jt. Bone Spine 2019, 86, 301–307. [Google Scholar] [CrossRef]
- Food and Drug Administration Home Page. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/008085s068lbl.pdf (accessed on 15 November 2020).
- Plant, D.; Maciejewski, M.; Smith, S.; Nair, N.; Hyrich, K.; Ziemek, D.; Barton, A.; Verstappen, S. Profiling of Gene Expression Biomarkers as a Classifier of Methotrexate Nonresponse in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 678–684. [Google Scholar] [CrossRef]
- Singh, A.; Patro, P.S.; Aggarwal, A. MicroRNA-132, MiR-146a, and MiR-155 as Potential Biomarkers of Methotrexate Response in Patients with Rheumatoid Arthritis. Clin. Rheumatol. 2019, 38, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Sode, J.; Krintel, S.B.; Carlsen, A.L.; Hetland, M.L.; Johansen, J.S.; Hørslev-Petersen, K.; Stengaard-Pedersen, K.; Ellingsen, T.; Burton, M.; Junker, P.; et al. Plasma MicroRNA Profiles in Patients with Early Rheumatoid Arthritis Responding to Adalimumab plus Methotrexate vs Methotrexate Alone: A Placebo-Controlled Clinical Trial. J. Rheumatol. 2018, 45, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Thorn, C.F.; Yang, J.J.; Ulrich, C.M.; French, D.; Zaza, G.; Dunnenberger, H.M.; Marsh, S.; McLeod, H.L.; Giacomini, K.; et al. PharmGKB Summary: Methotrexate Pathway. Pharmacogenet. Genom. 2011, 21, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Huang, J.; Lin, Y.; Shu, X.; Fan, H.; Tu, Z.; Zhou, Y.; Xiao, C. Polymorphisms and Pharmacogenomics for the Toxicity of Methotrexate Monotherapy in Patients with Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Medicine 2017, 96, e6337. [Google Scholar] [CrossRef] [PubMed]
- Giletti, A.; Esperon, P. Genetic Markers in Methotrexate Treatments. Pharmacogenom. J. 2018, 18, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Drozdzik, M.; Rudas, T.; Pawlik, A.; Gornik, W.; Kurzawski, M.; Herczynska, M. Reduced Folate Carrier-1 80G>A Polymorphism Affects Methotrexate Treatment Outcome in Rheumatoid Arthritis. Pharmacogenom. J. 2007, 7, 404–407. [Google Scholar] [CrossRef]
- Li, H.; Hu, M.; Li, W. The association between reduced folate carrier-1 gene 80G/A polymorphism and methotrexate efficacy or methotrexate related-toxicity in rheumatoid arthritis: A meta-analysis. Int. Immunopharmacol. 2016, 38, 8–15. [Google Scholar] [CrossRef]
- Bohanec Grabar, P.; Logar, D.; Lestan, B.; Dolzan, V. Genetic Determinants of Methotrexate Toxicity in Rheumatoid Arthritis Patients: A Study of Polymorphisms Affecting Methotrexate Transport and Folate Metabolism. Eur. J. Clin. Pharmacol. 2008, 64, 1057–1068. [Google Scholar] [CrossRef]
- Lima, A.; Bernardes, M.; Azevedo, R.; Monteiro, J.; Sousa, H.; Medeiros, R.; Seabra, V. SLC19A1, SLC46A1 and SLCO1B1 Polymorphisms as Predictors of Methotrexate-Related Toxicity in Portuguese Rheumatoid Arthritis Patients. Toxicol. Sci. 2014, 142, 196–209. [Google Scholar] [CrossRef]
- James, H.M.; Gillis, D.; Hissaria, P.; Lester, S.; Somogyi, A.A.; Cleland, L.G.; Proudman, S.M. Common Polymorphisms in the Folate Pathway Predict Efficacy of Combination Regimens Containing Methotrexate and Sulfasalazine in Early Rheumatoid Arthritis. J. Rheumatol. 2008, 35, 562–571. [Google Scholar]
- Hayashi, H.; Tazoe, Y.; Tsuboi, S.; Horino, M.; Morishita, M.; Arai, T.; Ohshima, M.; Matsuyama, T.; Kosuge, K.; Yamada, H.; et al. A Single Nucleotide Polymorphism of Reduced Folate Carrier 1 Predicts Methotrexate Efficacy in Japanese Patients with Rheumatoid Arthritis. Drug Metab. Pharmacokinet. 2013, 28, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Takatori, R.; Takahashi, K.A.; Tokunaga, D.; Hojo, T.; Fujioka, M.; Asano, T.; Hirata, T.; Kawahito, Y.; Satomi, Y.; Nishino, H.; et al. ABCB1 C3435T Polymorphism Influences Methotrexate Sensitivity in Rheumatoid Arthritis Patients. Clin. Exp. Rheumatol. 2006, 24, 546–554. [Google Scholar] [PubMed]
- Jekic, B.; Maksimovic, N.; Damnjanovic, T. Methotrexate Pharmacogenetics in the Treatment of Rheumatoid Arthritis. Pharmacogenomics 2019, 20, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.M.; Pratt, A.G.; Isaacs, J.D. Mechanism of Action of Methotrexate in Rheumatoid Arthritis, and the Search for Biomarkers. Nat. Rev. Rheumatol. 2016, 12, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Hamon, Y.; Chimini, G. The Human ATP-Binding Cassette (ABC) Transporter Superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef]
- Valeria, C.; Carmine, S.; Valentina, M.; Teresa, I.; Maria, C.; Martina, T.; Giancarlo, A.; Giovanna, N.; Graziamaria, C.; Amelia, F. The Need of a Multicomponent Guiding Approach to Personalize Clopidogrel Treatment. Pharmacogenom. J. 2020. [Google Scholar] [CrossRef]
- Guan, Z.-W.; Wu, K.-R.; Li, R.; Yin, Y.; Li, X.-L.; Zhang, S.-F.; Li, Y. Pharmacogenetics of Statins Treatment: Efficacy and Safety. J. Clin. Pharm. Ther. 2019, 44, 858–867. [Google Scholar] [CrossRef]
- Li, M.; Seiser, E.L.; Baldwin, R.M.; Ramirez, J.; Ratain, M.J.; Innocenti, F.; Kroetz, D.L. ABC Transporter Polymorphisms Are Associated with Irinotecan Pharmacokinetics and Neutropenia. Pharmacogenom. J. 2018, 18, 35–42. [Google Scholar] [CrossRef]
- Pawlik, A.; Wrzesniewska, J.; Fiedorowicz-Fabrycy, I.; Gawronska-Szklarz, B. The MDR1 3435 Polymorphism in Patients with Rheumatoid Arthritis. Int. J. Clin. Pharmacol. Ther. 2004, 42, 496–503. [Google Scholar] [CrossRef]
- He, X.; Sun, M.; Liang, S.; Li, M.; Li, L.; Yang, Y. Association between ABCB1 C3435T Polymorphism and Methotrexate Treatment Outcomes in Rheumatoid Arthritis Patients: A Meta-Analysis. Pharmacogenomics 2019, 20, 381–392. [Google Scholar] [CrossRef]
- Lee, Y.H.; Bae, S.-C.; Song, G.G. Association of the ABCB1 C3435T Polymorphism with Responsiveness to and Toxicity of DMARDs in Rheumatoid Arthritis: A Meta-Analysis. Z. Rheumatol. 2016, 75, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Tavakolpour, S.; Darvishi, M.; Ghasemiadl, M. Pharmacogenetics: A Strategy for Personalized Medicine for Autoimmune Diseases. Clin. Genet. 2018, 93, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Guo, M.; Wu, D.-Q.; Meng, L. Pharmacogenomics of Methotrexate: Current Status and Future Outlook. Curr. Drug Metab. 2018, 19, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Conti, V.; De Bellis, E.; Manzo, V.; Sabbatino, F.; Iannello, F.; Dal Piaz, F.; Izzo, V.; Charlier, B.; Stefanelli, B.; Torsiello, M.; et al. A Genotyping/Phenotyping Approach with Careful Clinical Monitoring to Manage the Fluoropyrimidines-Based Therapy: Clinical Cases and Systematic Review of the Literature. J. Personal. Med. 2020, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, T.; Ferraz, J.-M.; Zinzindohoué, F.; Loriot, M.-A.; Tregouet, D.-A.; Landi, B.; Berger, A.; Cugnenc, P.-H.; Jian, R.; Beaune, P.; et al. Thymidylate Synthase Gene Polymorphism Predicts Toxicity in Colorectal Cancer Patients Receiving 5-Fluorouracil-Based Chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 5880–5888. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Fan, H.; Qiu, Q.; Liu, K.; Lv, S.; Li, J.; Yang, H.; Shu, X.; Xu, Y.; Lu, X.; et al. Are Gene Polymorphisms Related to Adverse Events of Methotrexate in Patients with Rheumatoid Arthritis? A Retrospective Cohort Study Based on an Updated Meta-Analysis. Ther. Adv. Chronic Dis. 2020, 11, 2040622320916026. [Google Scholar] [CrossRef]
- Fisher, M.C.; Cronstein, B.N. Metaanalysis of Methylenetetrahydrofolate Reductase (MTHFR) Polymorphisms Affecting Methotrexate Toxicity. J. Rheumatol. 2009, 36, 539–545. [Google Scholar] [CrossRef]
- Lee, Y.H.; Song, G.G. Associations between the C677T and A1298C Polymorphisms of MTHFR and the Efficacy and Toxicity of Methotrexate in Rheumatoid Arthritis: A Meta-Analysis. Clin. Drug Investig. 2010, 30, 101–108. [Google Scholar] [CrossRef]
- Song, G.G.; Bae, S.-C.; Ati, Y.H. Association of the MTHFR C677T and A1298C Polymorphisms with Methotrexate Toxicity in Rheumatoid Arthritis: A Meta-Analysis. Clin. Rheumatol. 2014, 33, 1715–1724. [Google Scholar] [CrossRef]
- Mori, S.; Hirose, J.; Yonemura, K. Contribution of HLA-DRB1*04 Alleles and Anti-Cyclic Citrullinated Antibodies toDevelopment of Resistance to Disease-Modifying Antirheumatic Drugs in Early Rheumatoid Arthritis. Clin. Rheumatol. 2010, 29, 1357–1366. [Google Scholar] [CrossRef]
- Wessels, J.A.M.; Kooloos, W.M.; De Jonge, R.; De Vries-Bouwstra, J.K.; Allaart, C.F.; Linssen, A.; Collee, G.; De Sonnaville, P.; Lindemans, J.; Huizinga, T.W.J.; et al. Relationship between Genetic Variants in the Adenosine Pathway and Outcome of Methotrexate Treatment in Patients with Recent-Onset Rheumatoid Arthritis. Arthritis Rheum. 2006, 54, 2830–2839. [Google Scholar] [CrossRef] [PubMed]
- Weisman, M.H.; Furst, D.E.; Park, G.S.; Kremer, J.M.; Smith, K.M.; Wallace, D.J.; Caldwell, J.R.; Dervieux, T. Risk Genotypes in Folate-Dependent Enzymes and Their Association with Methotrexate-Related Side Effects in Rheumatoid Arthritis. Arthritis Rheum. 2006, 54, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Bae, S.-C. Association of the ATIC 347 C/G Polymorphism with Responsiveness to and Toxicity of Methotrexate in Rheumatoid Arthritis: A Meta-Analysis. Rheumatol. Int. 2016, 36, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.C.; Bongartz, T.; Massey, J.; Mifsud, B.; Spiliopoulou, A.; Scott, I.C.; Wang, J.; Morgan, M.; Plant, D.; Colombo, M.; et al. Genome-Wide Association Study of Response to Methotrexate in Early Rheumatoid Arthritis Patients. Pharmacogenom. J. 2018, 18, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Senapati, S.; Singh, S.; Das, M.; Kumar, A.; Gupta, R.; Kumar, U.; Jain, S.; Juyal, R.C.; Thelma, B.K. Genome-Wide Analysis of Methotrexate Pharmacogenomics in Rheumatoid Arthritis Shows Multiple Novel Risk Variants and Leads for TYMS Regulation. Pharmacogenet. Genom. 2014, 24, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Dziedziejko, V.; Kurzawski, M.; Safranow, K.; Chlubek, D.; Pawlik, A. The Effect of ESR1 and ESR2 Gene Polymorphisms on the Outcome of Rheumatoid Arthritis Treatment with Leflunomide. Pharmacogenomics 2011, 12, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Montagna, P.; Brizzolara, R.; Sulli, A.; Seriolo, B.; Villaggio, B.; Triolo, P.; Clerico, P.; Soldano, S. Sex Hormones Modulate the Effects of Leflunomide on Cytokine Production by Cultures of Differentiated Monocyte/Macrophages and Synovial Macrophages from Rheumatoid Arthritis Patients. J. Autoimmun. 2009, 32, 254–260. [Google Scholar] [CrossRef]
- Chamberlain, N.L.; Driver, E.D.; Miesfeld, R.L. The Length and Location of CAG Trinucleotide Repeats in the Androgen Receptor N-Terminal Domain Affect Transactivation Function. Nucleic Acids Res. 1994, 22, 3181–3186. [Google Scholar] [CrossRef]
- Dziedziejko, V.; Kurzawski, M.; Safranow, K.; Ossowski, A.; Piatek, J.; Parafiniuk, M.; Chlubek, D.; Pawlik, A. Lack of Association between CAG Repeat Polymorphism in the Androgen Receptor Gene and the Outcome of Rheumatoid Arthritis Treatment with Leflunomide. Eur. J. Clin. Pharmacol. 2012, 68, 371–377. [Google Scholar] [CrossRef]
- Giwercman, Y.L.; Xu, C.; Arver, S.; Pousette, A.; Reneland, R. No Association between the Androgen Receptor Gene CAG Repeat and Impaired Sperm Production in Swedish Men. Clin. Genet. 1998, 54, 435–436. [Google Scholar]
- Zitzmann, M.; Nieschlag, E. The CAG Repeat Polymorphism within the Androgen Receptor Gene and Maleness. Int. J. Androl. 2003, 26, 76–83. [Google Scholar] [CrossRef]
- Pawlik, A.; Herczynska, M.; Kurzawski, M.; Safranow, K.; Dziedziejko, V.; Drozdzik, M. The Effect of Exon (19C>A) Dihydroorotate Dehydrogenase Gene Polymorphism on Rheumatoid Arthritis Treatment with Leflunomide. Pharmacogenomics 2009, 10, 303–309. [Google Scholar] [CrossRef]
- Hopkins, A.M.; Wiese, M.D.; Proudman, S.M.; O’Doherty, C.E.; Upton, R.N.; Foster, D.J.R. Genetic Polymorphism of CYP1A2 but Not Total or Free Teriflunomide Concentrations Is Associated with Leflunomide Cessation in Rheumatoid Arthritis. Br. J. Clin. Pharmacol. 2016, 81, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Bohanec Grabar, P.; Rozman, B.; Tomsic, M.; Suput, D.; Logar, D.; Dolzan, V. Genetic Polymorphism of CYP1A2 and the Toxicity of Leflunomide Treatment in Rheumatoid Arthritis Patients. Eur. J. Clin. Pharmacol. 2008, 64, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M.D.; Alotaibi, N.; O’Doherty, C.; Sorich, M.J.; Suppiah, V.; Cleland, L.G.; Proudman, S.M. Pharmacogenomics of NAT2 and ABCG2 Influence the Toxicity and Efficacy of Sulphasalazine Containing DMARD Regimens in Early Rheumatoid Arthritis. Pharmacogenom. J. 2014, 14, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Salliot, C.; Finckh, A.; Katchamart, W.; Lu, Y.; Sun, Y.; Bombardier, C.; Keystone, E. Indirect Comparisons of the Efficacy of Biological Antirheumatic Agents in Rheumatoid Arthritis in Patients with an Inadequate Response to Conventional Disease-Modifying Antirheumatic Drugs or to an Anti-Tumour Necrosis Factor Agent: A Meta-Analysis. Ann. Rheum. Dis. 2011, 70, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Mugnier, B.; Balandraud, N.; Darque, A.; Roudier, C.; Roudier, J.; Reviron, D. Polymorphism at position -308 of the tumor necrosis factor alpha gene influences outcome of infliximab therapy in rheumatoid arthritis. Arthritis Rheum. 2003, 48, 1849–1852. [Google Scholar] [CrossRef] [PubMed]
- Guis, S.; Balandraud, N.; Bouvenot, J.; Auger, I.; Toussirot, E.; Wendling, D.; Mattei, J.P.; Nogueira, L.; Mugnier, B.; Legeron, P. Influence of -308 A/G polymorphism in the tumor necrosis factor alpha gene on etanercept treatment in rheumatoid arthritis. Arthritis Rheum. 2007, 57, 1426–1430. [Google Scholar] [CrossRef]
- Cuchacovich, M.; Soto, L.; Edwardes, M.; Gutierrez, M.; Llanos, C.; Pacheco, D.; Sabugo, F.; Alamo, M.; Fuentealba, C.; Villanueva, L.; et al. Tumour necrosis factor (TNF)alpha -308 G/G promoter polymorphism and TNFalpha levels correlate with a better response to adalimumab in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2006, 35, 435–440. [Google Scholar] [CrossRef]
- Zeng, Z.; Duan, Z.; Zhang, T.; Wang, S.; Li, G.; Gao, J.; Ye, D.; Xu, S.; Xu, J.; Zhang, L. Association between tumor necrosis factor-α (TNF-α) promoter -308 G/A and response to TNF-α blockers in rheumatoid arthritis: A meta-analysis. Mod. Rheumatol. 2013, 23, 489–495. [Google Scholar] [CrossRef]
- Maxwell, J.R.; Potter, C.; Hyrich, K.L.; Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate; Barton, A.; Worthington, J.; Isaacs, J.D.; Morgan, A.W.; Wilson, A.G. Association of the tumour necrosis factor-308 variant with differential response to anti-TNF agents in the treatment of rheumatoid arthritis. Hum. Mol Genet. 2008, 17, 3532–3538. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.P.; Lee, K.W.; Yoo, D.H.; Kang, C.; Bae, S.C. The influence of a polymorphism at position -857 of the tumour necrosis factor alpha gene on clinical response to etanercept therapy in rheumatoid arthritis. Rheumatology 2005, 44, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Miceli-Richard, C.; Comets, E.; Verstuyft, C.; Tamouza, R.; Loiseau, P.; Ravaud, P.; Kupper, H.; Becquemont, L.; Charron, D.; Mariette, X. A single tumour necrosis factor haplotype influences the response to adalimumab in rheumatoid arthritis. Ann. Rheum Dis. 2008, 67, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Ongaro, A.; De Mattei, M.; Pellati, A.; Caruso, A.; Ferretti, S.; Masieri, F.F.; Fotinidi, M.; Farina, I.; Trotta, F.; Padovan, M. Can tumor necrosis factor receptor II gene 676T>G polymorphism predict the response grading to anti-TNFalpha therapy in rheumatoid arthritis? Rheumatol Int. 2008, 28, 901–908. [Google Scholar] [CrossRef]
- Swierkot, J.; Bogunia-Kubik, K.; Nowak, B.; Bialowas, K.; Korman, L.; Gebura, K.; Kolossa, K.; Jeka, S.; Wiland, P. Analysis of associations between polymorphisms within genes coding for tumour necrosis factor (TNF)-alpha and TNF receptors and responsiveness to TNF-alpha blockers in patients with rheumatoid arthritis. Jt. Bone Spine 2015, 82, 94–99. [Google Scholar] [CrossRef]
- Canet, L.M.; Filipescu, I.; Cáliz, R.; Lupiañez, C.B.; Canhão, H.; Escudero, A.; Segura-Catena, J.; Soto-Pino, M.J.; Ferrer, M.A.; García, A.; et al. Genetic variants within the TNFRSF1B gene and susceptibility to rheumatoid arthritis and response to anti-TNF drugs: A multicenter study. Pharmacogenet. Genom. 2015, 25, 323–333. [Google Scholar] [CrossRef]
- Chen, W.; Xu, H.; Wang, X.; Gu, J.; Xiong, H.; Shi, Y. The tumor necrosis factor receptor superfamily member 1B polymorphisms predict response to anti-TNF therapy in patients with autoimmune disease: A meta-analysis. Int. Immunopharmacol. 2015, 28, 146–153. [Google Scholar] [CrossRef]
- Cui, J.; Stahl, E.A.; Saevarsdottir, S.; Miceli, C.; Diogo, D.; Trynka, G.; Raj, T.; Mirkov, M.U.; Canhao, H.; Ikari, K.; et al. Genome-Wide Association Study and Gene Expression Analysis Identifies CD84 as a Predictor of Response to Etanercept Therapy in Rheumatoid Arthritis. PLoS Genet. 2013, 9, e1003394. [Google Scholar] [CrossRef]
- Plant, D.; Bowes, J.; Potter, C.; Hyrich, K.L.; Morgan, A.W.; Wilson, A.G.; Isaacs, J.D.; Barton, A. Genome-Wide Association Study of Genetic Predictors of Anti-Tumor Necrosis Factor Treatment Efficacy in Rheumatoid Arthritis Identifies Associations with Polymorphisms at Seven Loci. Arthritis Rheum. 2011, 63, 645–653. [Google Scholar] [CrossRef]
- van Baarsen, L.G.; Wijbrandts, C.A.; Rustenburg, F.; Cantaert, T.; van der Pouw Kraan, T.C.; Baeten, D.L.; Dijkmans, B.A.; Tak, P.P.; Verweij, C.L. Regulation of IFN Response Gene Activity during Infliximab Treatment in Rheumatoid Arthritis Is Associated with Clinical Response to Treatment. Arthritis Res. Ther. 2010, 12, R11. [Google Scholar] [CrossRef]
- Guilliams, M.; Bruhns, P.; Saeys, Y.; Hammad, H.; Lambrecht, B.N. The Function of Fcγ Receptors in Dendritic Cells and Macrophages. Nat. Rev. Immunol. 2014, 14, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Cañete, J.D.; Suárez, B.; Hernández, M.V.; Sanmartí, R.; Rego, I.; Celis, R.; Moll, C.; Pinto, J.A.; Blanco, F.J.; Lozano, F. Influence of Variants of Fc Gamma Receptors IIA and IIIA on the American College of Rheumatology and European League Against Rheumatism Responses to Anti-Tumour Necrosis Factor Alpha Therapy in Rheumatoid Arthritis. Ann. Rheum. Dis. 2009, 68, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Montes, A.; Perez-Pampin, E.; Narváez, J.; Cañete, J.D.; Navarro-Sarabia, F.; Moreira, V.; Fernández-Nebro, A.; Del Carmen Ordóñez, M.; de la Serna, A.R.; Magallares, B.; et al. Association of FCGR2A with the Response to Infliximab Treatment of Patients with Rheumatoid Arthritis. Pharmacogenet. Genom. 2014, 24, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Criswell, L.A.; Lum, R.F.; Turner, K.N.; Woehl, B.; Zhu, Y.; Wang, J.; Tiwari, H.K.; Edberg, J.C.; Kimberly, R.P.; Moreland, L.W.; et al. The Influence of Genetic Variation in the HLA-DRB1 and LTA-TNF Regions on the Response to Treatment of Early Rheumatoid Arthritis with Methotrexate or Etanercept. Arthritis Rheum. 2004, 50, 2750–2756. [Google Scholar] [CrossRef] [PubMed]
- Avila-Pedretti, G.; Tornero, J.; Fernández-Nebro, A.; Blanco, F.; González-Alvaro, I.; Cañete, J.D.; Maymó, J.; Alperiz, M.; Fernández-Gutiérrez, B.; Olivé, A.; et al. Variation at FCGR2A and Functionally Related Genes Is Associated with the Response to Anti-TNF Therapy in Rheumatoid Arthritis. PLoS ONE 2015, 10, e0122088. [Google Scholar] [CrossRef]
- Bek, S.; Bojesen, A.B.; Nielsen, J.V.; Sode, J.; Bank, S.; Vogel, U.; Andersen, V. Systematic Review and Meta-Analysis: Pharmacogenetics of Anti-TNF Treatment Response in Rheumatoid Arthritis. Pharmacogenom. J. 2017, 17, 403–411. [Google Scholar] [CrossRef]
- Jiménez Morales, A.; Maldonado-Montoro, M.; Martínez de la Plata, J.E.; Pérez Ramírez, C.; Daddaoua, A.; Alarcón Payer, C.; Expósito Ruiz, M.; García Collado, C. FCGR2A/FCGR3A Gene Polymorphisms and Clinical Variables as Predictors of Response to Tocilizumab and Rituximab in Patients With Rheumatoid Arthritis. J. Clin. Pharmacol. 2019, 59, 517–531. [Google Scholar] [CrossRef]
- Mirouse, A.; Seror, R.; Vicaut, E.; Mariette, X.; Dougados, M.; Fauchais, A.-L.; Deroux, A.; Dellal, A.; Costedoat-Chalumeau, N.; Denis, G.; et al. Arthritis in Primary Sjögren’s Syndrome: Characteristics, Outcome and Treatment from French Multicenter Retrospective Study. Autoimmun. Rev. 2019, 18, 9–14. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| References | Pts (n) | Biomarkers | Conclusions of the Study |
|---|---|---|---|
| Lard et al. (2002) [27] | 361 | HLA class II antigens | An early and aggressive treatment with DMARDs, such as MTX, sulfasalazine, and chloroquine can regulate the immune process, maybe by preventing a secondary release of autoantigens, by modulating the autoantigen presentation by APCs to the CD4+ T cells, and/or by inhibiting the response of T cells. However, this approach is mainly effective in a subgroup of patients with shared epitope positivity. |
| de Moel et al. (2018) [52] | 399 | IgG, IgM, and IgA isotypes for ACPA and anti-CarP, IgM, and IgA RFs; AMPAs | The presence at baseline of multiple autoantibody types could improve the early response to drug therapy, reflecting a more active humoral immunity. The impact of the baseline autoantibody profile on treatment outcomes diminishes over time. |
| Pascual-Salcedo et al. (2011) [57] | 85 | ADA | The presence of ADA, like anti-infliximab Abs, may compromise efficacy and safety of the therapy. The formation of anti-infliximab Abs is associated with appearance of infusion reactions, discontinuation of therapy, and poor clinical response. |
| Hambardzumyan et al. (2019) [58] | 128 | ADA, RF | ADA development, low serum infliximab, and RF positivity were more common in females and were associated with treatment failure. |
| Quistrebert et al. (2019) [60] | 366 | ADA | In RA patients treated with adalimumab or infliximab and co-treated with MTX, longer disease duration, moderate disease activity, and lifetime smoking were all factors associated with ADA development. |
| Bartelds et al. (2011) [56] | 272 | ADA | The production of ADA is associated with a negative outcome. Patients with ADA against adalimumab discontinue therapy earlier and more frequently and less often achieved remission. The presence of ADA influenced serum adalimumab concentrations. |
| Bendtzen et al. (2006) [61] | 106 | ADA | Up to 44% of patients treated with infliximab developed ADA in the first 6 months of treatment. ADA development is associated with increased risk of infusion reaction and treatment failure. |
| Dore et al. (2007) [62] | 222 | ADA | ADA, all non-neutralizing Abs, were detected in few patients and their presence did not affect etanercept safety or efficacy, except for few cases of serious adverse events (6.3%) and serious infections (2.3%). |
| Garcês et al. (2012) [63] | 1801 | ADA | In patients treated with anti-TNFα, the development of ADA reduces the drug efficacy. This event can be avoided by administration of other immunosuppressive agents. |
| Moots et al. (2017) [64] | 595 | ADA | All patients treated with etanercept did not develop ADA, while ADA were present in patients treated with adalimumab and infliximab. Patients negative for ADA generally showed better clinical outcomes than those who were ADA-positive. |
| Pecoraro et al. (2017) [65] | 4273 | ADA | ADA reduced drug response in patients treated with anti-TNFα, especially infliximab or adalimumab. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conti, V.; Corbi, G.; Costantino, M.; De Bellis, E.; Manzo, V.; Sellitto, C.; Stefanelli, B.; Colucci, F.; Filippelli, A. Biomarkers to Personalize the Treatment of Rheumatoid Arthritis: Focus on Autoantibodies and Pharmacogenetics. Biomolecules 2020, 10, 1672. https://doi.org/10.3390/biom10121672
Conti V, Corbi G, Costantino M, De Bellis E, Manzo V, Sellitto C, Stefanelli B, Colucci F, Filippelli A. Biomarkers to Personalize the Treatment of Rheumatoid Arthritis: Focus on Autoantibodies and Pharmacogenetics. Biomolecules. 2020; 10(12):1672. https://doi.org/10.3390/biom10121672
Chicago/Turabian StyleConti, Valeria, Graziamaria Corbi, Maria Costantino, Emanuela De Bellis, Valentina Manzo, Carmine Sellitto, Berenice Stefanelli, Francesca Colucci, and Amelia Filippelli. 2020. "Biomarkers to Personalize the Treatment of Rheumatoid Arthritis: Focus on Autoantibodies and Pharmacogenetics" Biomolecules 10, no. 12: 1672. https://doi.org/10.3390/biom10121672
APA StyleConti, V., Corbi, G., Costantino, M., De Bellis, E., Manzo, V., Sellitto, C., Stefanelli, B., Colucci, F., & Filippelli, A. (2020). Biomarkers to Personalize the Treatment of Rheumatoid Arthritis: Focus on Autoantibodies and Pharmacogenetics. Biomolecules, 10(12), 1672. https://doi.org/10.3390/biom10121672