Different Regulation of Glut1 Expression and Glucose Uptake during the Induction and Chronic Stages of TGFβ1-Induced EMT in Breast Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture Conditions
2.2. Western Blot Analysis
2.3. Immunofluorescence Staining
2.4. Glucose Uptake Measurement
2.5. Quantitative Real-Time PCR Analysis
2.6. Proliferation Assays
2.7. RNA Interference
2.8. Stable Overexpression of Glut1
2.9. Statistical Analysis
3. Results
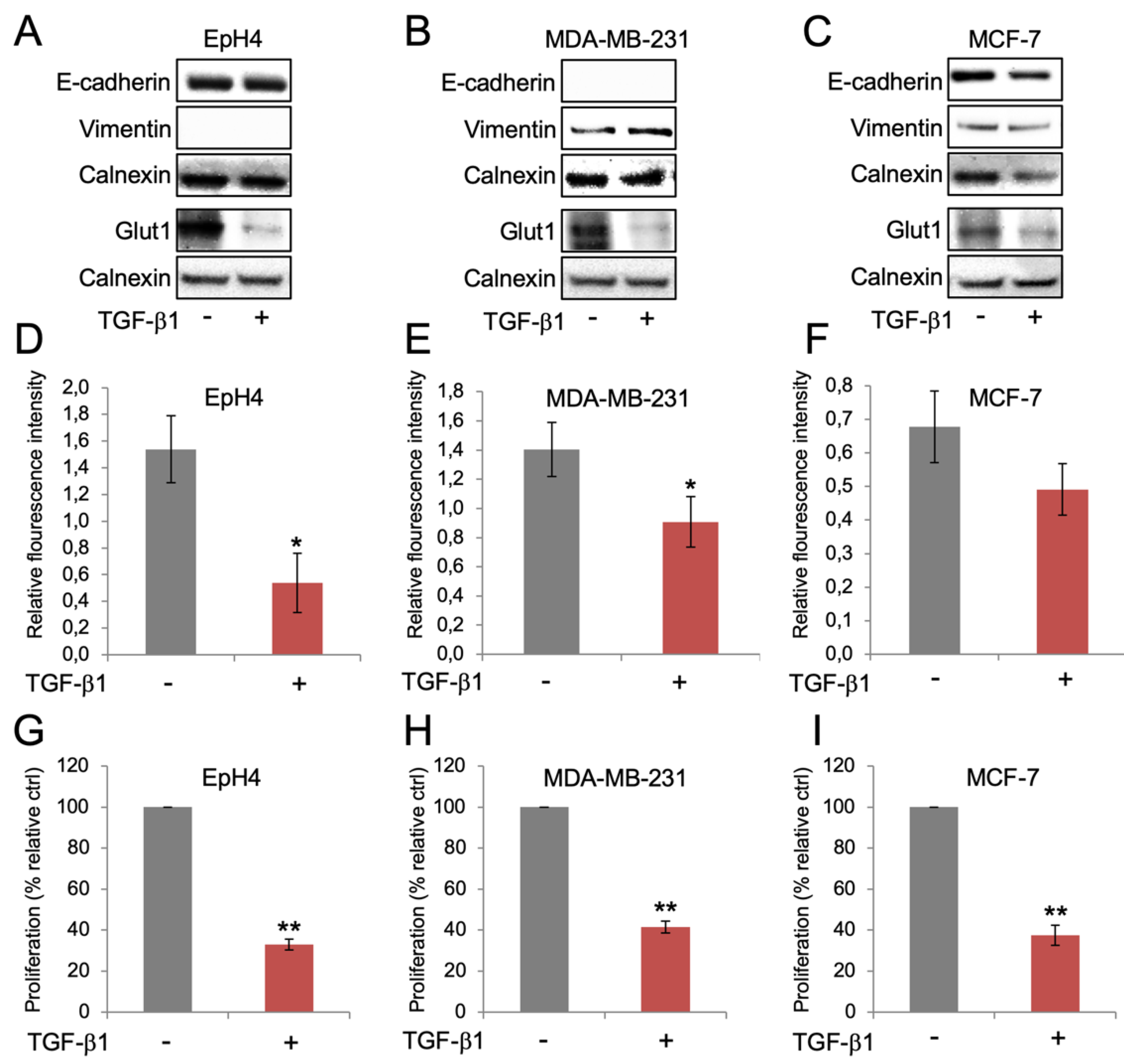
3.1. Reduced Glut1 Expression and Glucose Uptake during the Induction of EMT
3.2. Reduced Expression of Glut1 and Glucose Uptake Is Linked to Decreased Cell Proliferation but not EMT
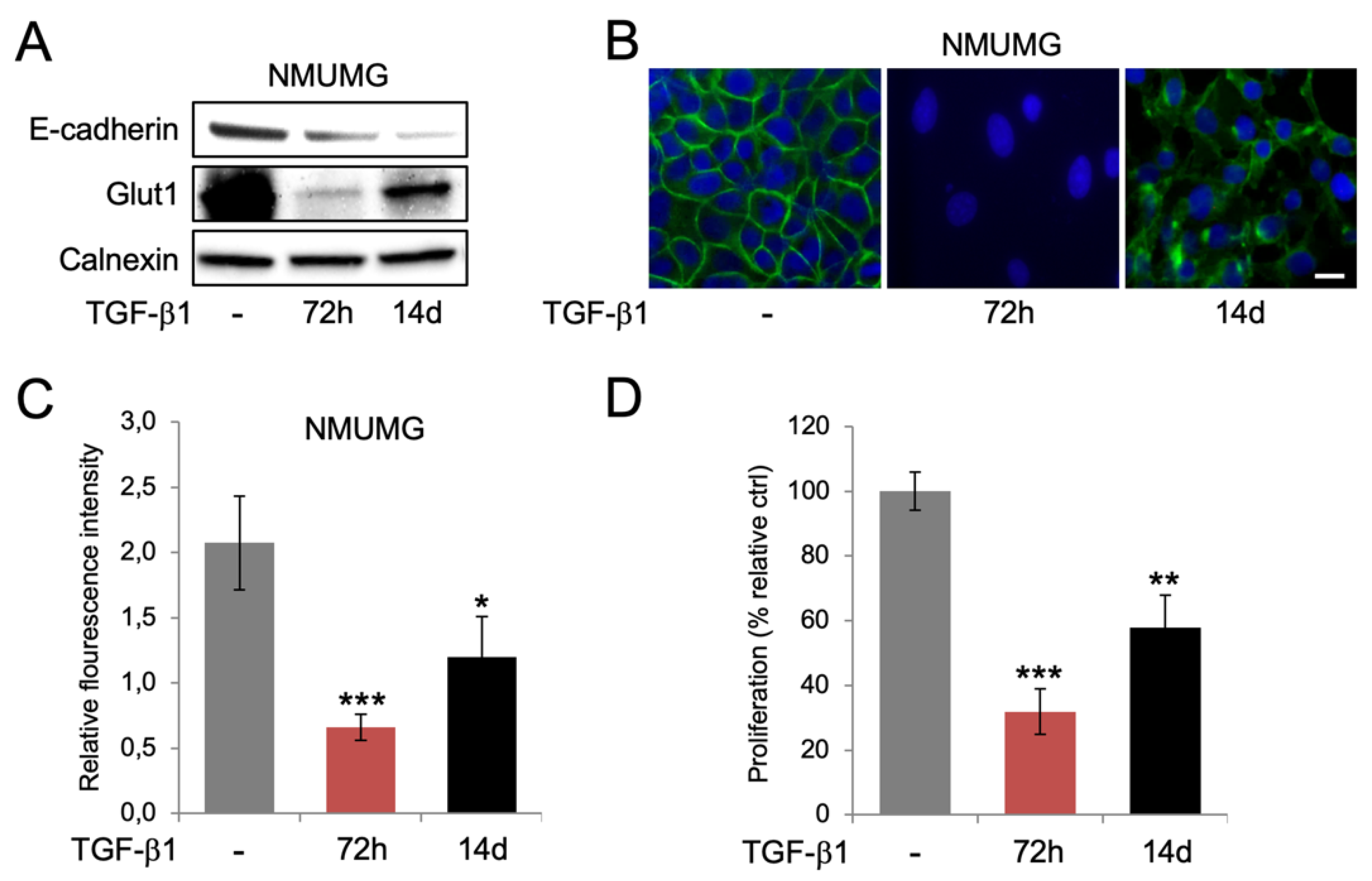
3.3. Re-Expression of Glut1 and Increased Glucose Uptake during Long-Term EMT
3.4. Glut1 Regulates Cell Proliferation and TGF-β1-Induced EMT
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuxe, J.; Karlsson, M.C. TGF-beta-induced epithelial-mesenchymal transition: A link between cancer and inflammation. Semin. Cancer Biol. 2012, 22, 455–461. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell. Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Fuxe, J.; Vincent, T.; Garcia de Herreros, A. Transcriptional crosstalk between TGF-beta and stem cell pathways in tumor cell invasion: Role of EMT promoting Smad complexes. Cell Cycle 2010, 9, 2363–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Katsuno, Y.; Meyer, D.S.; Zhang, Z.; Shokat, K.M.; Akhurst, R.J.; Miyazono, K.; Derynck, R. Chronic TGF-beta exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Nilchian, A.; Johansson, J.; Ghalali, A.; Asanin, S.T.; Santiago, A.; Rosencrantz, O.; Sollerbrant, K.; Vincent, C.T.; Sund, M.; Stenius, U.; et al. CXADR-Mediated Formation of an AKT Inhibitory Signalosome at Tight Junctions Controls Epithelial-Mesenchymal Plasticity in Breast Cancer. Cancer Res. 2019, 79, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Flores-Lopez, L.A.; Martinez-Hernandez, M.G.; Viedma-Rodriguez, R.; Diaz-Flores, M.; Baiza-Gutman, L.A. High glucose and insulin enhance uPA expression, ROS formation and invasiveness in breast cancer-derived cells. Cell Oncol. 2016, 39, 365–378. [Google Scholar] [CrossRef]
- Hua, W.; Ten Dijke, P.; Kostidis, S.; Giera, M.; Hornsveld, M. TGFbeta-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell. Mol. Life Sci. 2020, 77, 2103–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buller, C.L.; Loberg, R.D.; Fan, M.H.; Zhu, Q.; Park, J.L.; Vesely, E.; Inoki, K.; Guan, K.L.; Brosius, F.C., 3rd. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am. J. Physiol. Cell Physiol. 2008, 295, C836–C843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, J.; Tabor, V.; Wikell, A.; Jalkanen, S.; Fuxe, J. TGF-beta1-Induced Epithelial-Mesenchymal Transition Promotes Monocyte/Macrophage Properties in Breast Cancer Cells. Front. Oncol. 2015, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.A.; Aakre, M.E.; Gorska, A.E.; Price, J.O.; Eltom, S.E.; Pietenpol, J.A.; Moses, H.L. Induction by transforming growth factor-beta1 of epithelial to mesenchymal transition is a rare event in vitro. Breast Cancer Res. 2004, 6, R215–R231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, J.; Berg, T.; Kurzejamska, E.; Pang, M.F.; Tabor, V.; Jansson, M.; Roswall, P.; Pietras, K.; Sund, M.; Religa, P.; et al. MiR-155-mediated loss of C/EBPbeta shifts the TGF-beta response from growth inhibition to epithelial-mesenchymal transition, invasion and metastasis in breast cancer. Oncogene 2013, 32, 5614–5624. [Google Scholar] [CrossRef] [Green Version]
- Andrianifahanana, M.; Hernandez, D.M.; Yin, X.; Kang, J.H.; Jung, M.Y.; Wang, Y.; Yi, E.S.; Roden, A.C.; Limper, A.H.; Leof, E.B. Profibrotic up-regulation of glucose transporter 1 by TGF-beta involves activation of MEK and mammalian target of rapamycin complex 2 pathways. FASEB J. 2016, 30, 3733–3744. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wei, Z.; Liu, Y.; Li, H.; Ren, R.; Tang, Y. Increased 18F-FDG uptake and expression of Glut1 in the EMT transformed breast cancer cells induced by TGF-beta. Neoplasma 2010, 57, 234–240. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-beta signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Hefka Blahnova, V.; Dankova, J.; Rampichova, M.; Filova, E. Combinations of growth factors for human mesenchymal stem cell proliferation and osteogenic differentiation. Bone Joint Res. 2020, 9, 412–420. [Google Scholar] [CrossRef]
- Reynisdottir, I.; Polyak, K.; Iavarone, A.; Massague, J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev. 1995, 9, 1831–1845. [Google Scholar] [CrossRef] [Green Version]
- Scimeca, M.; Bonfiglio, R.; Menichini, E.; Albonici, L.; Urbano, N.; De Caro, M.T.; Mauriello, A.; Schillaci, O.; Gambacurta, A.; Bonanno, E. Microcalcifications Drive Breast Cancer Occurrence and Development by Macrophage-Mediated Epithelial to Mesenchymal Transition. Int. J. Mol. Sci. 2019, 20, 5633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scimeca, M.; Giocondo, R.; Montanaro, M.; Granaglia, A.; Bonfiglio, R.; Tancredi, V.; Mauriello, A.; Urbano, N.; Schillaci, O.; Bonanno, E. BMP-2 Variants in Breast Epithelial to Mesenchymal Transition and Microcalcifications Origin. Cells 2020, 9, 1381. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Kim, H.; Nam, K.; Shin, I. Glut1 promotes cell proliferation, migration and invasion by regulating epidermal growth factor receptor and integrin signaling in triple-negative breast cancer cells. BMB Rep. 2017, 50, 132–137. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nilchian, A.; Giotopoulou, N.; Sun, W.; Fuxe, J. Different Regulation of Glut1 Expression and Glucose Uptake during the Induction and Chronic Stages of TGFβ1-Induced EMT in Breast Cancer Cells. Biomolecules 2020, 10, 1621. https://doi.org/10.3390/biom10121621
Nilchian A, Giotopoulou N, Sun W, Fuxe J. Different Regulation of Glut1 Expression and Glucose Uptake during the Induction and Chronic Stages of TGFβ1-Induced EMT in Breast Cancer Cells. Biomolecules. 2020; 10(12):1621. https://doi.org/10.3390/biom10121621
Chicago/Turabian StyleNilchian, Azadeh, Nikolina Giotopoulou, Wenwen Sun, and Jonas Fuxe. 2020. "Different Regulation of Glut1 Expression and Glucose Uptake during the Induction and Chronic Stages of TGFβ1-Induced EMT in Breast Cancer Cells" Biomolecules 10, no. 12: 1621. https://doi.org/10.3390/biom10121621
APA StyleNilchian, A., Giotopoulou, N., Sun, W., & Fuxe, J. (2020). Different Regulation of Glut1 Expression and Glucose Uptake during the Induction and Chronic Stages of TGFβ1-Induced EMT in Breast Cancer Cells. Biomolecules, 10(12), 1621. https://doi.org/10.3390/biom10121621