Structural Insight into the Contributions of the N-Terminus and Key Active-Site Residues to the Catalytic Efficiency of Glutamine Synthetase 2

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Expression and Purification of DmGS2
2.1.2. Bacteria
2.2. Crystallisation, Data Collection, and Refinement
2.3. Enzyme Assays
2.4. Kinetic Analysis
2.5. Analytical Ultracentrifugation
2.6. Biophysical Properties of WT DmGS2 and R4D
3. Results and Discussion
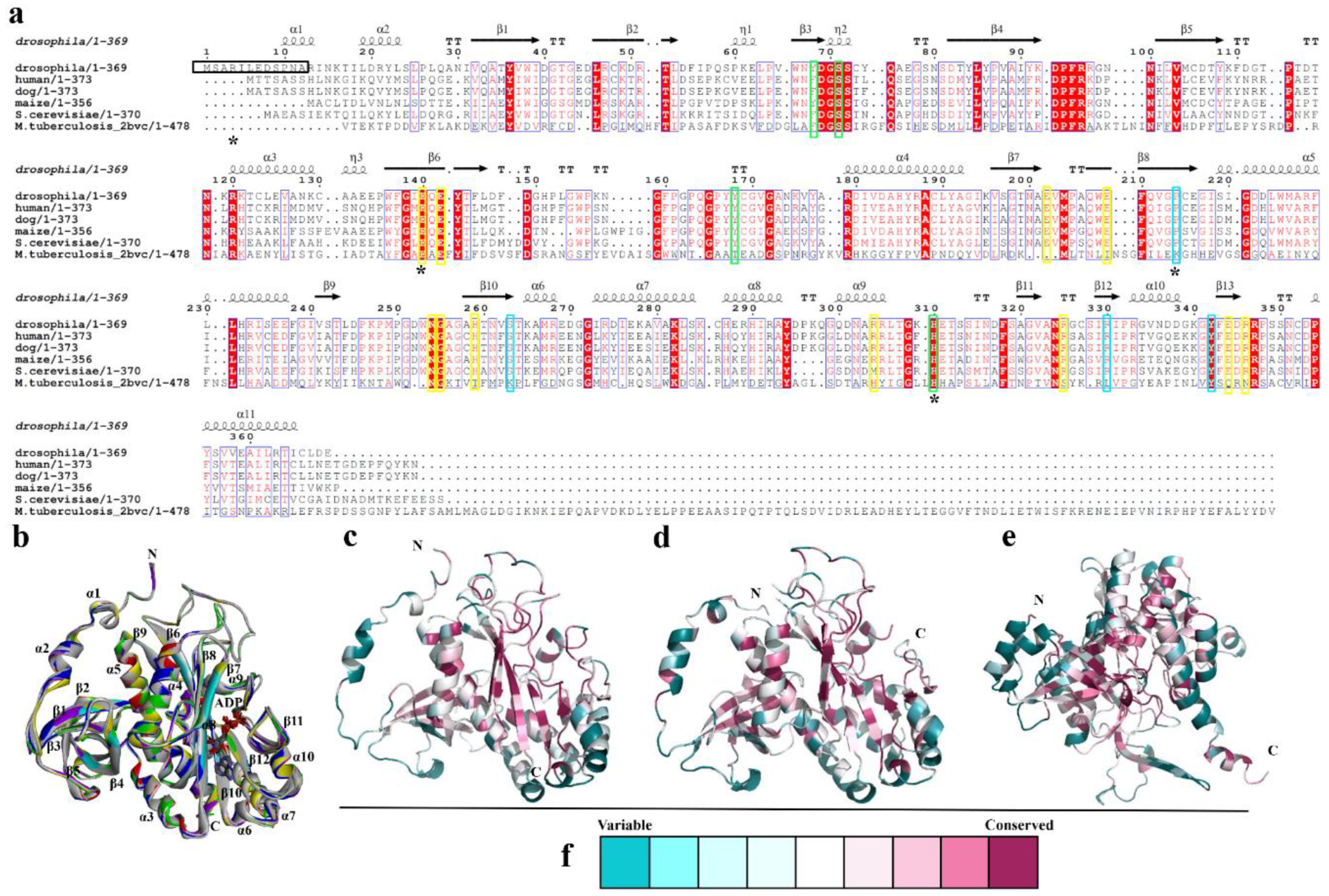
3.1. Overall structure of DmGS2
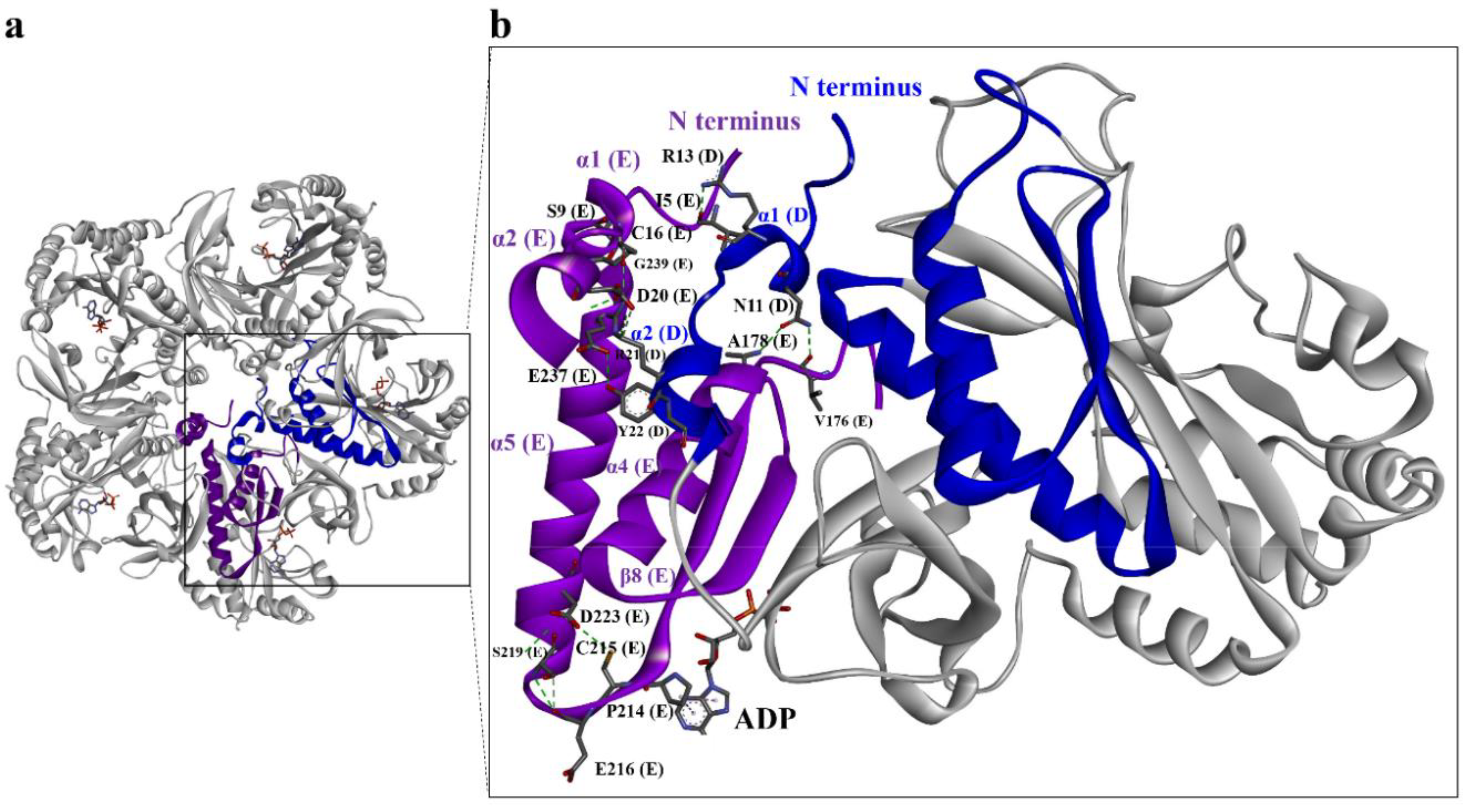
3.2. Quaternary Structure of DmGS2
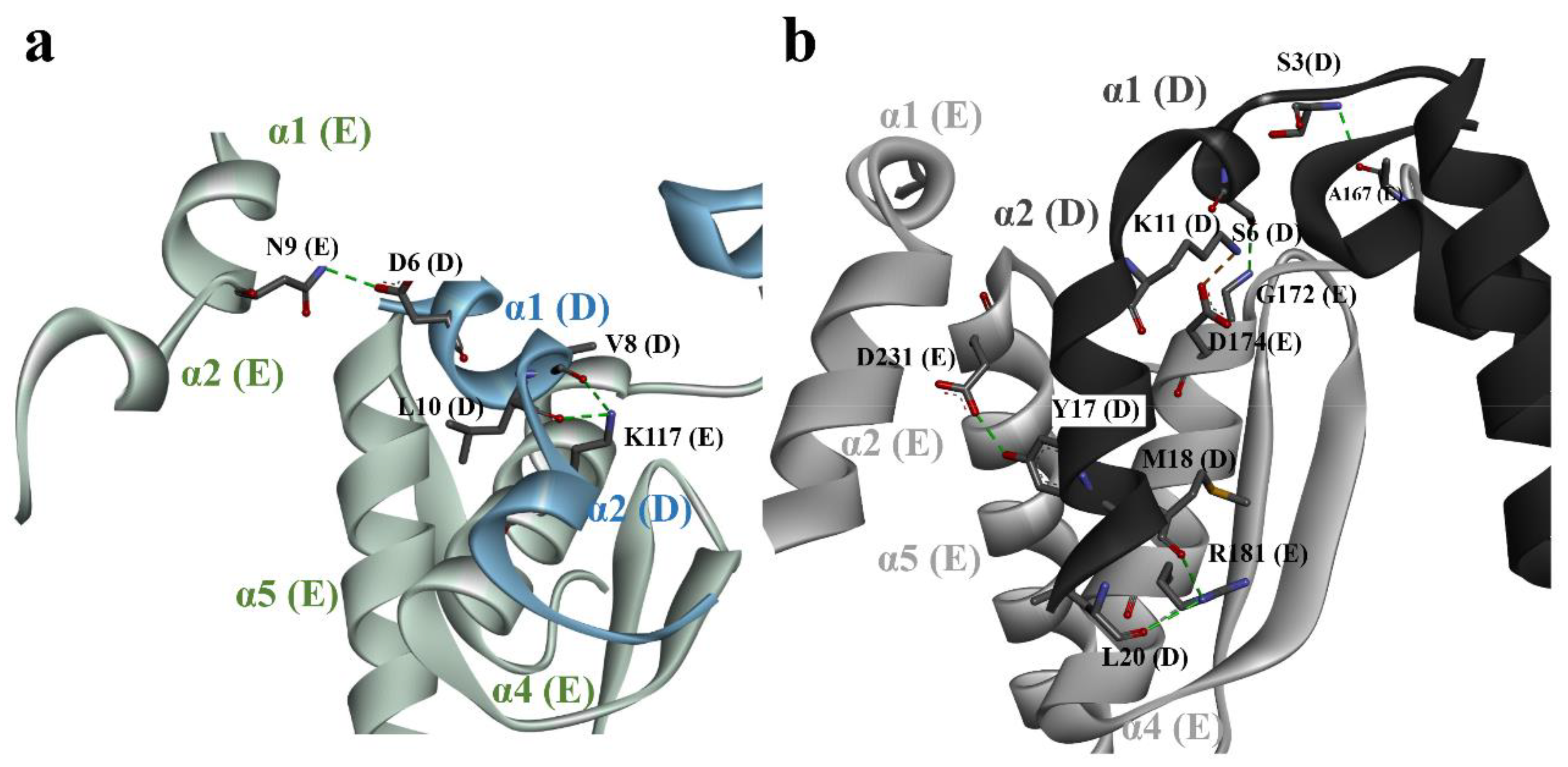
3.3. Structural Comparison of N-Terminal Meander Region of Maize and Human GSs
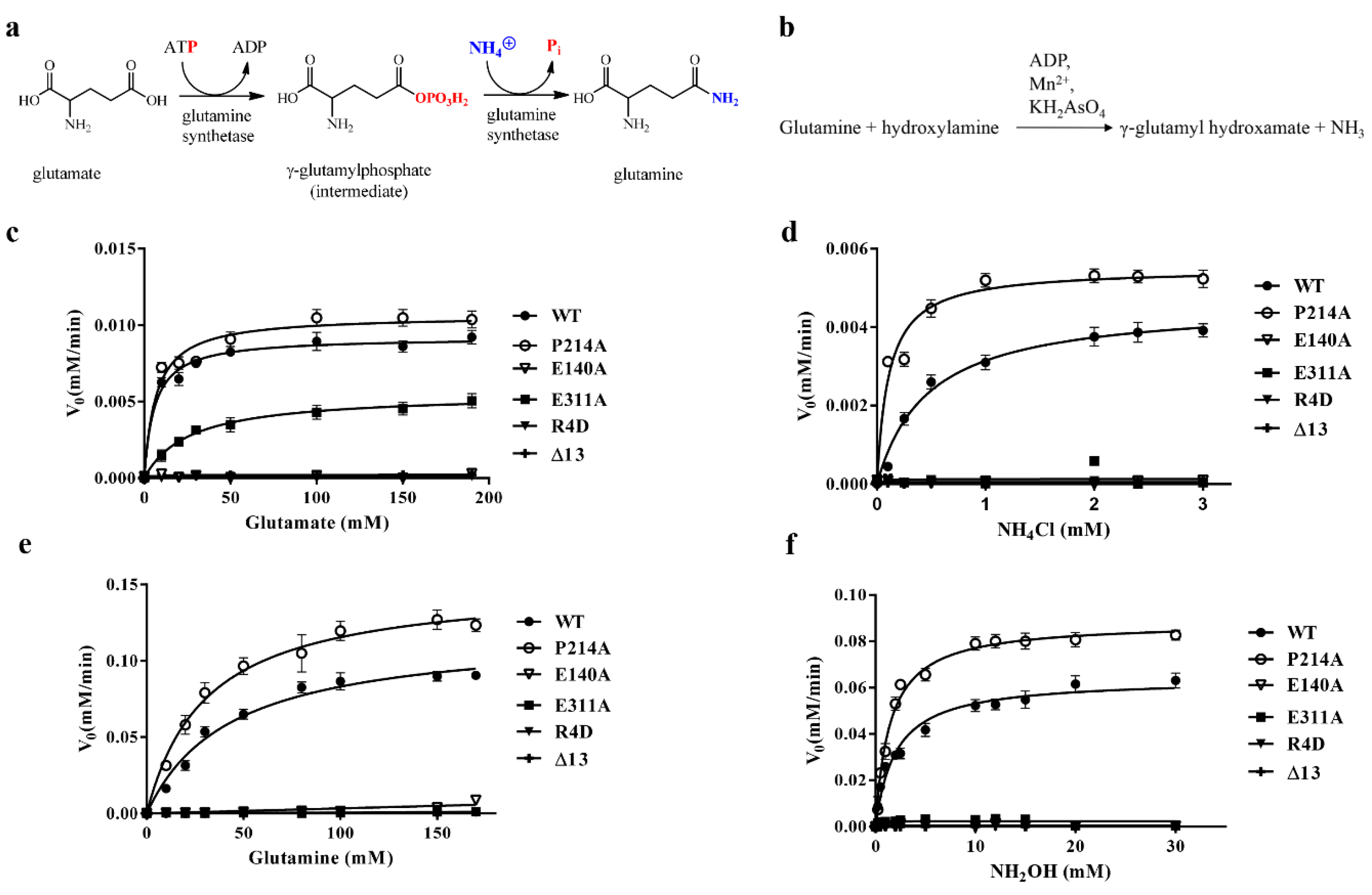
3.4. DmGS2 Mutants and Kinetic Assay
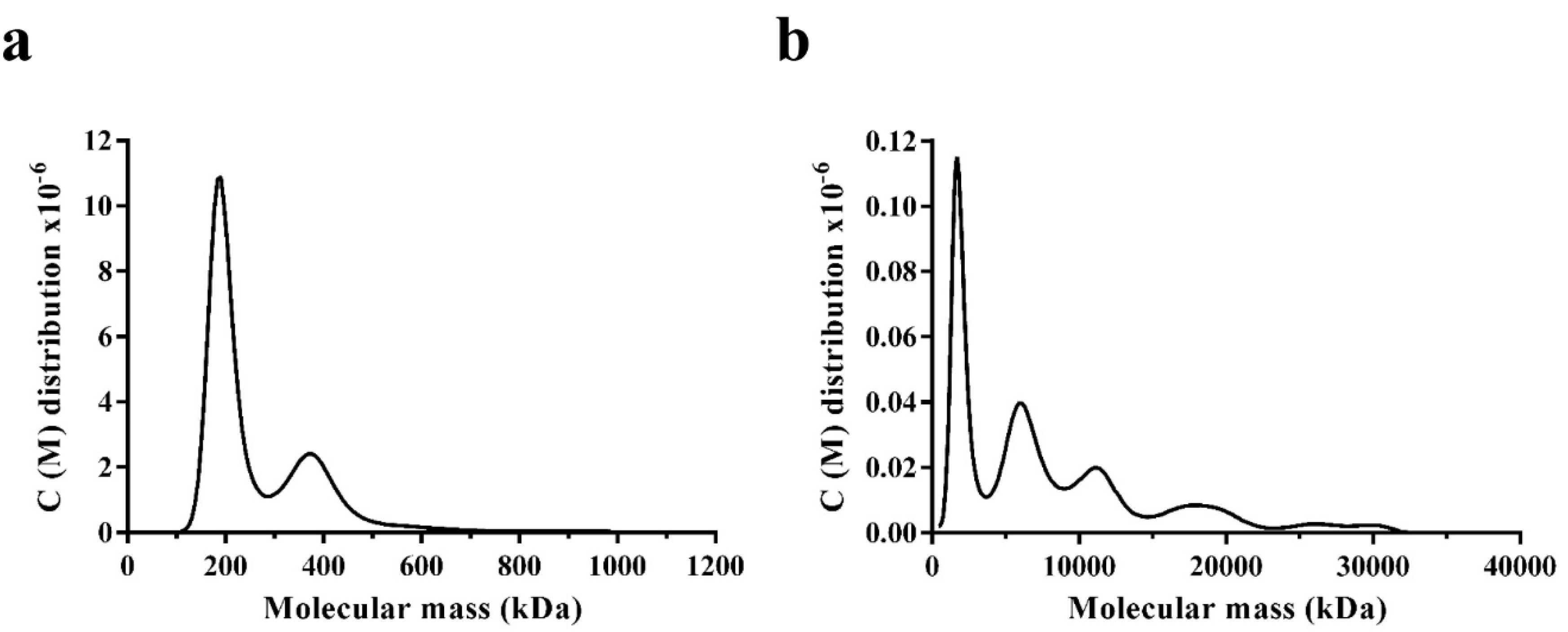
3.5. Oligomerisation States of WT DmGS2 and R4D
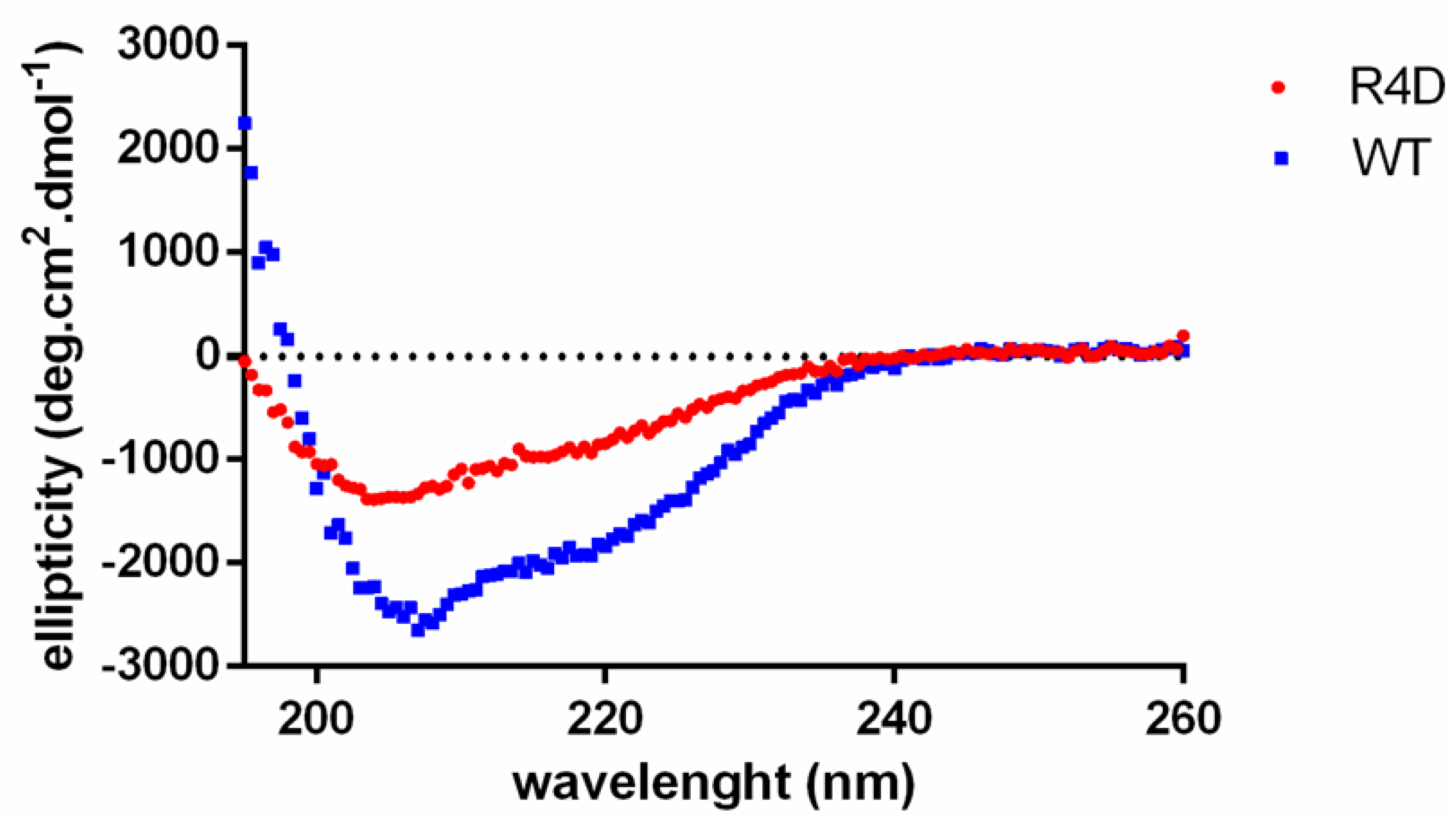
3.6. Biophysical Properties of Recombinant WT DmGS2 and R4D
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eisenberg, D.; Gill, H.S.; Pfluegl, G.M.; Rotstein, S.H. Structure-function relationships of glutamine synthetases. Biochim. Biophys. Acta 2000, 1477, 122–145. [Google Scholar] [CrossRef]
- Krajewski, W.W.; Collins, R.; Holmberg-Schiavone, L.; Jones, T.A.; Karlberg, T.; Mowbray, S.L. Crystal structures of mammalian glutamine synthetases illustrate substrate-induced conformational changes and provide opportunities for drug and herbicide design. J. Mol. Biol. 2008, 375, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Gunnersen, D.; Haley, B. Detection of glutamine synthetase in the cerebrospinal fluid of Alzheimer diseased patients: A potential diagnostic biochemical marker. Proc. Natl. Acad. Sci. USA 1992, 89, 11949–11953. [Google Scholar] [CrossRef] [PubMed]
- Kusnan, M.B.; Berger, M.G.; Fock, H.P. The involvement of glutamine synthetase/glutamate synthase in ammonia assimilation by Aspergillus nidulans. J. Gen. Microbiol. 1987, 133, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Magasanik, B. Genetic control of nitrogen assimilation in bacteria. Annu. Rev. Genet. 1982, 16, 135–168. [Google Scholar] [CrossRef]
- Caizzi, R.; Bozzetti, M.P.; Caggese, C.; Ritossa, F. Homologous nuclear genes encode cytoplasmic and mitochondrial glutamine synthetase in Drosophila melanogaster. J. Mol. Biol. 1990, 212, 17–26. [Google Scholar] [CrossRef]
- Caggese, C.; Caizzi, R.; Bozzetti, M.P.; Barsanti, P.; Ritossa, F. Genetic determinants of glutamine synthetase in Drosophila melanogaster: A gene for glutamine synthetase I resides in the 21B3-6 region. Biochem. Genet. 1988, 26, 571–584. [Google Scholar] [CrossRef]
- De Pinto, V.; Caggese, C.; Prezioso, G.; Ritossa, F. Purification of the glutamine synthetase II isozyme of Drosophila melanogaster and structural and functional comparison of glutamine synthetases I and II. Biochem. Genet. 1987, 25, 821–836. [Google Scholar] [CrossRef]
- Caggese, C.; Barsanti, P.; Viggiano, L.; Bozzetti, M.P.; Caizzi, R. Genetic, molecular and developmental analysis of the glutamine synthetase isozymes of Drosophila melanogaster. Genetica 1994, 94, 275–281. [Google Scholar] [CrossRef]
- Sinakevitch, I.; Grau, Y.; Strausfeld, N.J.; Birman, S. Dynamics of glutamatergic signaling in the mushroom body of young adult Drosophila. Neural Dev. 2010, 5, 10. [Google Scholar] [CrossRef]
- Heisenberg, M. Mushroom body memoir: From maps to models. Nat. Rev. Neurosci. 2003, 4, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Unno, H.; Uchida, T.; Sugawara, H.; Kurisu, G.; Sugiyama, T.; Yamaya, T.; Sakakibara, H.; Hase, T.; Kusunoki, M. Atomic structure of plant glutamine synthetase: A key enzyme for plant productivity. J. Biol. Chem. 2006, 281, 29287–29296. [Google Scholar] [CrossRef] [PubMed]
- He, Y.X.; Gui, L.; Liu, Y.Z.; Du, Y.; Zhou, Y.; Li, P.; Zhou, C.Z. Crystal structure of Saccharomyces cerevisiae glutamine synthetase Gln1 suggests a nanotube-like supramolecular assembly. Proteins 2009, 76, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.S.; Pfluegl, G.M.; Eisenberg, D. Preliminary crystallographic studies on glutamine synthetase from Mycobacterium tuberculosis. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 865–868. [Google Scholar] [CrossRef]
- Almassy, R.J.; Janson, C.A.; Hamlin, R.; Xuong, N.H.; Eisenberg, D. Novel subunit-subunit interactions in the structure of glutamine synthetase. Nature 1986, 323, 304–309. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Chen, Y.W.; Chou, H.C.; Lyu, P.C.; Yin, H.S.; Huang, F.L.; Chang, W.S.; Fan, C.Y.; Tu, I.F.; Lai, T.C.; Lin, S.T. Mitochondrial proteomics analysis of tumorigenic and metastatic breast cancer markers. Funct. Integr. Genom. 2011, 11, 225–239. [Google Scholar] [CrossRef]
- Collaborative Computational Project, N. The CCP4 suite: Programs for protein crystallography. Acta Cryst. D Biol. Cryst. 1994, 50, 760–763. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A. Overview of the CCP4 suite and current developments. Acta Cryst. D 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Cryst. D Biol. Cryst. 2010, 66, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. D Biol. Cryst. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. D Biol. Cryst. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.L.; MacArthur, M.W.; Hutchinson, E.G.; Thornton, J.M. Stereochemical quality of protein structure coordinates. Proteins 1992, 12, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Ronzio, R.A.; Wilk, S.; Rowe, W.B.; Meister, A. Preparation and studies on the characterization of sheep brain glutamine synthetase. Biochemistry 1969, 8, 2670–2674. [Google Scholar] [CrossRef]
- Gawronski, J.D.; Benson, D.R. Microtiter assay for glutamine synthetase biosynthetic activity using inorganic phosphate detection. Anal. Biochem. 2004, 327, 114–118. [Google Scholar] [CrossRef]
- Lebowitz, J.; Lewis, M.S.; Schuck, P. Modern analytical ultracentrifugation in protein science: A tutorial review. Protein Sci. 2002, 11, 2067–2079. [Google Scholar] [CrossRef]
- Greenfield, N.J. Analysis of circular dichroism data. Methods Enzymol. 2004, 383, 282–317. [Google Scholar]
- Vaguine, A.A.; Richelle, J.; Wodak, S.J. SFCHECK: A unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 191–205. [Google Scholar] [CrossRef]
- Hooft, R.W.; Sander, C.; Vriend, G. Objectively judging the quality of a protein structure from a Ramachandran plot. Comput. Appl. Biosci. 1997, 13, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Huang, W.; Liao, J.L. QM/MM investigation of ATP hydrolysis in aqueous solution. J. Phys. Chem. B 2015, 119, 3720–3726. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.H. Magnesium ion catalyzed ATP hydrolysis. J. Am. Chem. Soc. 2000, 122, 12023–12024. [Google Scholar] [CrossRef]
- Krajewski, W.W.; Jones, T.A.; Mowbray, S.L. Structure of Mycobacterium tuberculosis glutamine synthetase in complex with a transition-state mimic provides functional insights. Proc. Natl. Acad. Sci. USA 2005, 102, 10499–10504. [Google Scholar] [CrossRef]
- Murray, D.S.; Chinnam, N.; Tonthat, N.K.; Whitfill, T.; Wray, L.V., Jr.; Fisher, S.H.; Schumacher, M.A. Structures of the Bacillus subtilis glutamine synthetase dodecamer reveal large intersubunit catalytic conformational changes linked to a unique feedback inhibition mechanism. J. Biol. Chem. 2013, 288, 35801–35811. [Google Scholar] [CrossRef]
- Liaw, S.H.; Jun, G.; Eisenberg, D. Interactions of nucleotides with fully unadenylylated glutamine synthetase from Salmonella typhimurium. Biochemistry 1994, 33, 11184–11188. [Google Scholar] [CrossRef]
- Liaw, S.H.; Eisenberg, D. Structural model for the reaction mechanism of glutamine synthetase, based on five crystal structures of enzyme-substrate complexes. Biochemistry 1994, 33, 675–681. [Google Scholar] [CrossRef]
- Joo, H.K.; Park, Y.W.; Jang, Y.Y.; Lee, J.Y. Structural Analysis of Glutamine Synthetase from Helicobacter pylori. Sci. Rep. 2018, 8, 11657. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344-50. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Gouet, P.; Robert, X.; Courcelle, E. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003, 31, 3320–3323. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.T.; Krajewski, W.W.; Yellagunda, S.; Prabhumurthy, S.; Chamarahally, G.N.; Siddamadappa, C.; Srinivasa, B.R.; Yahiaoui, S.; Larhed, M.; Karlen, A. Structural basis for the inhibition of Mycobacterium tuberculosis glutamine synthetase by novel ATP-competitive inhibitors. J. Mol. Biol. 2009, 393, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Liaw, S.H.; Kuo, I.; Eisenberg, D. Discovery of the ammonium substrate site on glutamine synthetase, a third cation binding site. Protein Sci. 1995, 4, 2358–2365. [Google Scholar] [CrossRef]
- Liaw, S.H.; Pan, C.; Eisenberg, D. Feedback inhibition of fully unadenylylated glutamine synthetase from Salmonella typhimurium by glycine, alanine, and serine. Proc. Natl. Acad. Sci. USA 1993, 90, 4996–5000. [Google Scholar] [CrossRef]
- Spodenkiewicz, M.; Diez-Fernandez, C.; Rufenacht, V.; Gemperle-Britschgi, C.; Haberle, J. Minireview on Glutamine Synthetase Deficiency, an Ultra-Rare Inborn Error of Amino Acid Biosynthesis. Biology (Basel) 2016, 5, 40. [Google Scholar] [CrossRef]
- Pushkin, A.V.; Antoniuk, L.P.; Solovieva, N.A.; Shubin, V.V.; Evstigneeva, Z.G.; Kretovich, W.L.; Cherednikova, T.V.; Tsuprun, V.L.; Zograf, O.N.; Kiselev, N.A. Glutamine Synthetases of Pea Leaf and Seed Cytosol—Structure and Properties. Biochim. Biophys. Acta 1985, 828, 336–350. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DmGS2/ADP Complex | |
|---|---|
| Data collection: | |
| Wavelength (Å) | 1.0000 |
| Temperature (K) | 100 |
| Resolution (Å) | 30-2.12 (2.20–2.12) a |
| Space group | C121 |
| Unit cell dimensions | |
| a, b, c (Å) | 229.70, 120.92, 206.51 |
| α, β, γ (°) | 90.00, 120.09, 90.00 |
| Number of unique reflections | 234,758 |
| Number of observations | 637,222 |
| Redundancy | 2.8 (2.7) a |
| Completeness (%) | 98.0 (97.2) a |
| Mean I/σ (I) | 16.41 (2.50) a |
| No. of protein monomers | 10 |
| Matthew coefficient (Å3 Da−1) | 2.55 |
| Solvent content (%) | 51.38 |
| Wilson B-factor (Å2) | 42.2 |
| Rmerge | 0.071 (0.592) a |
| Refinement: | |
| Resolution range (Å) | 27.40–2.12 (2.20–2.12) a |
| Number of reflections used | 216,390 (11,245) a |
| Number of protein heavy atoms | 29,527 |
| Number of water molecules | 521 |
| Number of heterogeneous molecules | 280 |
| Average B-factor (Å2) | 50.32 |
| Rwork/Rfree (%) | 19.19/24.08 (28.9/32.2) a |
| Ramachandran plot statistics: | |
| Residues in favored region (%) | 95.9 |
| Residues in allowed region (%) | 4.0 |
| Residues in outlier region (%) | 0.1 |
| Average RMSD, bond length (Å) | 0.016 |
| Average RMSD, bond angle (°) | 1.801 |
| PDB entry | 7CPR |
| Protein Type | Substrate | Km (mM) | vmax (mM min−1) c | kcat (min−1) | kcat/Km (min−1 mM−1) |
|---|---|---|---|---|---|
| WT | Glutamate a | 5.9 ± 1.6 | 0.0092 ± 0.0004 | 1.84 ± 0.08 | 0.34 ± 0.08 |
| R4D | Glutamate | ND | ND | ND | ND |
| Δ13 | Glutamate | ND | ND | ND | ND |
| E140A | Glutamate | ND | ND | ND | ND |
| P214A | Glutamate | 6.8 ± 2.2 | 0.0106 ± 0.0006 | 2.13 ± 0.11 | 0.38 ± 0.13 |
| E311A | Glutamate | 26.2 ± 2.2 | 0.0055 ± 0.0004 | 1.10 ± 0.08 | 0.0420 ± 0.0005 |
| WT | Ammonium chloride b | 0.47 ± 0.03 | 0.0046 ± 0.0001 | 0.92 ± 0.02 | 1.96 ± 0.08 |
| R4D | Ammonium chloride | ND | ND | ND | ND |
| Δ13 | Ammonium chloride | ND | ND | ND | ND |
| E140A | Ammonium chloride | ND | ND | ND | ND |
| P214A | Ammonium chloride | 0.11 ± 0.03 | 0.0055 ± 0.0003 | 1.10 ± 0.06 | 10.6 ± 2.4 |
| E311A | Ammonium chloride | ND | ND | ND | ND |
| Protein Type | Substrate | Km (mM) | vmax (mM min−1) c | kcat (min−1) | kcat/Km (min−1 mM−1) |
|---|---|---|---|---|---|
| WT | Glutamine a | 43.1 ± 9.7 | 0.12 ± 0.01 | 24 ± 2 | 0.58 ± 0.08 |
| R4D | Glutamine | ND | ND | ND | ND |
| Δ13 | Glutamine | ND | ND | ND | ND |
| E140A | Glutamine | ND | ND | ND | ND |
| P214A | Glutamine | 30.6 ± 6.1 | 0.15 ± 0.01 | 30 ± 2 | 1.01 ± 0.14 |
| E311A | Glutamine | ND | ND | ND | ND |
| WT | Hydroxylamine b | 2.0 ± 0.4 | 0.064 ± 0.003 | 12.8 ± 0.6 | 0.033 ± 0.005 |
| R4D | Hydroxylamine | ND | ND | ND | ND |
| Δ13 | Hydroxylamine | ND | ND | ND | ND |
| E140A | Hydroxylamine | ND | ND | ND | ND |
| P214A | Hydroxylamine | 1.5 ± 0.2 | 0.088 ± 0.003 | 17.6 ± 0.6 | 0.059 ± 0.006 |
| E311A | Hydroxylamine | ND | ND | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.-T.; Yang, H.-Y.; Lin, C.-Y.; Lee, Y.-Z.; Ma, S.-C.; Chen, W.-C.; Yin, H.-S. Structural Insight into the Contributions of the N-Terminus and Key Active-Site Residues to the Catalytic Efficiency of Glutamine Synthetase 2. Biomolecules 2020, 10, 1671. https://doi.org/10.3390/biom10121671
Chen W-T, Yang H-Y, Lin C-Y, Lee Y-Z, Ma S-C, Chen W-C, Yin H-S. Structural Insight into the Contributions of the N-Terminus and Key Active-Site Residues to the Catalytic Efficiency of Glutamine Synthetase 2. Biomolecules. 2020; 10(12):1671. https://doi.org/10.3390/biom10121671
Chicago/Turabian StyleChen, Wen-Ting, Hsin-Yu Yang, Chih-Yu Lin, Yi-Zong Lee, Szu-Chin Ma, Wei-Cheng Chen, and Hsien-Sheng Yin. 2020. "Structural Insight into the Contributions of the N-Terminus and Key Active-Site Residues to the Catalytic Efficiency of Glutamine Synthetase 2" Biomolecules 10, no. 12: 1671. https://doi.org/10.3390/biom10121671
APA StyleChen, W.-T., Yang, H.-Y., Lin, C.-Y., Lee, Y.-Z., Ma, S.-C., Chen, W.-C., & Yin, H.-S. (2020). Structural Insight into the Contributions of the N-Terminus and Key Active-Site Residues to the Catalytic Efficiency of Glutamine Synthetase 2. Biomolecules, 10(12), 1671. https://doi.org/10.3390/biom10121671