Cannabinoid Receptor Interacting Protein 1a (CRIP1a) in Health and Disease

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
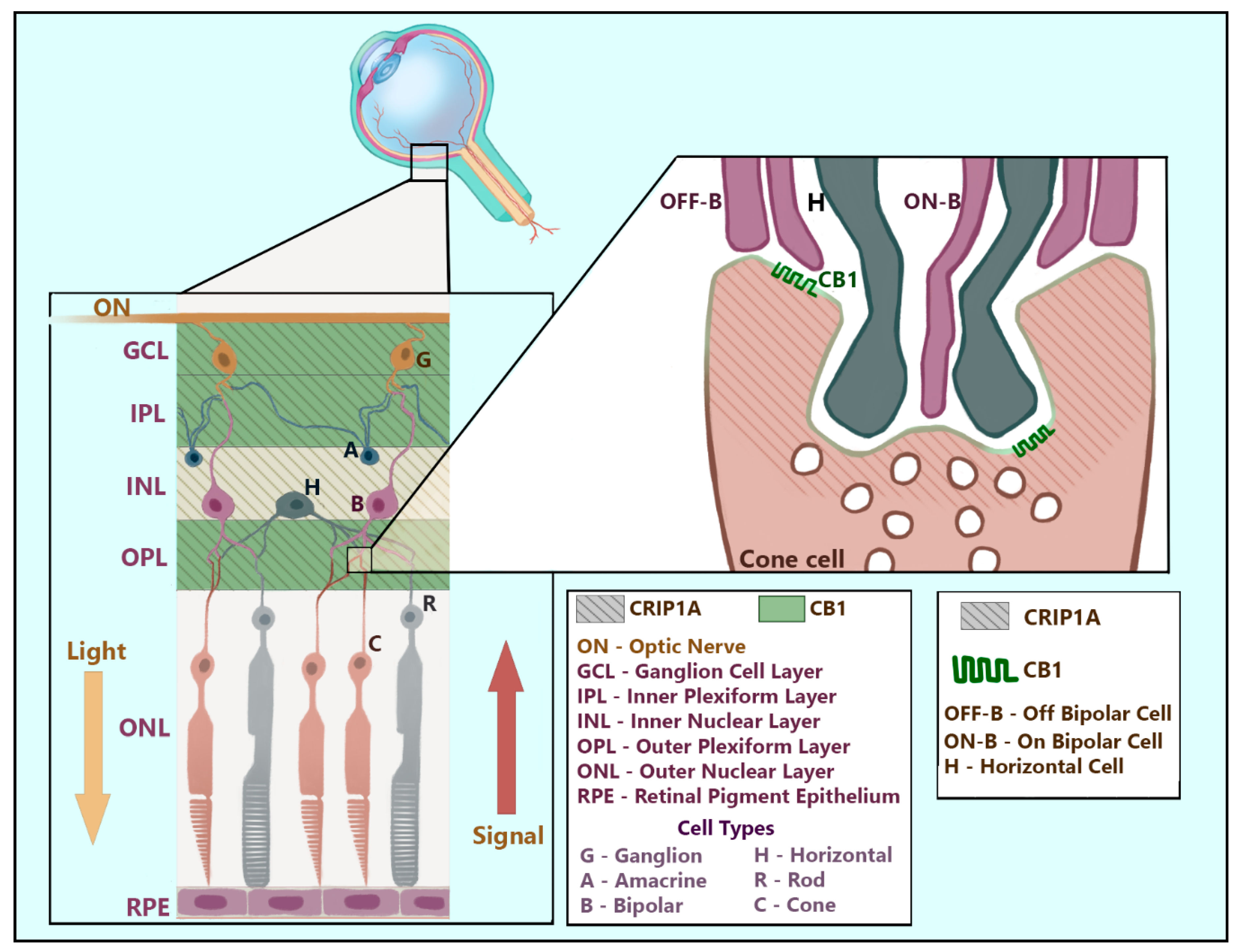{kind=link}
Abstract
1. Introduction
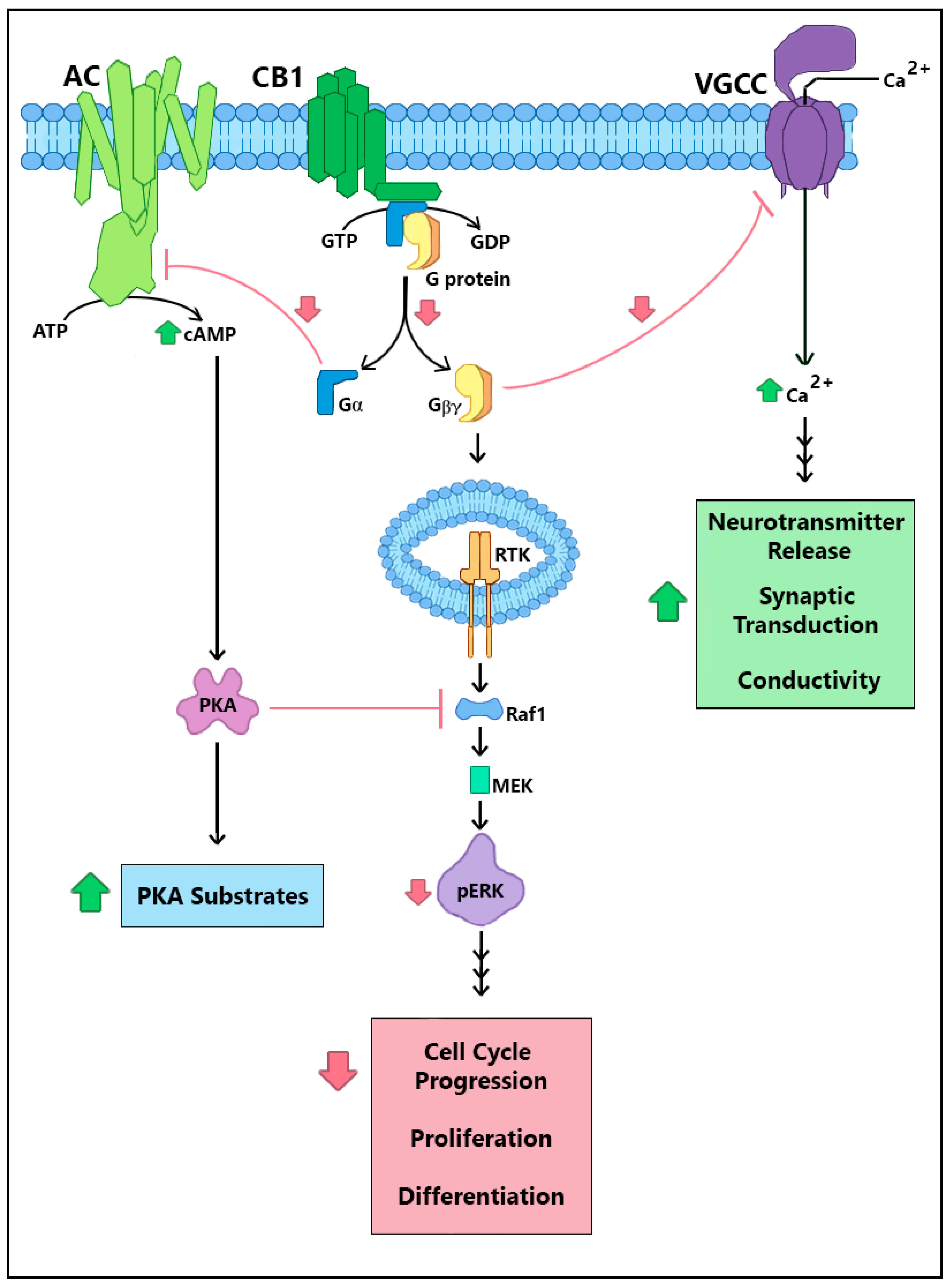
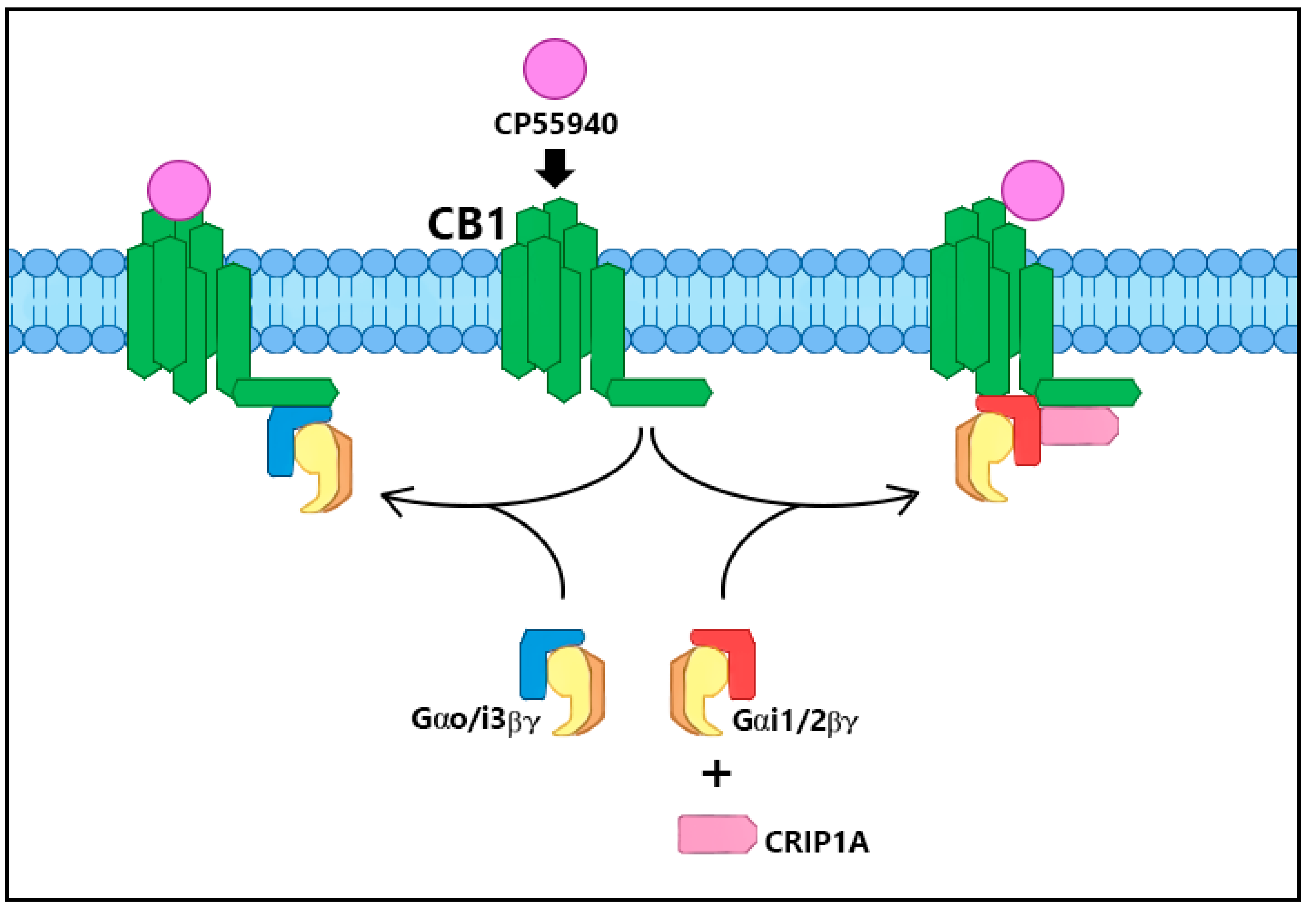
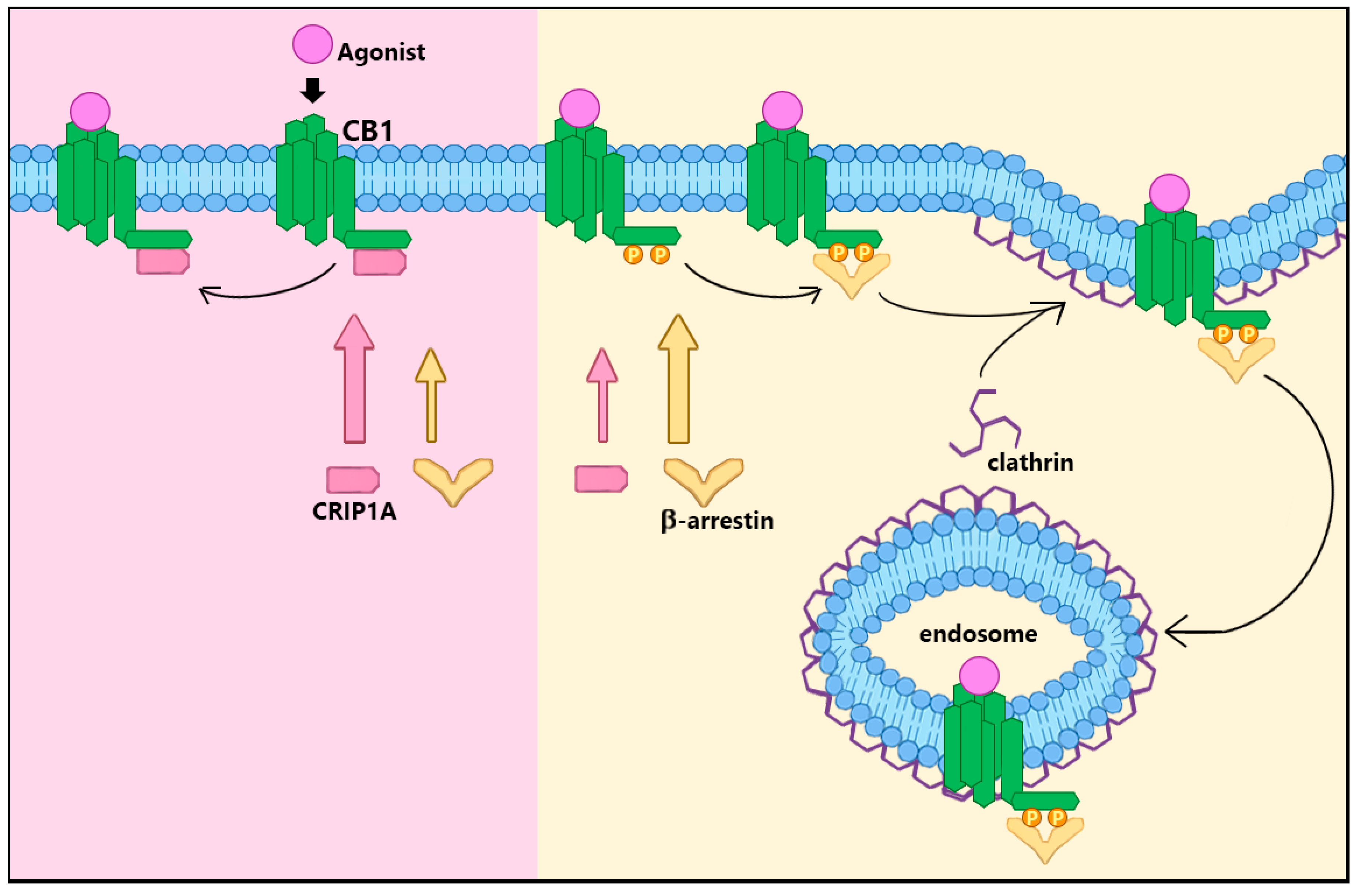
2. CRIP1a Cellular Mechanisms of Action
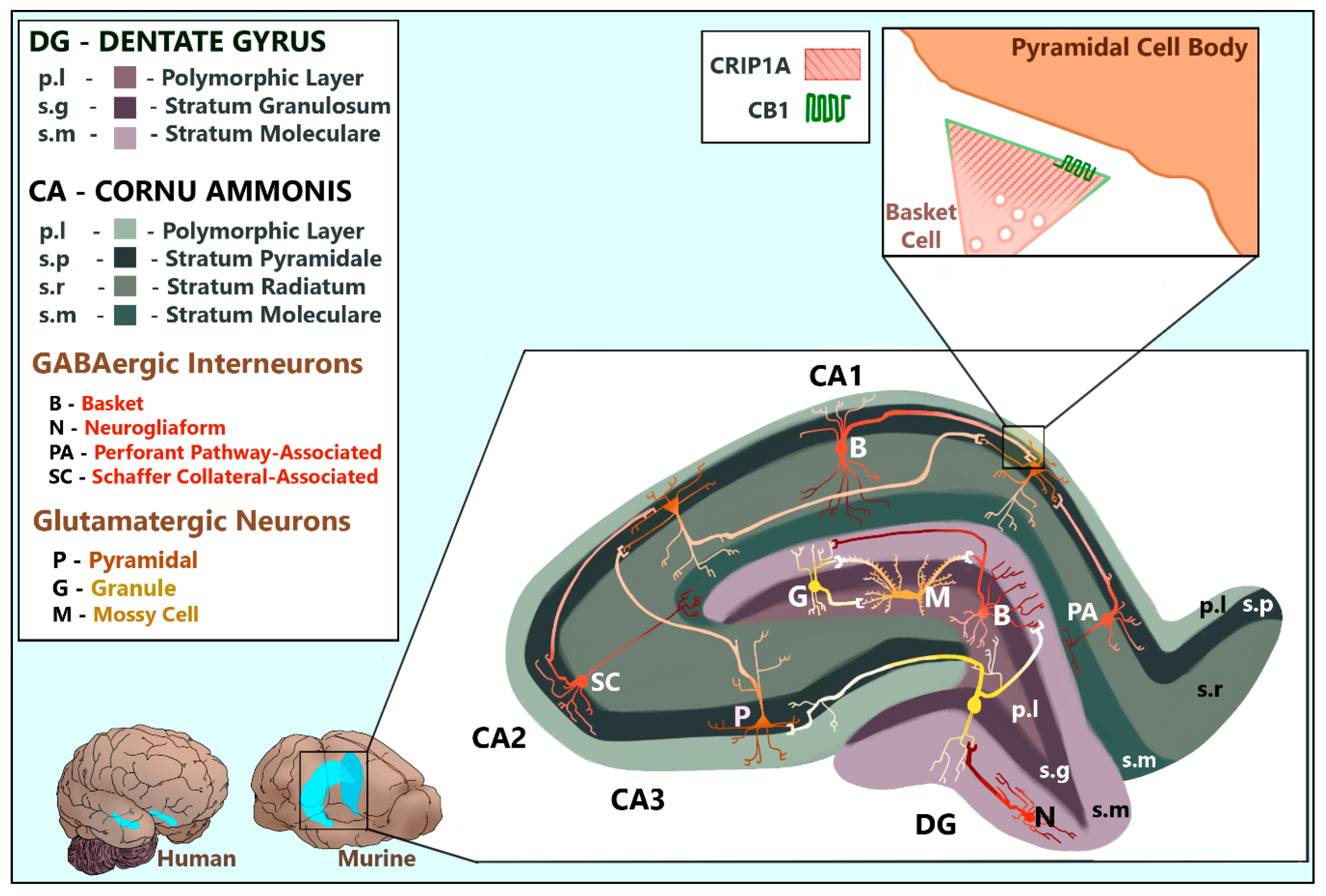
3. CRIP1a Distribution in the Mammalian Brain
4. CRIP1a in Development
5. CRIP1a and Sensory Systems
6. CRIP1a in Neurophysiology and Seizures
7. CRIP1a and Neuropsychiatric Diseases
8. CRIP1a within Cancer Epigenetics
9. Summary and Prospectus
Author Contributions
Funding
Conflicts of Interest
References
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef]
- Kendall, D.A.; Yudowski, G. Cannabinoid Receptors in the Central Nervous System: Their Signaling and Roles in Disease. Front. Cell. Neurosci. 2016, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Anderson, H.D. Cannabinoid signaling in health and disease. Can. J. Physiol. Pharmacol. 2017, 95, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Ligresti, A.; De Petrocellis, L.; Di Marzo, V. From Phytocannabinoids to Cannabinoid Receptors and Endocannabinoids: Pleiotropic Physiological and Pathological Roles through Complex Pharmacology. Physiol. Rev. 2016, 96, 1593–1659. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Hanuš, L.O.; Pertwee, R.G.; Howlett, A.C. Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat. Rev. Neurosci. 2014, 15, 757–764. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB1and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef]
- Howlett, A.C. International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Eldeeb, K.; Leone-Kabler, S.; Howlett, A.C. Mouse Neuroblastoma CB1 Cannabinoid Receptor-Stimulated [35S]GTPɣS Binding: Total and Antibody-Targeted Gα Protein-Specific Scintillation Proximity Assays. Methods Enzymol. 2017, 593, 1–21. [Google Scholar] [CrossRef]
- Turu, G.; Hunyady, L. Signal transduction of the CB1 cannabinoid receptor. J. Mol. Endocrinol. 2010, 44, 75–85. [Google Scholar] [CrossRef]
- Lovinger, D.M. Presynaptic Modulation by Endocannabinoids. Handb. Exp. Pharmacol. 2008, 2008, 435–477. [Google Scholar] [CrossRef]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-Mediated Control of Synaptic Transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef] [PubMed]
- Dalton, G.D.; Carney, S.T.; Marshburn, J.D.; Norford, D.C.; Howlett, A.C. CB1 Cannabinoid Receptors Stimulate Gβγ-GRK2-Mediated FAK Phosphorylation at Tyrosine 925 to Regulate ERK Activation Involving Neuronal Focal Adhesions. Front. Cell. Neurosci. 2020, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Dalton, G.D.; Peterson, L.J.; Howlett, A.C. CB1 cannabinoid receptors promote maximal FAK catalytic activity by stimulating cooperative signaling between receptor tyrosine kinases and integrins in neuronal cells. Cell Signal. 2013, 25, 1665–1677. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fletcher-Jones, A.; Hildick, K.L.; Evans, A.J.; Nakamura, Y.; Henley, J.M.; Wilkinson, K.A. Protein Interactors and Trafficking Pathways That Regulate the Cannabinoid Type 1 Receptor (CB1R). Front. Mol. Neurosci. 2020, 13, 108. [Google Scholar] [CrossRef]
- Howlett, A.; Blume, L.C.; Dalton, G.D. CB1 Cannabinoid Receptors and their Associated Proteins. Curr. Med. Chem. 2010, 17, 1382–1393. [Google Scholar] [CrossRef]
- Stadel, R.; Ahn, K.H.; Kendall, D.A. The cannabinoid type-1 receptor carboxyl-terminus, more than just a tail. J. Neurochem. 2011, 117, 1–18. [Google Scholar] [CrossRef]
- Houston, D.B.; Howlett, A.C. Differential Receptor–G-Protein Coupling Evoked by Dissimilar Cannabinoid Receptor Agonists. Cell Signal. 1998, 10, 667–674. [Google Scholar] [CrossRef]
- Blume, L.C.; Eldeeb, K.; Bass, C.E.; Selley, D.E.; Howlett, A.C. Cannabinoid receptor interacting protein (CRIP1a) attenuates CB1R signaling in neuronal cells. Cell Signal. 2015, 27, 716–726. [Google Scholar] [CrossRef]
- Blume, L.C.; Leone-Kabler, S.; Luessen, D.J.; Marrs, G.S.; Lyons, E.W.; Bass, C.E.; Chen, R.; Selley, D.E.; Howlett, A.C. Cannabinoid receptor interacting protein suppresses agonist-driven CB1 receptor internalization and regulates receptor replenishment in an agonist-biased manner. J. Neurochem. 2016, 139, 396–407. [Google Scholar] [CrossRef]
- Niehaus, J.L.; Liu, Y.; Wallis, K.T.; Egertová, M.; Bhartur, S.G.; Mukhopadhyay, S.; Shi, S.; He, H.; Selley, D.E.; Howlett, A.C.; et al. CB1 Cannabinoid Receptor Activity Is Modulated by the Cannabinoid Receptor Interacting Protein CRIP 1a. Mol. Pharmacol. 2007, 72, 1557–1566. [Google Scholar] [CrossRef]
- Smith, T.H.; Blume, L.C.; Straiker, A.; Cox, J.O.; David, B.G.; McVoy, J.R.S.; Sayers, K.W.; Poklis, J.L.; Abdullah, R.A.; Egertová, M.; et al. Cannabinoid receptor-interacting protein 1a modulates CB1 receptor signaling and regulation. Mol. Pharmacol. 2015, 87, 747–765. [Google Scholar] [CrossRef] [PubMed]
- Nogueras-Ortiz, C.; Yudowski, G.A. The Multiple Waves of Cannabinoid 1 Receptor Signaling. Mol. Pharmacol. 2016, 90, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Martini, L.; Waldhoer, M.; Pusch, M.; Kharazia, V.; Fong, J.; Lee, J.H.; Freissmuth, C.; Whistler, J.L. Ligand-induced down-regulation of the cannabinoid 1 receptor is mediated by the G-protein-coupled receptor-associated sorting protein GASP1. FASEB J. 2007, 21, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Tappe-Theodor, A.; Agarwal, N.; Katona, I.; Rubino, T.; Martini, L.; Swiercz, J.; Mackie, K.; Monyer, H.; Parolaro, D.; Whistler, J.; et al. A Molecular Basis of Analgesic Tolerance to Cannabinoids. J. Neurosci. 2007, 27, 4165–4177. [Google Scholar] [CrossRef]
- Mascia, F.; Klotz, L.; Lerch, J.; Ahmed, M.H.; Zhang, Y.; Enz, R. CRIP1a inhibits endocytosis of G-protein coupled receptors activated by endocannabinoids and glutamate by a common molecular mechanism. J. Neurochem. 2017, 141, 577–591. [Google Scholar] [CrossRef]
- Blume, L.C.; Patten, T.; Eldeeb, K.; Leone-Kabler, S.; Ilyasov, A.A.; Keegan, B.M.; O’Neal, J.E.; Bass, C.E.; Hantgan, R.R.; Lowther, W.T.; et al. Cannabinoid Receptor Interacting Protein 1a Competition with β-Arrestin for CB1 Receptor Binding Sites. Mol. Pharmacol. 2017, 91, 75–86. [Google Scholar] [CrossRef]
- Bakshi, K.; Mercier, R.W.; Pavlopoulos, S. Interaction of a fragment of the cannabinoid CB1 receptor C-terminus with arrestin-2. FEBS Lett. 2007, 581, 5009–5016. [Google Scholar] [CrossRef]
- Flores-Otero, J.; Ahn, K.H.; Delgado-Peraza, F.; Mackie, K.; Kendall, D.A.; Yudowski, G. Ligand-specific endocytic dwell times control functional selectivity of the cannabinoid receptor 1. Nat. Commun. 2014, 5, 4589. [Google Scholar] [CrossRef]
- Delgado-Peraza, F.; Ahn, K.H.; Nogueras-Ortiz, C.; Mungrue, I.N.; Mackie, K.; Kendall, D.A.; Yudowski, G.A. Mechanisms of Biased β-Arrestin-Mediated Signaling Downstream from the Cannabinoid 1 Receptor. Mol. Pharmacol. 2016, 89, 618–629. [Google Scholar] [CrossRef]
- Guggenhuber, S.; Alpár, A.; Chen, R.; Schmitz, N.; Wickert, M.; Mattheus, T.; Harasta, A.E.; Purrio, M.; Kaiser, N.; Elphick, M.R.; et al. Cannabinoid receptor-interacting protein Crip1a modulates CB1 receptor signaling in mouse hippocampus. Brain Struct. Funct. 2016, 221, 2061–2074. [Google Scholar] [CrossRef]
- Marsicano, G.; Lutz, B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur. J. Neurosci. 1999, 11, 4213–4225. [Google Scholar] [CrossRef] [PubMed]
- Tsou, K.; Mackie, K.; Sañudo-Peña, M.; Walker, J. Cannabinoid CB1 receptors are localized primarily on cholecystokinin-containing GABAergic interneurons in the rat hippocampal formation. Neuroscience 1999, 93, 969–975. [Google Scholar] [CrossRef]
- Monory, K.; Massa, F.; Egertová, M.; Eder, M.; Blaudzun, H.; Westenbroek, R.; Kelsch, W.; Jacob, W.; Marsch, R.; Ekker, M.; et al. The Endocannabinoid System Controls Key Epileptogenic Circuits in the Hippocampus. Neuron 2006, 51, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Münster-Wandowski, A.; Gómez-Lira, G.; Gutiérrez, R. Mixed neurotransmission in the hippocampal mossy fibers. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Caiati, M.D.; Sivakumaran, S.; Lanore, F.; Mulle, C.; Richard, E.; Verrier, D.; Marsicano, G.; Miles, R.; Cherubini, E. Developmental regulation of CB1-mediated spike-time dependent depression at immature mossy fiber-CA3 synapses. Sci. Rep. 2012, 2, 285. [Google Scholar] [CrossRef]
- Pelkey, K.A.; Chittajallu, R.; Craig, M.T.; Tricoire, L.; Wester, J.C.; McBain, C.J. Hippocampal GABAergic Inhibitory Interneurons. Physiol. Rev. 2017, 97, 1619–1747. [Google Scholar] [CrossRef]
- Scharfman, H.E. The enigmatic mossy cell of the dentate gyrus. Nat. Rev. Neurosci. 2016, 17, 562–575. [Google Scholar] [CrossRef]
- Scharfman, H.E.; Myers, C.E. Hilar mossy cells of the dentate gyrus: A historical perspective. Front. Neural Circuits 2013, 6, 106. [Google Scholar] [CrossRef]
- Spruston, N. Pyramidal neurons: Dendritic structure and synaptic integration. Nat. Rev. Neurosci. 2008, 9, 206–221. [Google Scholar] [CrossRef]
- Zhao, C.; Teng, E.M.; Summers, R.G.; Ming, G.-L.; Gage, F.H. Distinct Morphological Stages of Dentate Granule Neuron Maturation in the Adult Mouse Hippocampus. J. Neurosci. 2006, 26, 3–11. [Google Scholar] [CrossRef]
- Dudok, B.; Barna, L.; Ledri, M.; Szabó, S.I.; Szabadits, E.; Pintér, B.; Woodhams, S.G.; Henstridge, C.M.; Balla, G.Y.; Nyilas, R.; et al. Cell-specific STORM super-resolution imaging reveals nanoscale organization of cannabinoid signaling. Nat. Neurosci. 2015, 18, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Hashimotodani, Y.; Ohno-Shosaku, T.; Kano, M. Endocannabinoids and Synaptic Function in the CNS. Neuroscience 2007, 13, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Janz, R.; Südhof, T.C. SV2C is a synaptic vesicle protein with an unusually restricted localization: Anatomy of a synaptic vesicle protein family. Neuroscience 1999, 94, 1279–1290. [Google Scholar] [CrossRef]
- Bajjalieh, S.; Frantz, G.; Weimann, J.; McConnell, S.; Scheller, R. Differential expression of synaptic vesicle protein 2 (SV2) isoforms. J. Neurosci. 1994, 14, 5223–5235. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Suzuki, T.; Takahashi, C.; Nishida, E.; Kusakabe, M. cnrip1is a regulator of eye and neural development inXenopus laevis. Genes Cells 2015, 20, 324–339. [Google Scholar] [CrossRef] [PubMed]
- Fin, L.; Bergamin, G.; Steiner, R.A.; Hughes, S.M. The Cannabinoid Receptor Interacting Proteins 1 of zebrafish are not required for morphological development, viability or fertility. Sci. Rep. 2017, 7, 4858. [Google Scholar] [CrossRef]
- Jung, H.Y.; Kim, D.W.; Nam, S.M.; Kim, J.W.; Chung, J.Y.; Won, M.-H.; Seong, J.K.; Yoon, Y.S.; Yoo, D.Y.; Hwang, I.K. Pyridoxine improves hippocampal cognitive function via increases of serotonin turnover and tyrosine hydroxylase, and its association with CB1 cannabinoid receptor-interacting protein and the CB1 cannabinoid receptor pathway. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 3142–3153. [Google Scholar] [CrossRef]
- Gerdes, J.; Lemke, H.; Baisch, H.; Wacker, H.H.; Schwab, U.; Stein, H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J. Immunol. 1984, 133, 1710–1715. [Google Scholar]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Brown, J.P.; Couillard-Després, S.; Cooper-Kuhn, C.M.; Winkler, J.; Aigner, L.; Kuhn, H.G. Transient expression of doublecortin during adult neurogenesis. J. Comp. Neurol. 2003, 467, 1–10. [Google Scholar] [CrossRef]
- Gendrel, A.-V.; Attia, M.; Chen, C.-J.; Diabangouaya, P.; Servant, N.; Barillot, E.; Heard, E. Developmental Dynamics and Disease Potential of Random Monoallelic Gene Expression. Dev. Cell 2014, 28, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.L.; Plotkin, J.L.; Venø, M.T.; Von Schimmelmann, M.; Feinberg, P.; Mann, S.; Handler, A.; Kjems, J.; Surmeier, D.J.; O’Carroll, D.; et al. MicroRNA-128 Governs Neuronal Excitability and Motor Behavior in Mice. Science 2013, 342, 1254–1258. [Google Scholar] [CrossRef] [PubMed]
- Yazulla, S. Endocannabinoids in the retina: From marijuana to neuroprotection. Prog. Retin. Eye Res. 2008, 27, 501–526. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.S.-J.; Arnold, A.; Hutchens, J.M.; Radicke, J.; Cravatt, B.F.; Wager-Miller, J.; Mackie, K.; Straiker, A. Architecture of cannabinoid signaling in mouse retina. J. Comp. Neurol. 2010, 518, 3848–3866. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, P.; Rajnicek, A.M.; Morozov, Y.M.; Ross, R.A.; Mulder, J.; Urbán, G.M.; Monory, K.; Marsicano, G.; Matteoli, M.; Canty, A.; et al. Hardwiring the Brain: Endocannabinoids Shape Neuronal Connectivity. Science 2007, 316, 1212–1216. [Google Scholar] [CrossRef]
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A.; Matias, I.; Schiano-Moriello, A.; Paul, P.; Williams, E.-J.; et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 2003, 163, 463–468. [Google Scholar] [CrossRef]
- Katona, I.; Urbán, G.M.; Wallace, M.; Ledent, C.; Jung, K.M.; Piomelli, D.; Mackie, K.; Freund, T.F. Molecular composition of the endocannabinoid system at glutamatergic synapses. J. Neurosci. 2006, 26, 5628–5637. [Google Scholar] [CrossRef]
- Mátyás, F.; Urbán, G.M.; Watanabe, M.; Mackie, K.; Zimmer, A.; Freund, T.F.; Katona, I. Identification of the sites of 2-arachidonoylglycerol synthesis and action imply retrograde endocannabinoid signaling at both GABAergic and glutamatergic synapses in the ventral tegmental area. Neuropharmacology 2008, 54, 95–107. [Google Scholar] [CrossRef]
- Koulen, P.; Fletcher, E.L.; Craven, S.E.; Bredt, D.S.; Wässle, H. Immunocytochemical Localization of the Postsynaptic Density Protein PSD-95 in the Mammalian Retina. J. Neurosci. 1998, 18, 10136–10149. [Google Scholar] [CrossRef]
- Schreiner, D.; Jande, S.; Lawson, D. Target Cells of Vitamin D in the Vertebrate Retina. Acta Anat. 1985, 121, 153–162. [Google Scholar] [CrossRef]
- Thomasset, M.; Parkes, C.O.; Clavel, M.C. Immunocytochemical detection of 28000-MW calcium-binding protein in horizontal cells of the rat retina. Cell Tissue Res. 1985, 240, 493–496. [Google Scholar] [CrossRef]
- Röhrenbeck, J.; Wässle, H.; Heizmann, C.W. Immunocytochemical labelling of horizontal cells in mammalian retina using antibodies against calcium-binding proteins. Neurosci. Lett. 1987, 77, 255–260. [Google Scholar] [CrossRef]
- Chapot, C.A.; Euler, T.; Schubert, T. How do horizontal cells ‘talk’ to cone photoreceptors? Different levels of complexity at the cone-horizontal cell synapse. J. Physiol. 2017, 595, 5495–5506. [Google Scholar] [CrossRef] [PubMed]
- Purves, D.; Augustine, G.A.; Fitzpatrick, D.; Katz, L.C.; LaMantia, A.-S.; McNamara, J.O.; Williams, S.M. Neuroscience, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Lezirovitz, K.; Vieira-Silva, G.A.; Batissoco, A.C.; Levy, D.; Kitajima, J.P.; Trouillet, A.; Ouyang, E.; Zebajardi, N.; Sampaio-Silva, J.; Pedroso-Campos, V.; et al. A rare genomic duplication in 2p14 underlies autosomal dominant hearing loss DFNA58. Hum. Mol. Genet. 2020, 29, 1520–1536. [Google Scholar] [CrossRef]
- Benbadis, S.R. Localization-Related (Focal) Epilepsy: Causes and Clinical Features; UpToDate: Waltham, MA, USA, 2018. [Google Scholar]
- Ben-Ari, Y.; Cossart, R. Kainate, a double agent that generates seizures: Two decades of progress. Trends Neurosci. 2000, 23, 580–587. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Limbic seizure and brain damage produced by kainic acid: Mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 1985, 14, 375–403. [Google Scholar] [CrossRef]
- Nadler, J.V. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci. 1981, 29, 2031–2042. [Google Scholar] [CrossRef]
- Cronin, J.; Dudek, F.E. Chronic seizures and collateral sprouting of dentate mossy fibers after kainic acid treatment in rats. Brain Res. 1988, 474, 181–184. [Google Scholar] [CrossRef]
- Represa, A.; Tremblay, E.; Ben-Ari, Y. Kainate binding sites in the hippocampal mossy fibers: Localization and plasticity. Neuroscience 1987, 20, 739–748. [Google Scholar] [CrossRef]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Gutiérrez, S.O.; Van Der Stelt, M.; et al. CB1 Cannabinoid Receptors and On-Demand Defense against Excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef]
- Schauwecker, P.E.; Steward, O. Genetic determinants of susceptibility to excitotoxic cell death: Implications for gene targeting approaches. Proc. Natl. Acad. Sci. USA 1997, 94, 4103–4108. [Google Scholar] [CrossRef] [PubMed]
- Bojnik, E.; Turunc, E.; Armagan, G.; Kanıt, L.; Benyhe, S.; Yalçın, A.; Borsodi, A. Changes in the cannabinoid (CB1) receptor expression level and G-protein activation in kainic acid induced seizures. Epilepsy Res. 2012, 99, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, B.; Wallis, K.T.; Wilson, S.P.; Egertová, M.; Elphick, M.R.; Lewis, D.L.; Hardy, L.R. CRIP1a switches cannabinoid receptor agonist/antagonist-mediated protection from glutamate excitotoxicity. Neurosci. Lett. 2011, 503, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Ludányi, A.; Erőss, L.; Czirják, S.; Vajda, J.; Halász, P.; Watanabe, M.; Palkovits, M.; Maglóczky, Z.; Freund, T.F.; Katona, I. Downregulation of the CB1 Cannabinoid Receptor and Related Molecular Elements of the Endocannabinoid System in Epileptic Human Hippocampus. J. Neurosci. 2008, 28, 2976–2990. [Google Scholar] [CrossRef]
- Kovasznay, B.; Fleischer, J.; Tanenberg-Karant, M.; Jandorf, L.; Miller, A.D.; Bromet, E. Substance Use Disorder and the Early Course of Illness in Schizophrenia and Affective Psychosis. Schizophr. Bull. 1997, 23, 195–201. [Google Scholar] [CrossRef][Green Version]
- Ayalew, M.; Le-Niculescu, H.; Levey, D.F.; Jain, N.K.; Changala, B.; Patel, S.D.; Winiger, E.; Breier, A.; Shekhar, A.; Amdur, R.L.; et al. Convergent functional genomics of schizophrenia: From comprehensive understanding to genetic risk prediction. Mol. Psychiatry 2012, 17, 887–905. [Google Scholar] [CrossRef]
- Dalton, V.S.; Long, L.E.; Weickert, C.S.; Zavitsanou, K. Paranoid Schizophrenia is Characterized by Increased CB1 Receptor Binding in the Dorsolateral Prefrontal Cortex. Neuropsychopharmacology 2011, 36, 1620–1630. [Google Scholar] [CrossRef]
- Dean, B.; Sundram, S.; Bradbury, R.; Scarr, E.; Copolov, D. Studies on [3H]CP-55940 binding in the human central nervous system: Regional specific changes in density of cannabinoid-1 receptors associated with schizophrenia and cannabis use. Neuroscience 2001, 103, 9–15. [Google Scholar] [CrossRef]
- Zavitsanou, K.; Garrick, T.; Huang, X.-F. Selective antagonist [3H]SR141716A binding to cannabinoid CB1 receptors is increased in the anterior cingulate cortex in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 355–360. [Google Scholar] [CrossRef]
- Blume, L.C.; Bass, C.E.; Childers, S.R.; Dalton, G.D.; Roberts, D.C.S.; Richardson, J.M.; Xiao, R.; Selley, D.E.; Howlett, A.C. Striatal CB1and D2receptors regulate expression of each other, CRIP1A and delta opioid systems. J. Neurochem. 2013, 124, 808–820. [Google Scholar] [CrossRef]
- Wockner, L.F.; Noble, E.P.; Lawford, B.R.; Young, R.; Morris, C.; Whitehall, V.L.J.; Voisey, J. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl. Psychiatry 2014, 4, e339. [Google Scholar] [CrossRef] [PubMed]
- Lodge, D.J.; Grace, A.A. Gestational methylazoxymethanol acetate administration: A developmental disruption model of schizophrenia. Behav. Brain Res. 2009, 204, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Cattabeni, F.; Di Luca, M. Developmental models of brain dysfunctions induced by targeted cellular ablations with methylazoxymethanol. Physiol. Rev. 1997, 77, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.M.; Aguilar, D.D.; Neary, J.L.; Carless, M.A.; Giuffrida, A.; Lodge, D.J. Schizophrenia-Like Phenotype Inherited by the F2 Generation of a Gestational Disruption Model of Schizophrenia. Neuropsychopharmacology 2016, 41, 477–486. [Google Scholar] [CrossRef]
- Perez, S.M.; Donegan, J.J.; Boley, A.M.; Aguilar, D.D.; Giuffrida, A.; Lodge, D.J. Ventral hippocampal overexpression of Cannabinoid Receptor Interacting Protein 1 (CNRIP1) produces a schizophrenia-like phenotype in the rat. Schizophr. Res. 2019, 206, 263–270. [Google Scholar] [CrossRef]
- Robison, A.; Thakkar, K.N.; Diwadkar, V.A. Cognition and Reward Circuits in Schizophrenia: Synergistic, Not Separate. Biol. Psychiatry 2020, 87, 204–214. [Google Scholar] [CrossRef]
- Salling, M.C.; Faccidomo, S.P.; Li, C.; Psilos, K.; Galunas, C.; Spanos, M.; Agoglia, A.E.; Kash, T.L.; Hodge, C.W. Moderate Alcohol Drinking and the Amygdala Proteome: Identification and Validation of Calcium/Calmodulin Dependent Kinase II and AMPA Receptor Activity as Novel Molecular Mechanisms of the Positive Reinforcing Effects of Alcohol. Biol. Psychiatry 2016, 79, 430–442. [Google Scholar] [CrossRef]
- Javid, F.A.; Phillips, R.M.; Afshinjavid, S.; Verde, R.; Ligresti, A. Cannabinoid pharmacology in cancer research: A new hope for cancer patients? Eur. J. Pharmacol. 2016, 775, 1–14. [Google Scholar] [CrossRef]
- Sun, Z.; Qi, X.; Zhang, Y. Bioinformatics Analysis of the Expression of ATP binding cassette subfamily C member 3 (ABCC3) in Human Glioma. Open Med. 2020, 15, 107–113. [Google Scholar] [CrossRef]
- Lind, G.E.; Danielsen, S.A.; Ahlquist, T.; Merok, M.A.; Andresen, K.; Skotheim, R.I.; Hektoen, M.; Rognum, T.O.; Meling, G.I.; Hoff, G.; et al. Identification of an epigenetic biomarker panel with high sensitivity and specificity for colorectal cancer and adenomas. Mol. Cancer 2011, 10, 85. [Google Scholar] [CrossRef]
- Øster, B.; Thorsen, K.; Lamy, P.; Wojdacz, T.K.; Hansen, L.L.; Birkenkamp-Demtröder, K.; Sørensen, K.D.; Laurberg, S.; Ørntoft, T.F.; Andersen, C.L. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. Int. J. Cancer 2011, 129, 2855–2866. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Yang, Q.; Mo, M.; Ye, X.; Zhang, J.; Zhang, L.; Chen, B.; Li, J.; Cai, C.; Yang, Q. Screening of exon methylation biomarkers for colorectal cancer via LC-MS/MS strategy. J. Mass Spectrom. 2017, 52, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Andresen, K.; Boberg, K.M.; Vedeld, H.M.; Honne, H.; Jebsen, P.; Hektoen, M.; Wadsworth, C.A.; Clausen, O.P.; Lundin, K.E.; Paulsen, V.; et al. Four DNA methylation biomarkers in biliary brush samples accurately identify the presence of cholangiocarcinoma. Hepatology 2015, 61, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Galvan, A.; Frullanti, E.; Anderlini, M.; Manenti, G.; Noci, S.; Dugo, M.; Ambrogi, F.; De Cecco, L.; Spinelli, R.; Piazza, R.; et al. Gene expression signature of non-involved lung tissue associated with survival in lung adenocarcinoma patients. Carcinogenesis 2013, 34, 2767–2773. [Google Scholar] [CrossRef] [PubMed]
- Bethge, N.; Lothe, R.A.; Honne, H.; Andresen, K.; Trøen, G.; Eknæs, M.; Liestøl, K.; Holte, H.; Delabie, J.; Smeland, E.B.; et al. Colorectal cancer DNA methylation marker panel validated with high performance in Non-Hodgkin lymphoma. Epigenetics 2014, 9, 428–436. [Google Scholar] [CrossRef]
- Chong, Y.; Mia-Jan, K.; Ryu, H.; Abdul-Ghafar, J.; Munkhdelger, J.; Lkhagvadorj, S.; Jung, S.; Lee, M.; Ji, S.-Y.; Choi, E.; et al. DNA methylation status of a distinctively different subset of genes is associated with each histologic Lauren classification subtype in early gastric carcinogenesis. Oncol. Rep. 2014, 31, 2535–2544. [Google Scholar] [CrossRef]
- Liu, D.; Zhou, B.; Liu, R. A transcriptional co-expression network-based approach to identify prognostic biomarkers in gastric carcinoma. PeerJ 2020, 8, e8504. [Google Scholar] [CrossRef]
- Zhang, T.; Cui, G.; Yao, Y.-L.; Wang, Q.-C.; Gu, H.-G.; Li, X.-N.; Zhang, H.; Feng, W.-M.; Shi, Q.-L.; Cui, W. Value of CNRIP1 promoter methylation in colorectal cancer screening and prognosis assessment and its influence on the activity of cancer cells. Arch. Med. Sci. 2017, 13, 1281–1294. [Google Scholar] [CrossRef]
- Sun, X.; Chen, D.; Jin, Z.; Chen, T.; Lin, A.; Jin, H.; Zhu, Y.; Lai, M. Genome-wide methylation and expression profiling identify methylation-associated genes in colorectal cancer. Epigenomics 2020, 12, 19–36. [Google Scholar] [CrossRef]
- Noort, S.; Wander, P.; Alonzo, T.A.; Smith, J.; Ries, R.E.; Gerbing, R.B.; Dolman, M.E.M.; Locatelli, F.; Reinhardt, D.; Baruchel, A.; et al. The clinical and biological characteristics of NUP98-KDM5A in pediatric acute myeloid leukemia. Haematologica 2020. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Booth, W.; Walker, N.; Lowther, W.T.; Howlett, A.C. Cannabinoid Receptor Interacting Protein 1a (CRIP1a): Function and Structure. Molecules 2019, 24, 3672. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliver, E.E.; Hughes, E.K.; Puckett, M.K.; Chen, R.; Lowther, W.T.; Howlett, A.C. Cannabinoid Receptor Interacting Protein 1a (CRIP1a) in Health and Disease. Biomolecules 2020, 10, 1609. https://doi.org/10.3390/biom10121609
Oliver EE, Hughes EK, Puckett MK, Chen R, Lowther WT, Howlett AC. Cannabinoid Receptor Interacting Protein 1a (CRIP1a) in Health and Disease. Biomolecules. 2020; 10(12):1609. https://doi.org/10.3390/biom10121609
Chicago/Turabian StyleOliver, Emily E., Erin K. Hughes, Meaghan K. Puckett, Rong Chen, W. Todd Lowther, and Allyn C. Howlett. 2020. "Cannabinoid Receptor Interacting Protein 1a (CRIP1a) in Health and Disease" Biomolecules 10, no. 12: 1609. https://doi.org/10.3390/biom10121609
APA StyleOliver, E. E., Hughes, E. K., Puckett, M. K., Chen, R., Lowther, W. T., & Howlett, A. C. (2020). Cannabinoid Receptor Interacting Protein 1a (CRIP1a) in Health and Disease. Biomolecules, 10(12), 1609. https://doi.org/10.3390/biom10121609