Glycosaminoglycans and Contrast Agents: The Role of Hyaluronic Acid as MRI Contrast Enhancer



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
2.3. Time-Domain Relaxometry at 20 MHz and 60 MHz
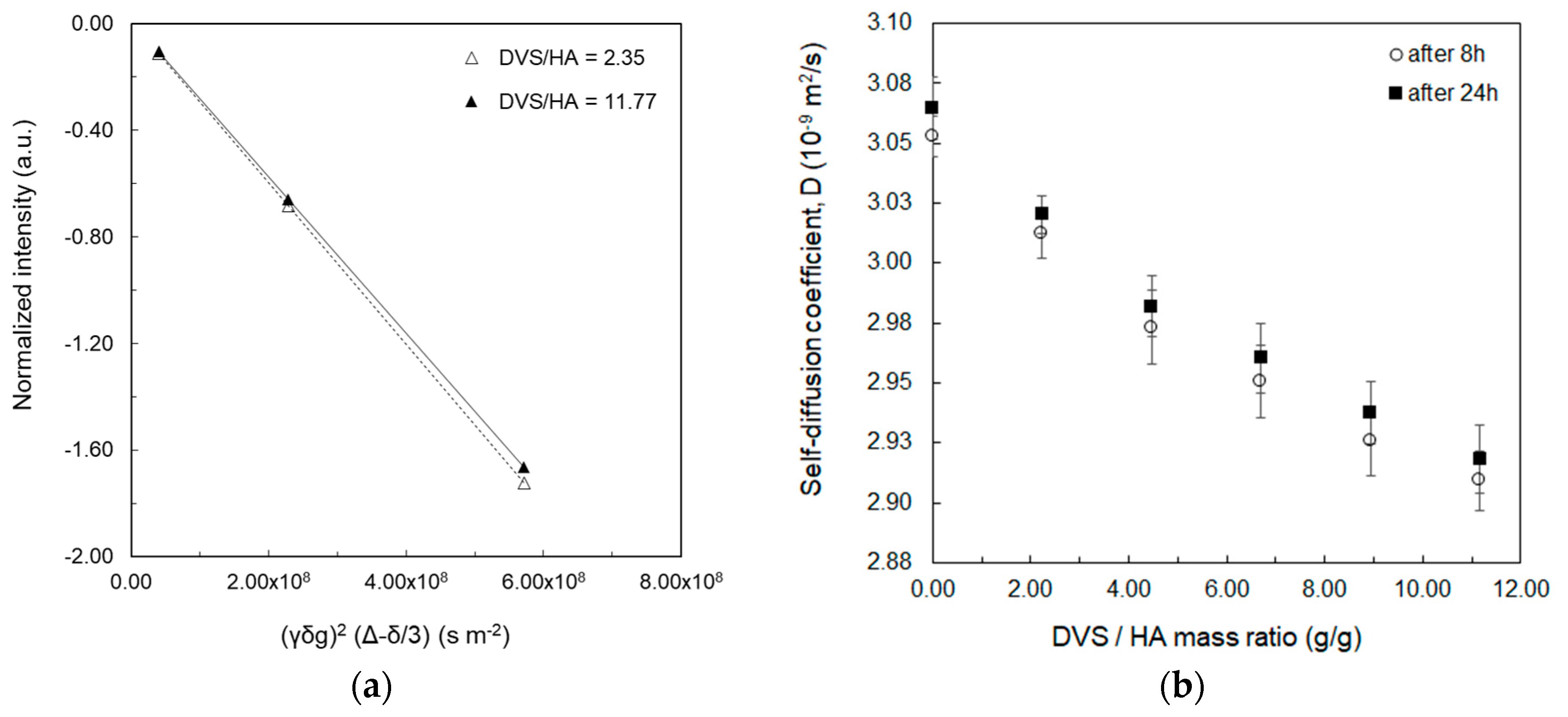
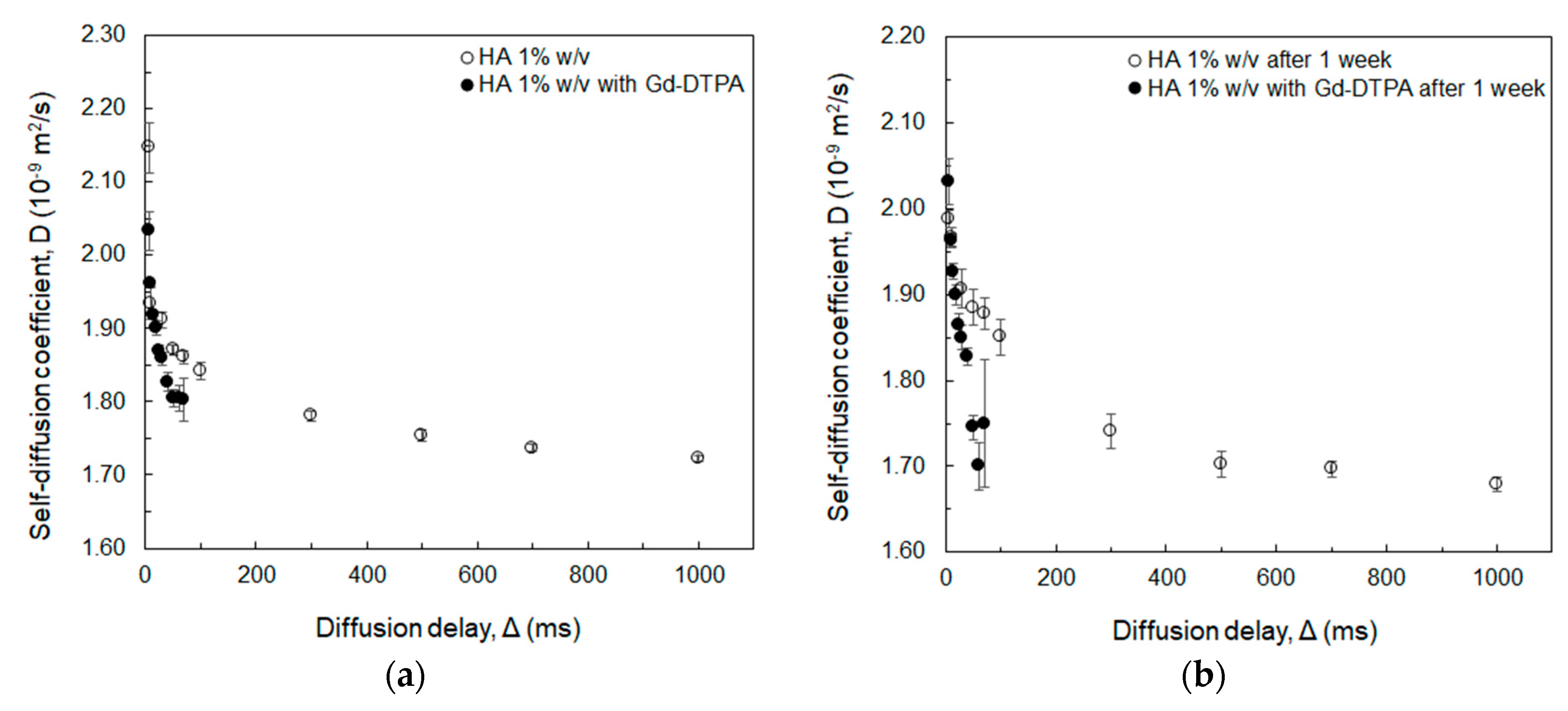
2.4. Measurement of Water Self-Diffusion Coefficient at 20 MHz
2.5. Diffusion-Ordered NMR Spectroscopy (DOSY) at 600 MHz
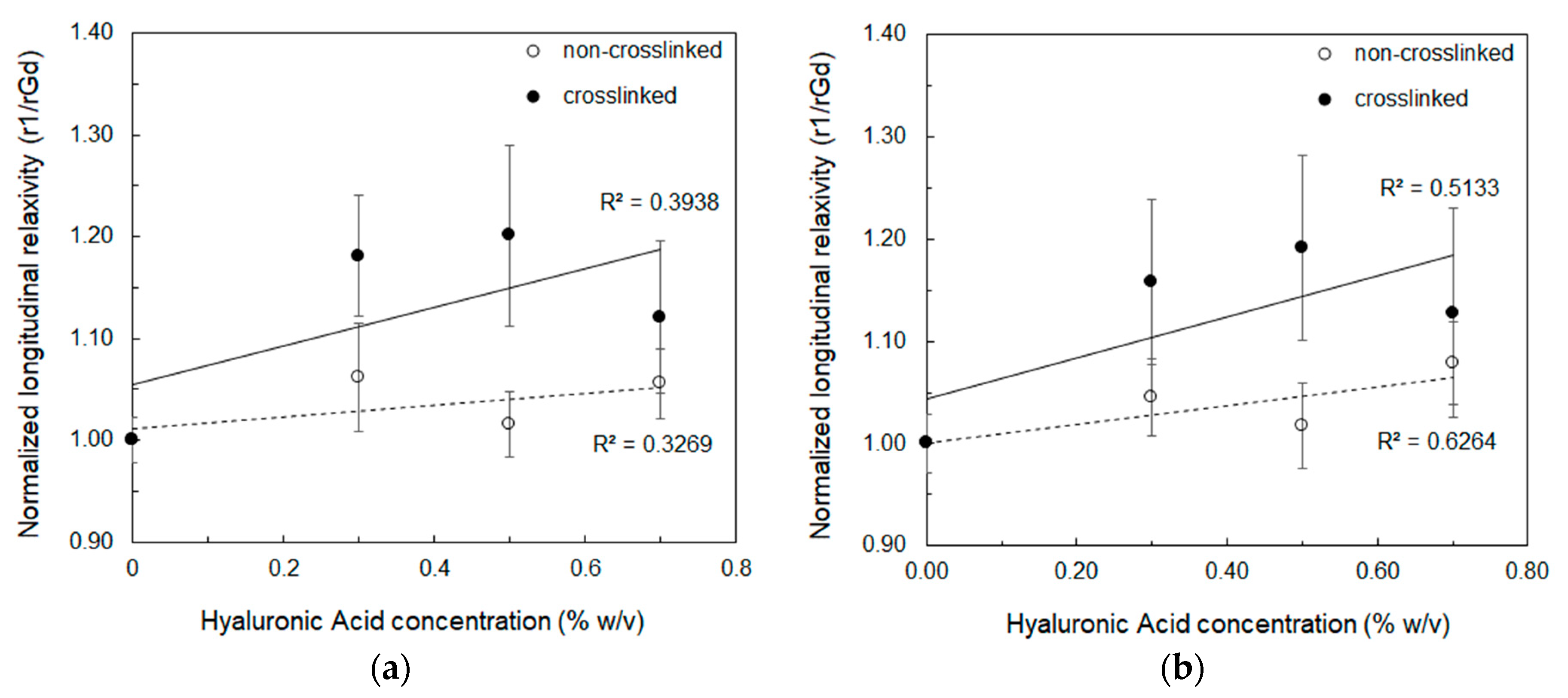
2.6. Isothermal Titration Calorimetry
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Acronyms
| DVS | Divinyl Sulfone |
| GAG | Glycosaminoglycan |
| HA | Hyaluronic Acid |
| IR | Inversion Recovery |
| ITC | Isothermal Titration Calorimetry |
| MRI | Magnetic Resonance Imaging |
| NMR | Nuclear Magnetic Resonance |
| NMR-DOSY | Diffusion-ordered NMR Spectroscopy |
| NPs | Nanoparticles |
| PFG-SE | Pulsed-Field Gradient Spin Echo |
| RT | Room Temperature |
| SR | Saturation Recovery |
References
- Scott, R.A.; Panitch, A. Glycosaminoglycans in biomedicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Huang, H. Review on Magnetic Natural Polymer Constructed Hydrogels as Vehicles for Drug Delivery. Biomacromolecules 2020, 21, 2574–2594. [Google Scholar] [CrossRef] [PubMed]
- Albertsson, A.-C.; Percec, S. Future of Biomacromolecules at a Crossroads of Polymer Science and Biology. Biomacromolecules 2020, 21, 1–6. [Google Scholar] [CrossRef]
- Swierczewska, M.; Han, H.S.; Kim, K.; Park, J.H.; Lee, S. Polysaccharide-based Nanoparticles for Theranostic Nanomedicine. Adv. Drug Deliv. Rev. 2016, 99, 70–84. [Google Scholar] [CrossRef]
- Jian, W.-H.; Wang, H.-C.; Kuan, C.-H.; Chen, M.-H.; Wu, H.-C.; Sun, J.-S.; Wang, T.-W. Glycosaminoglycan-based hybrid hydrogel encapsulated with polyelectrolyte complex nanoparticles for endogenous stem cell regulation in central nervous system regeneration. Biomaterials 2018, 174, 17–30. [Google Scholar] [CrossRef]
- Oommen, O.P.; Duehrkop, C.; Nilsson, B.; Hilborn, J.; Varghese, O.P. Multifunctional Hyaluronic Acid and Chondroitin Sulfate Nanoparticles: Impact of Glycosaminoglycan Presentation on Receptor Mediated Cellular Uptake and Immune Activation. ACS Appl. Mater. Interfaces 2016, 8, 20614–20624. [Google Scholar] [CrossRef]
- Zhong, L.; Liu, Y.; Xu, L.; Li, Q.; Zhao, D.; Li, Z.; Zhang, H.; Zhang, H.; Kan, Q.; Sun, J.; et al. Exploring the relationship of hyaluronic acid molecular weight and active targeting efficiency for designing hyaluronic acid-modified nanoparticles. Asian J. Pharm. Sci. 2019, 14, 521–530. [Google Scholar] [CrossRef]
- Huang, G.; Huang, H. Application of hyaluronic acid as carriers in drug delivery. Drug Deliv. 2018, 25, 766–772. [Google Scholar] [CrossRef]
- Kim, K.; Choi, H.; Choi, E.S.; Park, M.-H.; Ryu, J.-H. Hyaluronic Acid-Coated Nanomedicine for Targeted Cancer Therapy. Pharmaceutics 2019, 11. [Google Scholar] [CrossRef]
- Patil-Sen, Y.; Torino, E.; Sarno, F.D.; Ponsiglione, A.M.; Chhabria, V.N.; Ahmed, W.; Mercer, T. Biocompatible superparamagnetic core-shell nanoparticles for potential use in hyperthermia-enabled drug release and as an enhanced contrast agent. Nanotechnology 2020. [Google Scholar] [CrossRef]
- Ori, G.; Villemot, F.; Viau, L.; Vioux, A.; Coasne, B. Ionic liquid confined in silica nanopores: molecular dynamics in the isobaric–isothermal ensemble. Mol. Phys. 2014, 112, 1350–1361. [Google Scholar] [CrossRef]
- Debroye, E.; Parac-Vogt, T.N. Towards polymetallic lanthanide complexes as dual contrast agents for magnetic resonance and optical imaging. Chem. Soc. Rev. 2014, 43, 8178–8192. [Google Scholar] [CrossRef]
- Russo, M.; Bevilacqua, P.; Netti, P.A.; Torino, E. A Microfluidic Platform to design crosslinked Hyaluronic Acid Nanoparticles (cHANPs) for enhanced MRI. Sci. Rep. 2016, 6, 37906. [Google Scholar] [CrossRef]
- Torino, E.; Russo, M.; Ponsiglione, A.M. Chapter 6—Lab-on-a-chip preparation routes for organic nanomaterials for drug delivery. In Microfluidics for Pharmaceutical Applications; Santos, H.A., Liu, D., Zhang, H., Eds.; Micro and Nano Technologies; Elsevier: Amsterdam, The Netherlands, 2019; pp. 137–153. ISBN 978-0-12-812659-2. [Google Scholar]
- Capretto, L.; Carugo, D.; Mazzitelli, S.; Nastruzzi, C.; Zhang, X. Microfluidic and lab-on-a-chip preparation routes for organic nanoparticles and vesicular systems for nanomedicine applications. Adv. Drug Deliv. Rev. 2013, 65, 1496–1532. [Google Scholar] [CrossRef]
- Capretto, L.; Cheng, W.; Carugo, D.; Katsamenis, O.L.; Hill, M.; Zhang, X. Mechanism of co-nanoprecipitation of organic actives and block copolymers in a microfluidic environment. Nanotechnology 2012, 23, 375602. [Google Scholar] [CrossRef]
- Bally, F.; Garg, D.K.; Serra, C.A.; Hoarau, Y.; Anton, N.; Brochon, C.; Parida, D.; Vandamme, T.; Hadziioannou, G. Improved size-tunable preparation of polymeric nanoparticles by microfluidic nanoprecipitation. Polymer 2012, 53, 5045–5051. [Google Scholar] [CrossRef]
- Bazban-Shotorbani, S.; Dashtimoghadam, E.; Karkhaneh, A.; Hasani-Sadrabadi, M.M.; Jacob, K.I. Microfluidic Directed Synthesis of Alginate Nanogels with Tunable Pore Size for Efficient Protein Delivery. Langmuir 2016, 32, 4996–5003. [Google Scholar] [CrossRef]
- Dashtimoghadam, E.; Mirzadeh, H.; Taromi, F.A.; Nyström, B. Microfluidic self-assembly of polymeric nanoparticles with tunable compactness for controlled drug delivery. Polymer 2013, 54, 4972–4979. [Google Scholar] [CrossRef]
- Maimouni, I.; Cejas, C.M.; Cossy, J.; Tabeling, P.; Russo, M. Microfluidics Mediated Production of Foams for Biomedical Applications. Micromachines 2020, 11, 83. [Google Scholar] [CrossRef]
- Yoon, H.Y.; Koo, H.; Choi, K.Y.; Lee, S.J.; Kim, K.; Kwon, I.C.; Leary, J.F.; Park, K.; Yuk, S.H.; Park, J.H.; et al. Tumor-targeting hyaluronic acid nanoparticles for photodynamic imaging and therapy. Biomaterials 2012, 33, 3980–3989. [Google Scholar] [CrossRef]
- Thomas, R.G.; Moon, M.; Lee, S.; Jeong, Y.Y. Paclitaxel loaded hyaluronic acid nanoparticles for targeted cancer therapy: In vitro and in vivo analysis. Int. J. Biol. Macromol. 2015, 72, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhang, H.; Wei, Y.; Cong, F. Hyaluronan-Inorganic Nanohybrid Materials for Biomedical Applications. Biomacromolecules 2017, 18, 1677–1696. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Huang, X.; Xu, Z. Development of Novel Nano Hyaluronic Acid Carrier for Diagnosis and Therapy of Atherosclerosis. J. Clust. Sci. 2020. [Google Scholar] [CrossRef]
- De Sarno, F.; Ponsiglione, A.M.; Torino, E. Emerging use of nanoparticles in diagnosis of atherosclerosis disease: A review. AIP Conf. Proc. 2018, 1990, 020021. [Google Scholar] [CrossRef]
- El-Dakdouki, M.H.; Zhu, D.C.; El-Boubbou, K.; Kamat, M.; Chen, J.; Li, W.; Huang, X. Development of Multifunctional Hyaluronan-Coated Nanoparticles for Imaging and Drug Delivery to Cancer Cells. Biomacromolecules 2012, 13, 1144–1151. [Google Scholar] [CrossRef]
- Torino, E.; Auletta, L.; Vecchione, D.; Orlandella, F.M.; Salvatore, G.; Iaccino, E.; Fiorenza, D.; Grimaldi, A.M.; Sandomenico, A.; Albanese, S.; et al. Multimodal imaging for a theranostic approach in a murine model of B-cell lymphoma with engineered nanoparticles. Nanomedicine Nanotechnol. Biol. Med. 2018, 14, 483–491. [Google Scholar] [CrossRef]
- Vecchione, D.; Aiello, M.; Cavaliere, C.; Nicolai, E.; Netti, P.A.; Torino, E. Hybrid core shell nanoparticles entrapping Gd-DTPA and 18F-FDG for simultaneous PET/MRI acquisitions. Nanomedicine 2017, 12, 2223–2231. [Google Scholar] [CrossRef]
- Russo, M.; Bevilacqua, P.; Netti, P.A.; Torino, E. Commentary on “A Microfluidic Platform to Design Crosslinked Hyaluronic Acid Nanoparticles (cHANPs) for Enhanced MRI.”. Mol. Imaging 2017, 16. [Google Scholar] [CrossRef]
- Russo, M.; Grimaldi, A.M.; Bevilacqua, P.; Tammaro, O.; Netti, P.A.; Torino, E. PEGylated crosslinked hyaluronic acid nanoparticles designed through a microfluidic platform for nanomedicine. Nanomedicine 2017, 12, 2211–2222. [Google Scholar] [CrossRef]
- Zheng, S.; Xia, Y. The impact of the relaxivity definition on the quantitative measurement of glycosaminoglycans in cartilage by the MRI dGEMRIC method. Magn. Reson. Med. 2010, 63, 25–32. [Google Scholar] [CrossRef]
- Bashir, A.; Gray, M.L.; Hartke, J.; Burstein, D. Nondestructive imaging of human cartilage glycosaminoglycan concentration by MRI. Magn. Reson. Med. 1999, 41, 857–865. [Google Scholar] [CrossRef]
- Fakhari, A.; Berkland, C. Applications and Emerging Trends of Hyaluronic Acid in Tissue Engineering, as a Dermal Filler, and in Osteoarthritis Treatment. Acta Biomater. 2013, 9, 7081–7092. [Google Scholar] [CrossRef]
- Gupta, R.C.; Lall, R.; Srivastava, A.; Sinha, A. Hyaluronic Acid: Molecular Mechanisms and Therapeutic Trajectory. Front. Vet. Sci. 2019, 6, 192. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, G.; Presutti, L.; Molteni, G. Injection Laryngoplasty; Springer International Publishing: Cham, Switzerland, 2015; ISBN 978-3-319-20143-6. [Google Scholar]
- Fragai, M.; Ravera, E.; Tedoldi, F.; Luchinat, C.; Parigi, G. Relaxivity of Gd-Based MRI Contrast Agents in Crosslinked Hyaluronic Acid as a Model for Tissues. ChemPhysChem 2019, 20, 2204–2209. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Park, K.; Shalaby, W.S.W. Biodegradable Hydrogels for Drug Delivery; CRC Press (Taylor and Francis Group): Boca Raton, FL, USA, 2011; ISBN 978-1-4398-9296-1. [Google Scholar]
- Fallacara, A.; Baldini, E.; Manfredini, S.; Vertuani, S. Hyaluronic Acid in the Third Millennium. Polymers 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.C.; Bergfeld, W.F.; Belsito, D.V.; Klaassen, C.D.; Marks, J.G.; Shank, R.C.; Slaga, T.J.; Snyder, P.W.; Cosmetic Ingredient Review Expert Panel; Andersen, F.A. Final report of the safety assessment of hyaluronic acid, potassium hyaluronate, and sodium hyaluronate. Int. J. Toxicol. 2009, 28, 5–67. [Google Scholar] [CrossRef] [PubMed]
- Caro, C.; García-Martín, M.L.; Pernia Leal, M. Manganese-Based Nanogels as pH Switches for Magnetic Resonance Imaging. Biomacromolecules 2017, 18, 1617–1623. [Google Scholar] [CrossRef]
- Russo, M.; Ponsiglione, A.M.; Forte, E.; Netti, P.A.; Torino, E. Hydrodenticity to enhance relaxivity of gadolinium-DTPA within crosslinked hyaluronic acid nanoparticles. Nanomedicine 2017, 12, 2199–2210. [Google Scholar] [CrossRef]
- Ponsiglione, A.M.; Russo, M.; Netti, P.A.; Torino, E. Impact of biopolymer matrices on relaxometric properties of contrast agents. Interface Focus 2016, 6. [Google Scholar] [CrossRef]
- De Sarno, F.; Ponsiglione, A.M.; Grimaldi, A.M.; Netti, P.A.; Torino, E. Effect of crosslinking agent to design nanostructured hyaluronic acid-based hydrogels with improved relaxometric properties. Carbohydr. Polym. 2019, 222, 114991. [Google Scholar] [CrossRef]
- De Sarno, F.; Ponsiglione, A.M.; Russo, M.; Grimaldi, A.M.; Forte, E.; Netti, P.A.; Torino, E. Water-Mediated Nanostructures for Enhanced MRI: Impact of Water Dynamics on Relaxometric Properties of Gd-DTPA. Theranostics 2019, 9, 1809–1824. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, D.; Grimaldi, A.M.; Forte, E.; Bevilacqua, P.; Netti, P.A.; Torino, E. Hybrid Core-Shell (HyCoS) Nanoparticles produced by Complex Coacervation for Multimodal Applications. Sci. Rep. 2017, 7, 45121. [Google Scholar] [CrossRef] [PubMed]
- Tammaro, O.; Costagliola di Polidoro, A.; Romano, E.; Netti, P.A.; Torino, E. A Microfluidic Platform to design Multimodal PEG—crosslinked Hyaluronic Acid Nanoparticles (PEG-cHANPs) for diagnostic applications. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Oh, E.J.; Kang, S.-W.; Kim, B.-S.; Jiang, G.; Cho, I.H.; Hahn, S.K. Control of the molecular degradation of hyaluronic acid hydrogels for tissue augmentation. J. Biomed. Mater. Res. A 2008, 86, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Stejskal, E.O.; Tanner, J.E. Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef]
- Horstmann, M.; Urbani, M.; Veeman, W.S. Self-Diffusion of Water in Block Copoly(ether−ester) Polymers: An NMR Study. Macromolecules 2003, 36, 6797–6806. [Google Scholar] [CrossRef]
- Lucas, L.H.; Larive, C.K. Measuring ligand-protein binding using NMR diffusion experiments. Concepts Magn. Reson. Part A 2004, 20A, 24–41. [Google Scholar] [CrossRef]
- Puibasset, J.; Porion, P.; Grosman, A.; Rolley, E. Structure and Permeability of Porous Silicon Investigated by Self-Diffusion NMR Measurements of Ethanol and Heptane. Oil Gas Sci. Technol.—Rev. D’IFP Energ. Nouv. 2016, 71, 54. [Google Scholar] [CrossRef]
- Ori, G.; Massobrio, C.; Pradel, A.; Ribes, M.; Coasne, B. Structure and Dynamics of Ionic Liquids Confined in Amorphous Porous Chalcogenides. Langmuir 2015, 31, 6742–6751. [Google Scholar] [CrossRef]
- Maiz-Fernández, S.; Pérez-Álvarez, L.; Ruiz-Rubio, L.; Pérez González, R.; Sáez-Martínez, V.; Ruiz Pérez, J.; Vilas-Vilela, J.L. Synthesis and Characterization of Covalently Crosslinked pH-Responsive Hyaluronic Acid Nanogels: Effect of Synthesis Parameters. Polymers 2019, 11. [Google Scholar] [CrossRef]
- Shimojo, A.A.M.; Pires, A.M.B.; Lichy, R.; Santana, M.H.A.; Shimojo, A.A.M.; Pires, A.M.B.; Lichy, R.; Santana, M.H.A. The Performance of Crosslinking with Divinyl Sulfone as Controlled by the Interplay Between the Chemical Modification and Conformation of Hyaluronic Acid. J. Braz. Chem. Soc. 2015, 26, 506–512. [Google Scholar] [CrossRef]
- Collins, M.N.; Birkinshaw, C. Physical properties of crosslinked hyaluronic acid hydrogels. J. Mater. Sci. Mater. Med. 2008, 19, 3335–3343. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.R.R.; Laverty, G.; Donnelly, R.; Laverty, G.; Donnelly, R. Hydrogels: Design, Synthesis and Application in Drug Delivery and Regenerative Medicine; CRC Press (Taylor and Francis Group): Boca Raton, FL, USA, 2018; ISBN 978-1-315-15222-6. [Google Scholar]
- Omidian, H.; Park, K. Introduction to Hydrogels. In Biomedical Applications of Hydrogels Handbook; Ottenbrite, R.M., Park, K., Okano, T., Eds.; Springer: New York, NY, USA, 2010; pp. 1–6. ISBN 978-1-4419-5919-5. [Google Scholar]
- Lüsse, S.; Arnold, K. Water Binding of PolysaccharidesNMR and ESR Studies. Macromolecules 1998, 31, 6891–6897. [Google Scholar] [CrossRef]
- Strain, S.M.; Fesik, S.W.; Armitage, I.M. Structure and metal-binding properties of lipopolysaccharides from heptoseless mutants of Escherichia coli studied by 13C and 31P nuclear magnetic resonance. J. Biol. Chem. 1983, 258, 13466–13477. [Google Scholar] [PubMed]
- Prudêncio, M.; Rohovec, J.; Peters, J.A.; Tocheva, E.; Boulanger, M.J.; Murphy, M.E.P.; Hupkes, H.-J.; Kosters, W.; Impagliazzo, A.; Ubbink, M. A caged lanthanide complex as a paramagnetic shift agent for protein NMR. Chem. Weinh. Bergstr. Ger. 2004, 10, 3252–3260. [Google Scholar] [CrossRef]
- Heatley, F. 18—Dynamics of Chains in Solutions by NMR Spectroscopy. In Comprehensive Polymer Science and Supplements; Allen, G., Bevington, J.C., Eds.; Pergamon: Amsterdam, The Netherlands, 1989; pp. 377–396. ISBN 978-0-08-096701-1. [Google Scholar]
- Nordlund, T.M. Quantitative Understanding of Biosystems: An Introduction to Biophysics; CRC Press (Taylor and Francis Group): Boca Raton, FL, USA, 2011; ISBN 978-1-4200-8973-8. [Google Scholar]
- Chen, F.J. Progress in Brain Mapping Research; Nova Publishers Inc.: Hauppauge, NY, USA, 2006; ISBN 978-1-59454-580-1. [Google Scholar]
- Knauss, R.; Schiller, J.; Fleischer, G.; Kärger, J.; Arnold, K. Self-diffusion of water in cartilage and cartilage components as studied by pulsed field gradient NMR. Magn. Reson. Med. 1999, 41, 285–292. [Google Scholar] [CrossRef]
- Burstein, D.; Gray, M.L.; Hartman, A.L.; Gipe, R.; Foy, B.D. Diffusion of small solutes in cartilage as measured by nuclear magnetic resonance (NMR) spectroscopy and imaging. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 1993, 11, 465–478. [Google Scholar] [CrossRef]
- Lüsse, S.; Arnold, K. The Interaction of Poly(ethylene glycol) with Water Studied by 1H and 2H NMR Relaxation Time Measurements. Macromolecules 1996, 29, 4251–4257. [Google Scholar] [CrossRef]
- Majonis, D.; Herrera, I.; Ornatsky, O.; Schulze, M.; Lou, X.; Soleimani, M.; Nitz, M.; Winnik, M.A. Synthesis of a functional metal-chelating polymer and steps toward quantitative mass cytometry bioassays. Anal. Chem. 2010, 82, 8961–8969. [Google Scholar] [CrossRef]
- Gouin, S.; Winnik, F.M. Quantitative assays of the amount of diethylenetriaminepentaacetic acid conjugated to water-soluble polymers using isothermal titration calorimetry and colorimetry. Bioconjug. Chem. 2001, 12, 372–377. [Google Scholar] [CrossRef]
- Dutta, A.K.; Rösgen, J.; Rajarathnam, K. Using isothermal titration calorimetry to determine thermodynamic parameters of protein-glycosaminoglycan interactions. Methods Mol. Biol. Clifton NJ 2015, 1229, 315–324. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, C.K.; Lee, Y.M.; Kim, I.Y.; Kim, S.I. Electrical/pH-sensitive swelling behavior of polyelectrolyte hydrogels prepared with hyaluronic acid–poly(vinyl alcohol) interpenetrating polymer networks. React. Funct. Polym. 2003, 55, 291–298. [Google Scholar] [CrossRef]
- Termühlen, F.; Kuckling, D.; Schönhoff, M. Isothermal Titration Calorimetry to Probe the Coil-to-Globule Transition of Thermoresponsive Polymers. J. Phys. Chem. B 2017, 121, 8611–8618. [Google Scholar] [CrossRef]
- Ivanov, D.; Neamtu, A. Molecular dynamics evaluation of hyaluronan interactions with dimethylsilanediol in aqueous solution. Rev. Roum. Chim. 2013, 58, 229–238. [Google Scholar]
- Santos, H.A.; Manzanares, J.A.; Murtomäki, L.; Kontturi, K. Thermodynamic analysis of binding between drugs and glycosaminoglycans by isothermal titration calorimetry and fluorescence spectroscopy. Eur. J. Pharm. Sci. 2007, 32, 105–114. [Google Scholar] [CrossRef]
- Othman, M.; Bouchemal, K.; Couvreur, P.; Gref, R. Microcalorimetric investigation on the formation of supramolecular nanoassemblies of associative polymers loaded with gadolinium chelate derivatives. Int. J. Pharm. 2009, 379, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Multi-Effect-Coupling Thermal-Stimulus (MECtherm) Model for Temperature-Sensitive Hydrogel. In Smart Hydrogel Modelling; Li, H., Ed.; Springer: Berlin, Germany, 2009; pp. 219–293. ISBN 978-3-642-02368-2. [Google Scholar]
- Ruggiero, F.; Netti, P.A.; Torino, E. Experimental Investigation and Thermodynamic Assessment of Phase Equilibria in the PLLA/Dioxane/Water Ternary System for Applications in the Biomedical Field. Langmuir 2015, 31, 13003–13010. [Google Scholar] [CrossRef] [PubMed]
- Hargittai, I.; Hargittai, M. Molecular structure of hyaluronan: an introduction. Struct. Chem. 2008, 19, 697–717. [Google Scholar] [CrossRef]
- Schlenoff, J.B.; Rmaile, A.H.; Bucur, C.B. Hydration Contributions to Association in Polyelectrolyte Multilayers and Complexes: Visualizing Hydrophobicity. J. Am. Chem. Soc. 2008, 130, 13589–13597. [Google Scholar] [CrossRef]
- Boros, E.; Srinivas, R.; Kim, H.-K.; Raitsimring, A.M.; Astashkin, A.V.; Poluektov, O.G.; Niklas, J.; Horning, A.D.; Tidor, B.; Caravan, P. Intramolecular Hydrogen Bonding Restricts Gd–Aqua-Ligand Dynamics. Angew. Chem. 2017, 129, 5695–5698. [Google Scholar] [CrossRef]
- Peitzsch, R.M.; Reed, W.F. High osmotic stress behavior of hyaluronate and heparin. Biopolymers 1992, 32, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, K.; Si, Y.; Guo, X.; Xu, Y. Protein–Polyelectrolyte Interaction: Thermodynamic Analysis Based on the Titration Method †. Polymers 2019, 11, 82. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
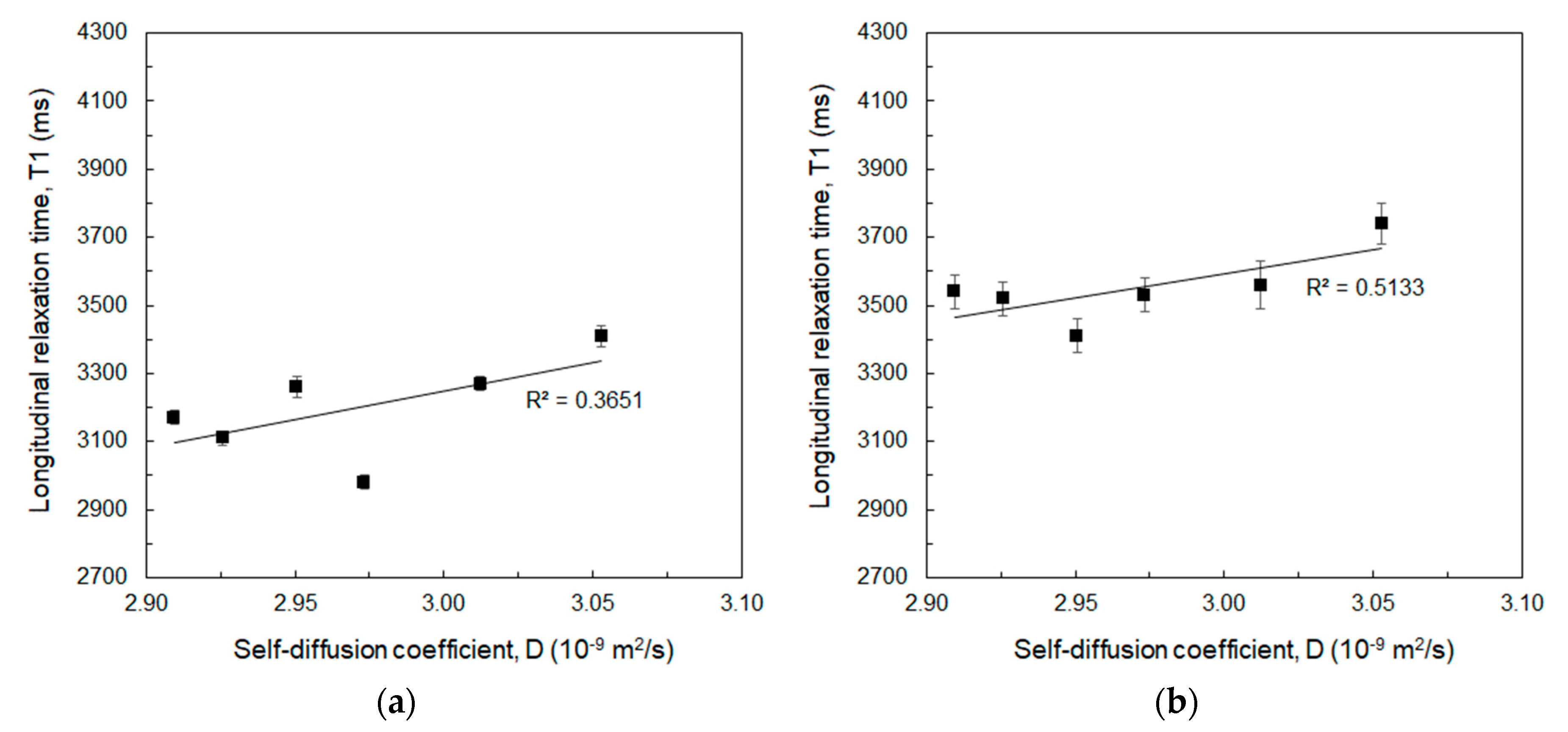
| HA (%w/v) | DVS/HA (g/g) | T1 (ms) 1 Mean ± std | T1 (ms) 2 Mean ± std |
|---|---|---|---|
| 0.25 | 0 | 3410 ± 30 | 3740 ± 60 |
| 0.25 | 2.35 | 3270 ± 20 | 3560 ± 70 |
| 0.25 | 4.70 | 2980 ± 20 | 3530 ± 50 |
| 0.25 | 7.06 | 3260 ± 30 | 3410 ± 50 |
| 0.25 | 9.42 | 3110 ± 20 | 3520 ± 50 |
| 0.25 | 11.77 | 3170 ± 20 | 3540 ± 50 |
| HA (%w/v) | Blank 1 Mean ± std 2 | n 1 Mean ± std 2 | Kd (*10−6 M) 1 Mean ± std 2 | ΔH (kJ/mol) 1 Mean ± std 2 | T∙ΔS (kJ/mol) 1 | ΔG (kJ/mol) 3 |
|---|---|---|---|---|---|---|
| 0.1 | −25.43 ± 24.36 | 9.99 ± 3.77 | 26.30 ± 722.3 | 24.04 ± 49.60 | 50.15 | -26.11 |
| 0.2 | 5.59 ± 4.02 | 0.71 ± 0.37 | 7.25 ± 0.58 | −10.12 ± 53.83 | 19.21 | 29.33 |
| 0.3 | 7.76 ± 1.21 | 0.30 ± 0.11 | 3.62 ± 0.12 | −10.56 ± 20.03 | 20.49 | 31.05 |
| 0.4 | 7.37 ± 0.96 | 0.33 ± 0.038 | 3.71 ± 3.84 | −11.54 ± 2.84 | 19.44 | 30.98 |
| 0.5 | −2.18 ± 1.16 | 0.18 ± 0.040 | 1.60 ± 7.98 | −10.06 ± 9.43 | 23.01 | 33.07 |
| 0.6 | 9.97 ± 22.97 | 0.39 ± 0.53 | 0.24 ± 0.013 | −23.84 ± 64.48 | 2.48 | 26.31 |
| 0.7 | 1.56 ± 1.71 | 0.20 ± 0.031 | 1.96 ± 5.16 | −11.79 ± 5.11 | 20.78 | 32.57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponsiglione, A.M.; Russo, M.; Torino, E. Glycosaminoglycans and Contrast Agents: The Role of Hyaluronic Acid as MRI Contrast Enhancer. Biomolecules 2020, 10, 1612. https://doi.org/10.3390/biom10121612
Ponsiglione AM, Russo M, Torino E. Glycosaminoglycans and Contrast Agents: The Role of Hyaluronic Acid as MRI Contrast Enhancer. Biomolecules. 2020; 10(12):1612. https://doi.org/10.3390/biom10121612
Chicago/Turabian StylePonsiglione, Alfonso Maria, Maria Russo, and Enza Torino. 2020. "Glycosaminoglycans and Contrast Agents: The Role of Hyaluronic Acid as MRI Contrast Enhancer" Biomolecules 10, no. 12: 1612. https://doi.org/10.3390/biom10121612
APA StylePonsiglione, A. M., Russo, M., & Torino, E. (2020). Glycosaminoglycans and Contrast Agents: The Role of Hyaluronic Acid as MRI Contrast Enhancer. Biomolecules, 10(12), 1612. https://doi.org/10.3390/biom10121612