Kidney Ischemia-Reperfusion Decreases Hydrogen Sulfide and Increases Oxidative Stress in the Heart

 ,
,  and
and 
Abstract
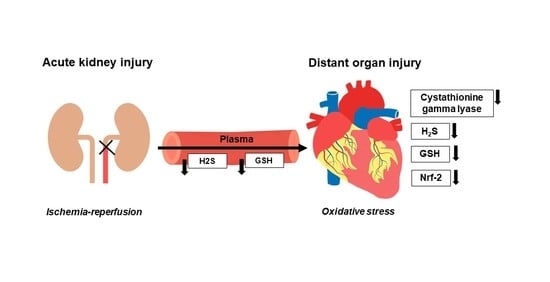
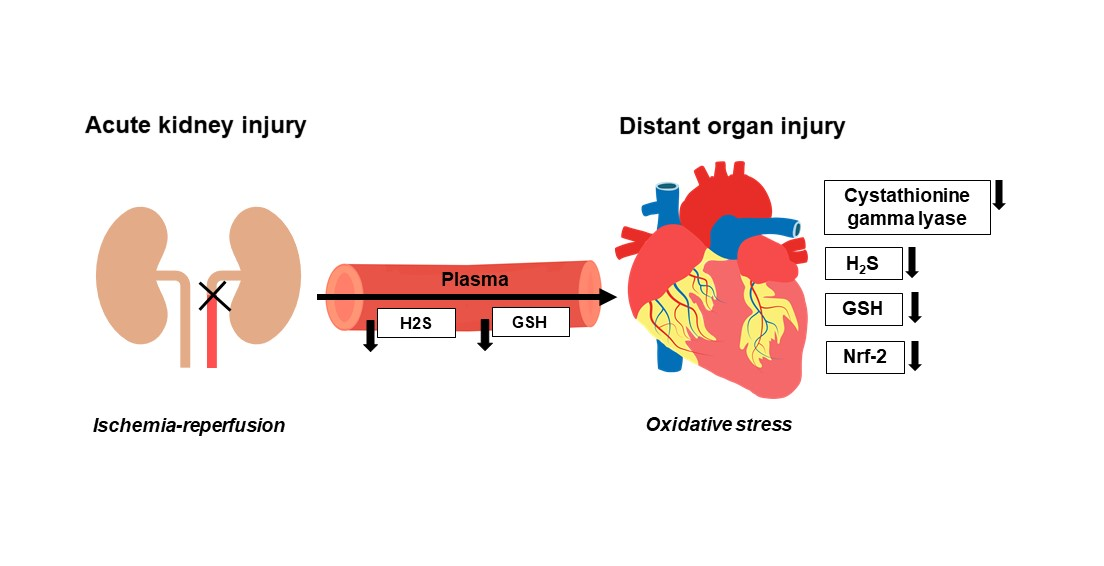
1. Introduction
2. Materials and Methods
2.1. Kidney Ischemia-Reperfusion
2.2. Biochemical Analysis
2.3. Histological Examination
2.4. Western Immunoblotting Analysis
2.5. Real-Time PCR Analysis
2.6. Statistical Analysis
3. Results
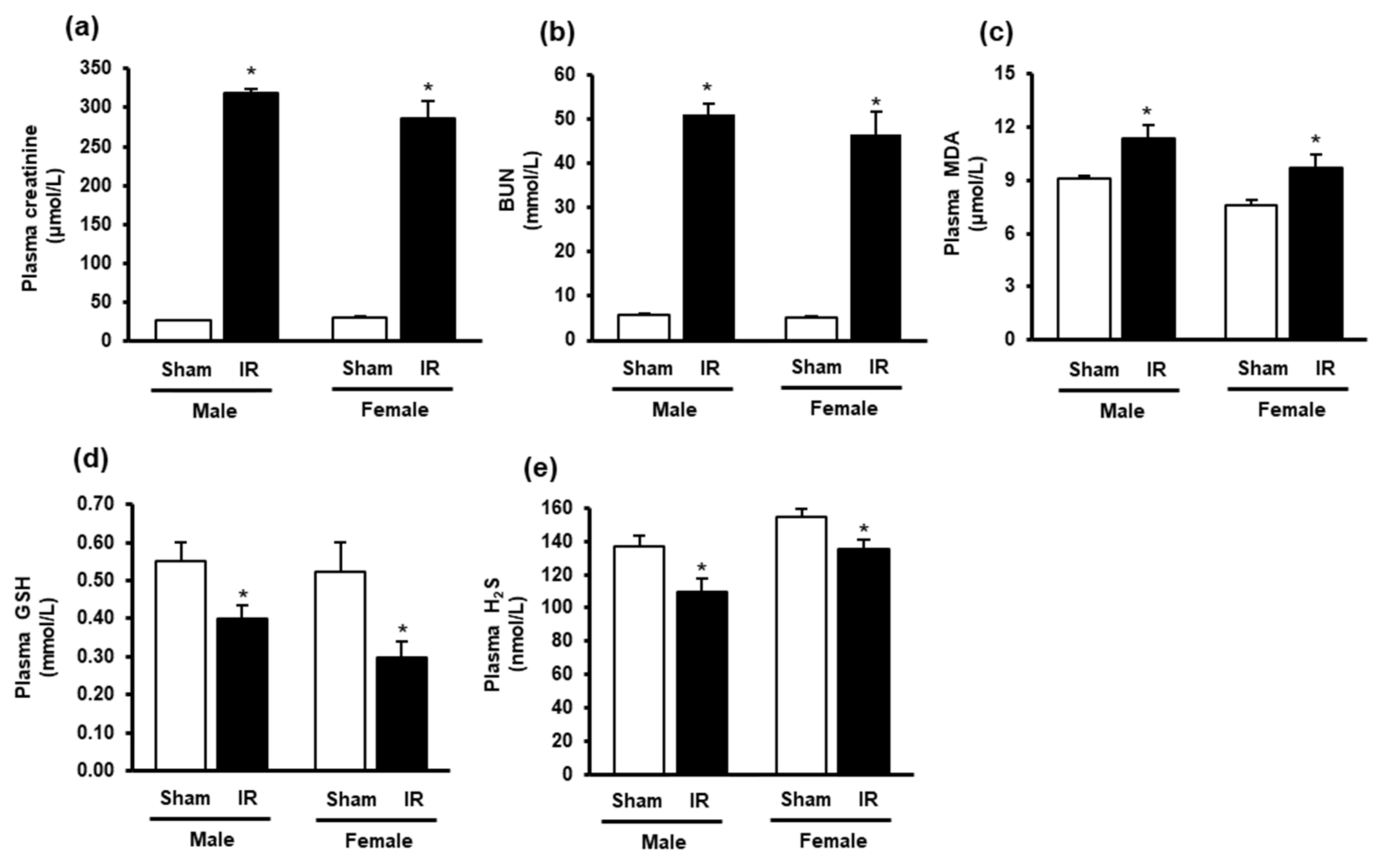
3.1. Ischemia-Reperfusion Impaired Kidney Function and Altered Oxidative Stress Biomarkers in the Plasma
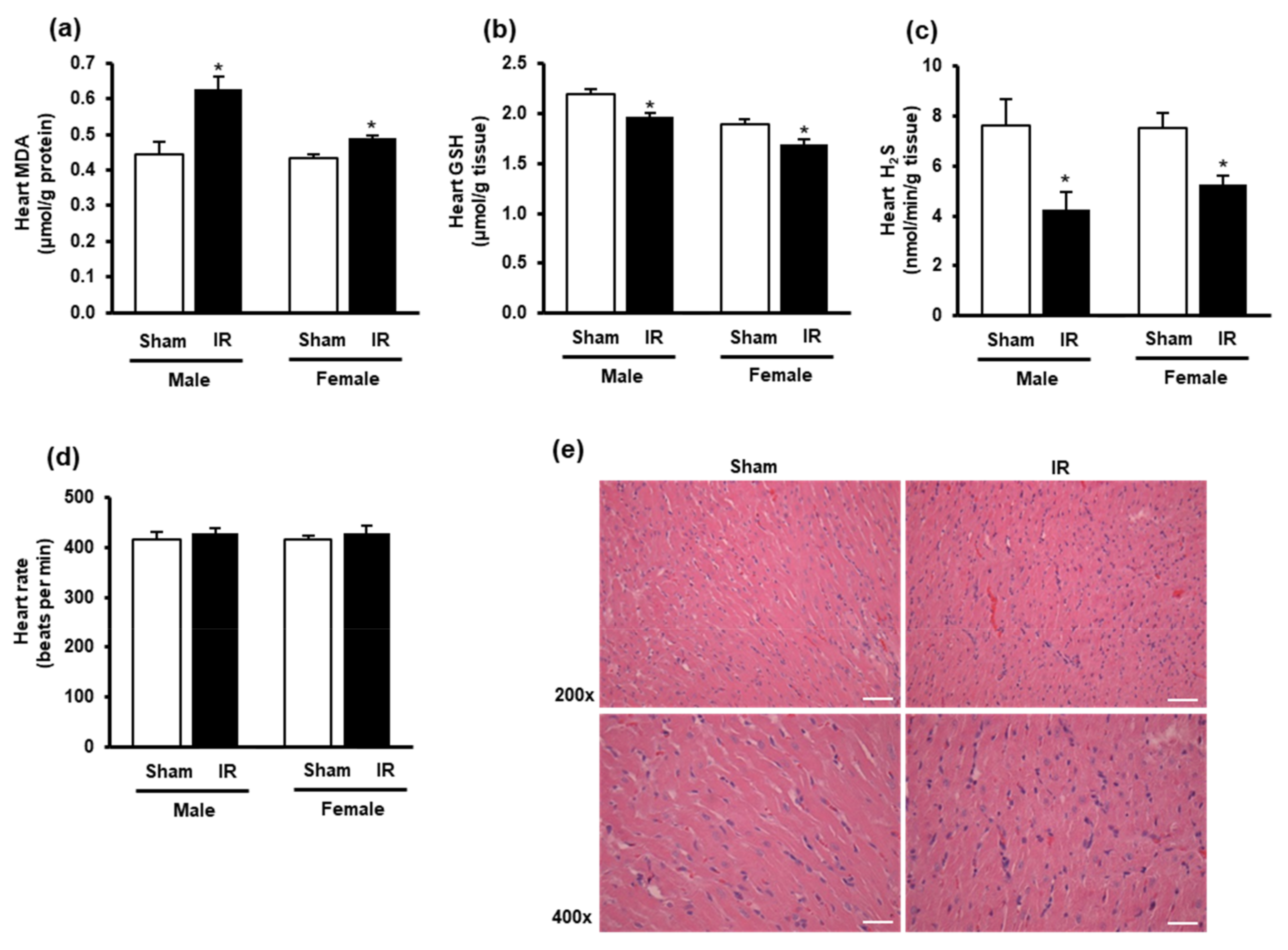
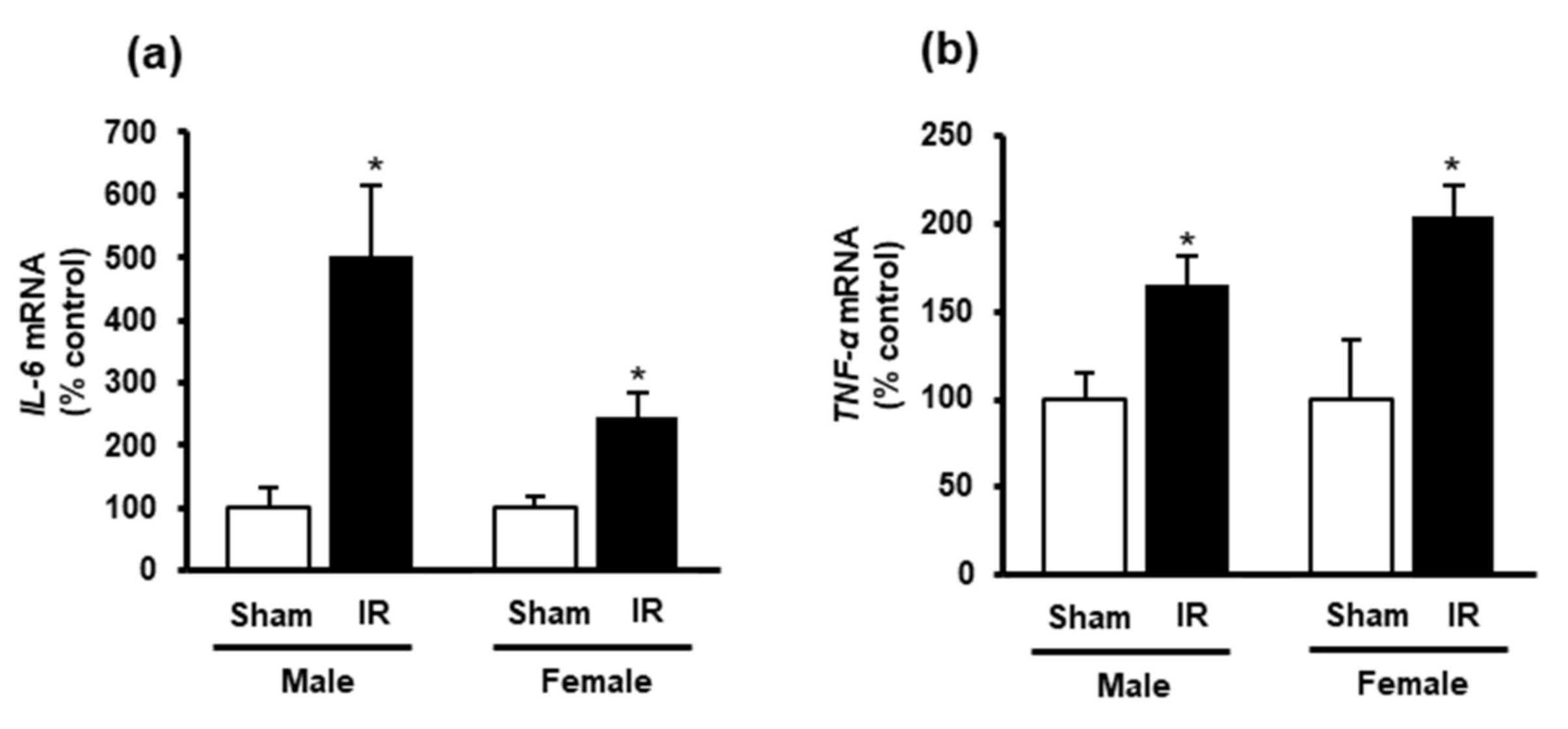
3.2. Kidney Ischemia-Reperfusion Increased Lipid Peroxidation, Decreased Glutathione and Hydrogen Sulfide Levels in the Heart
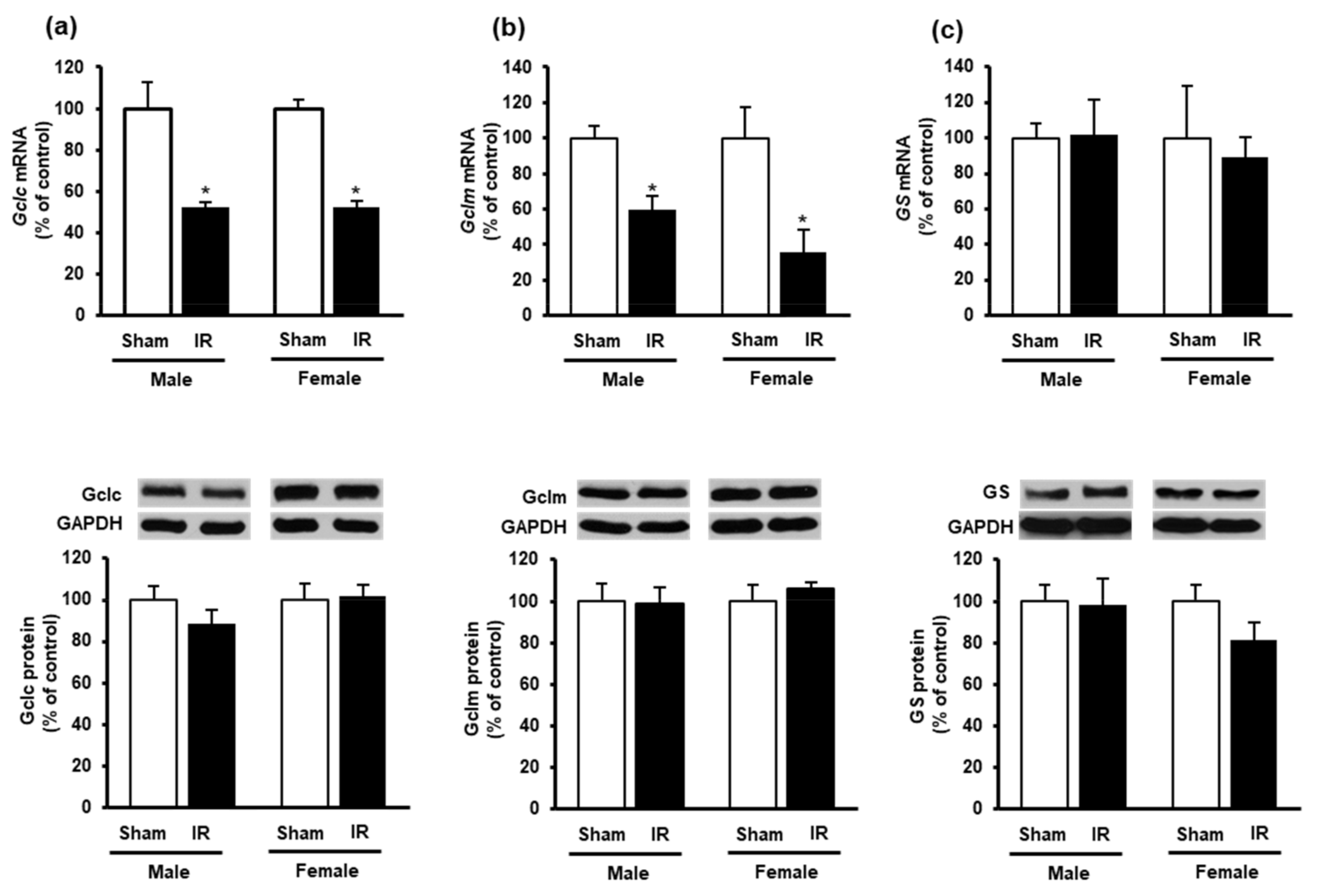
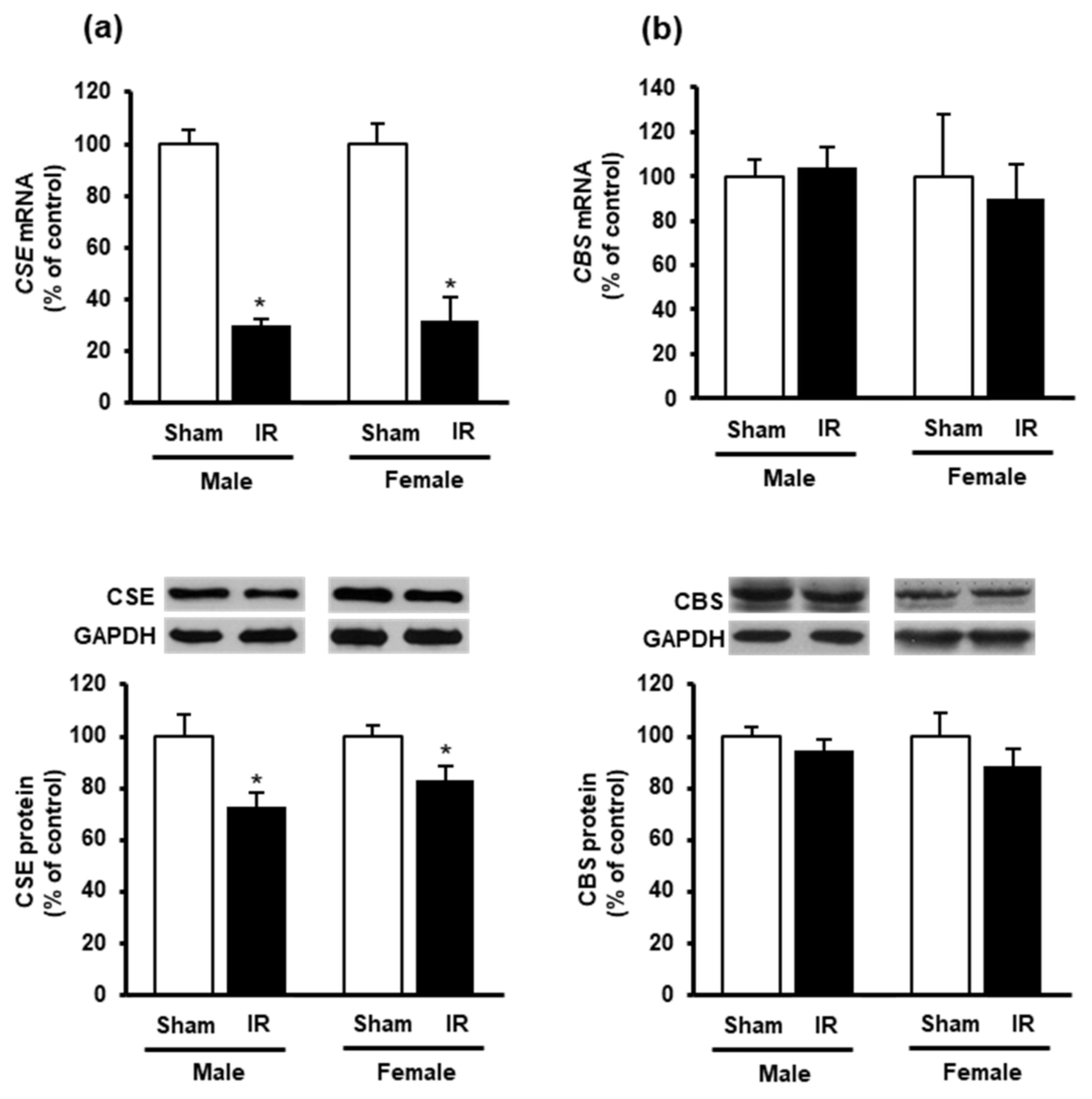
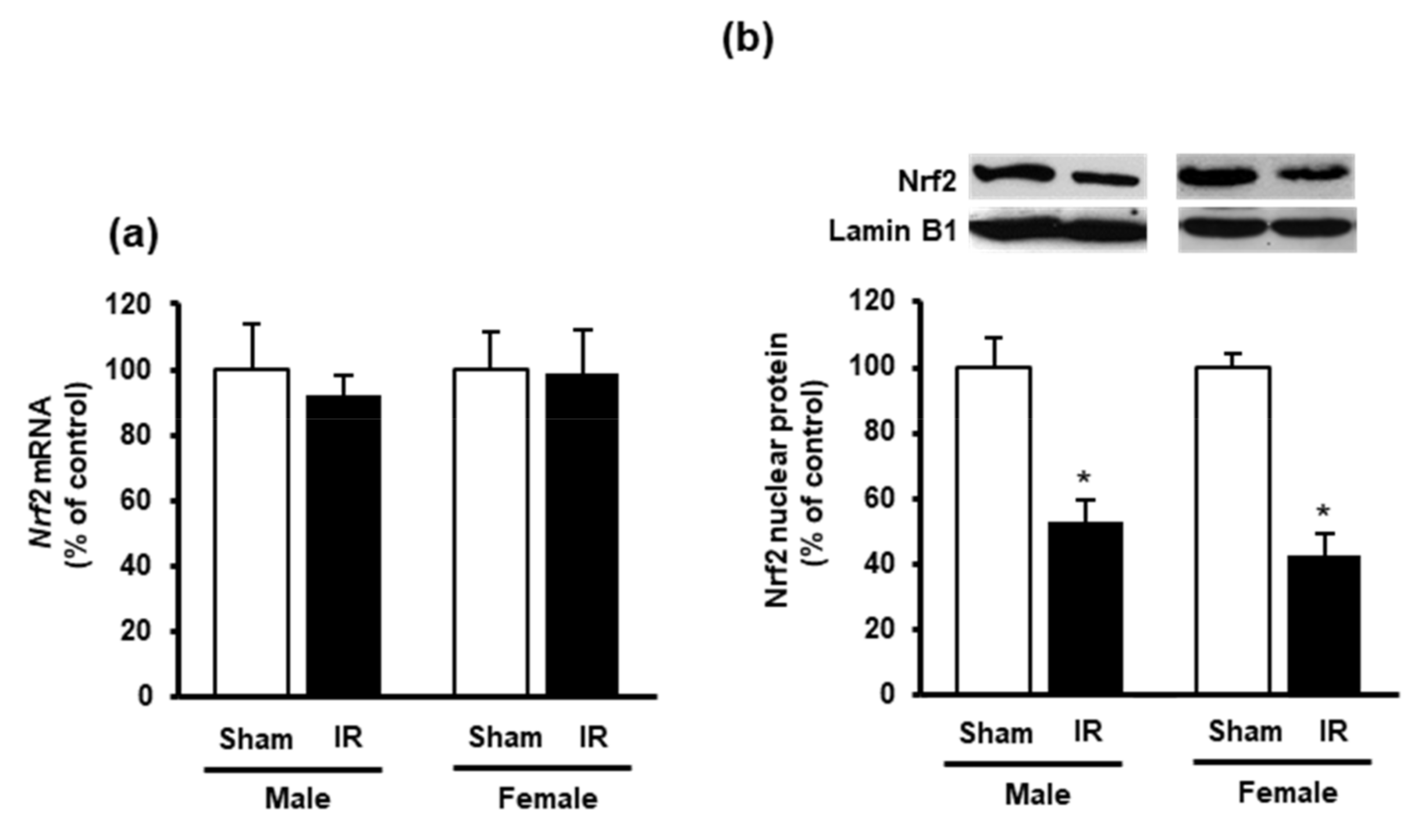
3.3. Effect of Kidney Ischemia-Reperfusion on Glutathione and Hydrogen Sulfide Synthesizing Enzyme Expression in the Heart
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shang, Y.; Hewage, S.M.; Wijerathne, C.U.; Siow, Y.L.; Isaak, C.K.; O, K. Kidney ischemia-reperfusion elicits acute liver injury and inflammatory response. Front. Med. 2020, 7, 201. [Google Scholar] [CrossRef]
- Lee, S.A.; Cozzi, M.; Bush, E.L.; Rabb, H. Distant organ dysfunction in acute kidney injury: A review. Am. J. Kidney. Dis. 2018, 72, 846–856. [Google Scholar] [CrossRef]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Hypertension 2003, 42, 1050–1065. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Hsu, C.-Y.; Yang, J.; Tan, T.C.; Zheng, S.; Ordonez, J.D.; Liu, K.D. Acute kidney injury and risk of heart failure and atherosclerotic events. Clin. J. Am. Soc. Nephrol. 2018, 13, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Shirali, A.C.; Bia, M.J. Management of cardiovascular disease in renal transplant recipients. Clin. J. Am. Soc. Nephrol. 2008, 3, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-J.; Zhou, D.; Barbosa, A.C.S.; Niu, Y.; Guan, X.; Xu, M.; Ren, S.; Nolin, T.D.; Liu, Y.; Xie, W. Activation of Constitutive Androstane Receptor Ameliorates Renal Ischemia-Reperfusion-Induced Kidney and Liver Injury. Mol. Pharmacol. 2018, 93, 239–250. [Google Scholar] [CrossRef]
- Ruparelia, N.; Chai, J.T.; Fisher, E.A.; Choudhury, R.P. Inflammatory processes in cardiovascular disease: A route to targeted therapies. Nat. Rev. Cardiol. 2017, 14, 133–144. [Google Scholar] [CrossRef]
- Förstermann, U. Oxidative stress in vascular disease: Causes, defense mechanisms and potential therapies. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 338–349. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Bajic, V.P.; Van Neste, C.; Obradovic, M.; Zafirovic, S.; Radak, D.; Bajic, V.B.; Essack, M.; Isenovic, E.R. Glutathione “redox homeostasis” and its relation to cardiovascular disease. Oxid. Med. Cell. Longev. 2019, 2019, 5028181. [Google Scholar] [CrossRef]
- Lefer, D.J. A new gaseous signaling molecule emerges: Cardioprotective role of hydrogen sulfide. Proc. Natl. Acad. Sci. USA 2007, 104, 17907–17908. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Shen, Z.; Luo, S.; Guo, W.; Zhu, Y.Z. The cardioprotective effects of hydrogen sulfide in heart diseases: From molecular mechanisms to therapeutic potential. Oxid. Med. Cell. Longev. 2015, 2015, 925167. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Shen, X.; Kevil, C.G. Reactive Sulfur Species: A New Redox Player in Cardiovascular Pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 874–884. [Google Scholar] [CrossRef]
- Yan, X.; Wu, H.; Wu, Z.; Hua, F.; Liang, D.; Sun, H.; Yang, Y.; Huang, D.; Bian, J.-S. The new synthetic H2S-releasing SDSS protects MC3T3-E1 osteoblasts against H2O2-induced apoptosis by suppressing oxidative stress, inhibiting MAPKs, and activating the PI3K/Akt pathway. Front. Pharmacol. 2017, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef]
- Aminzadeh, M.A.; Vaziri, N.D. Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrol. Dial. Transplant. 2012, 27, 498–504. [Google Scholar] [CrossRef]
- Kondo, K.; Bhushan, S.; King, A.L.; Prabhu, S.D.; Hamid, T.; Koenig, S.; Murohara, T.; Predmore, B.L.; Gojon Sr, G.; Gojon, G., Jr.; et al. H2S protects against pressure overload–induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 2013, 127, 1116–1127. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine γ-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef]
- Corsello, T.; Komaravelli, N.; Casola, A. Role of hydrogen sulfide in NRF2-and sirtuin-dependent maintenance of cellular redox balance. Antioxidants 2018, 7, 129. [Google Scholar] [CrossRef]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef]
- Zakkar, M.; Van der Heiden, K.; Luong, L.A.; Chaudhury, H.; Cuhlmann, S.; Hamdulay, S.S.; Krams, R.; Edirisinghe, I.; Rahman, I.; Carlsen, H.; et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Zhang, J.; Strom, J.; Lee, S.; Chen, Q.M. Myocardial ischemic reperfusion induces de novo Nrf2 protein translation. Biochim. Biophys. Acta 2014, 1842, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Donnarumma, E.; Bhushan, S.; Bradley, J.M.; Otsuka, H.; Donnelly, E.L.; Lefer, D.J.; Islam, K.N. Nitrite therapy ameliorates myocardial dysfunction via H2S and nuclear factor-erythroid 2-related factor 2 (Nrf2)-dependent signaling in chronic heart failure. J. Am. Heart Assoc. 2016, 5, e003551. [Google Scholar] [CrossRef] [PubMed]
- Grams, M.E.; Rabb, H. The distant organ effects of acute kidney injury. Kidney Int. 2012, 81, 942–948. [Google Scholar] [CrossRef]
- Prathapasinghe, G.A.; Siow, Y.L.; O, K. Detrimental role of homocysteine in renal ischemia-reperfusion injury. Am. J. Physiol. Renal. Physiol. 2007, 292, F1354–F1363. [Google Scholar] [CrossRef]
- Wu, N.; Siow, Y.L.; O, K. Ischemia/reperfusion reduces transcription factor Sp1-mediated cystathionine β-synthase expression in the kidney. J. Biol. Chem. 2010, 285, 18225–18233. [Google Scholar] [CrossRef]
- Xu, Z.; Prathapasinghe, G.; Wu, N.; Hwang, S.-Y.; Siow, Y.L.; O, K. Ischemia-reperfusion reduces cystathionine-β-synthase-mediated hydrogen sulfide generation in the kidney. Am. J. Physiol. Renal. Physiol. 2009, 297, F27–F35. [Google Scholar] [CrossRef]
- Wang, P.; Isaak, C.; Siow, Y.; O, K. Downregulation of cystathionine b-synthase and cystathionine c-lyase expression stimulates inflammation in kidney ischemia-reperfusion injury. Physiol. Rep. 2014, 2, e12251. [Google Scholar] [CrossRef]
- Shang, Y.; Siow, Y.L.; Isaak, C.K.; O, K. Downregulation of glutathione biosynthesis contributes to oxidative stress and liver dysfunction in acute kidney injury. Oxid. Med. Cell. Longev. 2016, 2016, 9707292. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef]
- Prathapasinghe, G.A.; Siow, Y.L.; Xu, Z.; O, K. Inhibition of cystathionine-β-synthase activity during renal ischemia-reperfusion: Role of pH and nitric oxide. Am. J. Physiol. Renal. Physiol. 2008, 295, F912–F922. [Google Scholar] [CrossRef] [PubMed]
- Okonko, D.O.; Shah, A.M. Mitochondrial dysfunction and oxidative stress in CHF. Nat. Rev. Cardiol. 2015, 12, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Oxid. Med. Cell. Longev. 2012, 111, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Voigt, A.; Rahnefeld, A.; Kloetzel, P.M.P.; Krüger, E. Cytokine-induced oxidative stress in cardiac inflammation and heart failure—How the ubiquitin proteasome system targets this vicious cycle. Front. Physiol. 2013, 4, 42. [Google Scholar] [CrossRef]
- Chen, T.-H.; Yang, Y.-C.; Wang, J.-C.; Wang, J.-J. Curcumin Treatment Protects Against Renal Ischemia and Reperfusion Injury–Induced Cardiac Dysfunction and Myocardial Injury. Transplant. Proc. 2013, 45, 3546–3549. [Google Scholar] [CrossRef]
- Amini, N.; Sarkaki, A.; Dianat, M.; Mard, S.A.; Ahangarpour, A.; Badavi, M. Protective effects of naringin and trimetazidine on remote effect of acute renal injury on oxidative stress and myocardial injury through Nrf-2 regulation. Pharmacol. Rep. 2019, 71, 1059–1066. [Google Scholar] [CrossRef]
- Mitaka, C.; Si, M.K.H.; Tulafu, M.; Yu, Q.; Uchida, T.; Abe, S.; Kitagawa, M.; Ikeda, S.; Eishi, Y.; Tomita, M. Effects of atrial natriuretic peptide on inter-organ crosstalk among the kidney, lung, and heart in a rat model of renal ischemia-reperfusion injury. Intensive Care Med. Exp. 2014, 2, 1–17. [Google Scholar] [CrossRef]
- Singh, A.; Lee, K.J.; Lee, C.; Goldfarb, R.D.; Tsan, M.-F. Relation between myocardial glutathione content and extent of ischemia-reperfusion injury. Circulation 1989, 80, 1795–1804. [Google Scholar] [CrossRef]
- Damy, T.; Kirsch, M.; Khouzami, L.; Caramelle, P.; Le Corvoisier, P.; Roudot-Thoraval, F.; Dubois-Randé, J.-L.; Hittinger, L.; Pavoine, C.; Pecker, F. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PLoS ONE 2009, 4, e4871. [Google Scholar] [CrossRef]
- Cheung, P.-Y.; Wang, W.; Schulz, R. Glutathione protects against myocardial ischemia–reperfusion injury by detoxifying peroxynitrite. J. Mol. Cell. Cardiol. 2000, 32, 1669–1678. [Google Scholar] [CrossRef]
- Fox, B.M.; Gil, H.-W.; Kirkbride-Romeo, L.; Bagchi, R.A.; Wennersten, S.A.; Haefner, K.R.; Skrypnyk, N.I.; Brown, C.N.; Soranno, D.E.; Gist, K.M.; et al. Metabolomics assessment reveals oxidative stress and altered energy production in the heart after ischemic acute kidney injury in mice. Kidney Int. 2019, 95, 590–610. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Sulistyoningrum, D.C.; Glier, M.B.; Verchere, C.B.; Devlin, A.M. Altered glutathione homeostasis in heart augments cardiac lipotoxicity associated with diet-induced obesity in mice. J. Biol. Chem. 2011, 286, 42483–42493. [Google Scholar] [CrossRef] [PubMed]
- Bos, E.M.; Wang, R.; Snijder, P.M.; Boersema, M.; Damman, J.; Fu, M.; Moser, J.; Hillebrands, J.L.; Ploeg, R.J.; Yang, G.; et al. Cystathionine gamma-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J. Am. Soc. Nephrol. 2013, 24, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Sojitra, B.; Bulani, Y.; Putcha, U.K.; Kanwal, A.; Gupta, P.; Kuncha, M.; Banerjee, S.K. Nitric oxide synthase inhibition abrogates hydrogen sulfide-induced cardioprotection in mice. Mol. Cell. Biochem. 2012, 360, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef]
- Zhu, H.; Itoh, K.; Yamamoto, M.; Zweier, J.L.; Li, Y. Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: Protection against reactive oxygen and nitrogen species-induced cell injury. FEBS Lett. 2005, 579, 3029–3036. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef]
- Barrett-Connor, E. Sex differences in coronary heart disease. Why are women so superior? The 1995 Ancel Keys Lecture. Circulation 1997, 95, 252–264. [Google Scholar] [CrossRef]
- Luczak, E.D.; Leinwand, L.A. Sex-based cardiac physiology. Annu. Rev. Physiol. 2009, 71, 1–18. [Google Scholar] [CrossRef]
- Walli-Attaei, M.; Joseph, P.; Rosengren, A.; Chow, C.K.; Rangarajan, S.; Lear, S.A.; AlHabib, K.F.; Davletov, K.; Dans, A.; Lanas, F.; et al. Variations between women and men in risk factors, treatments, cardiovascular disease incidence, and death in 27 high-income, middle-income, and low-income countries (PURE): A prospective cohort study. Lancet 2020, 396, 97–109. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Primer (5′–3′) | Reverse Primer (5′–3′) | Accession Number | Size (bp) |
|---|---|---|---|---|
| CSE | GTTGGGTTTGTGGGTGTTTC | GTATGGAGGCACCAACAGGT | XM_008761574.2 | 150 |
| CBS | TCGTGATGCCAGAGAAGATG | TTGGGGATTTCGTTCTTCAG | NM_012522.2 | 148 |
| Gclc | GCCCAATTGTTATGGCTTTG | AGTCCTCTCTCCTCCCGTGT | NM_012815.2 | 124 |
| Gclm | CGAGGAGCTTCGAGACTGTAT | ACTGCATGGGACATGGTACA | NM_017305.2 | 114 |
| GS | ACAACGAGCGAGTTGGGAT | TGAGGGGAAGAGCGTGAATG | NM_012962.1 | 182 |
| IL-6 | CCGGAGAGGAGACTTCACAG | ACAGTGCATCATCGCTGTTC | NM_012589.2 | 161 |
| TNF-α Nrf2 | CCCAGACCCTCACACTCAGAT CTGTCAGCTACTCCCAGGTTG | TTGTCCCTTGAAGAGAACCTG GCGACTCATGGTCATCTACAA | XM_008772775.2 NM_031789.2 | 215 111 |
| GAPDH | AGAGAAGGCAGCCCTGGT | GCTCTCTGCTCCTCCCTGT | NM_017008.4 | 138 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wijerathne, C.U.B.; Madduma Hewage, S.; Siow, Y.L.; O, K. Kidney Ischemia-Reperfusion Decreases Hydrogen Sulfide and Increases Oxidative Stress in the Heart. Biomolecules 2020, 10, 1565. https://doi.org/10.3390/biom10111565
Wijerathne CUB, Madduma Hewage S, Siow YL, O K. Kidney Ischemia-Reperfusion Decreases Hydrogen Sulfide and Increases Oxidative Stress in the Heart. Biomolecules. 2020; 10(11):1565. https://doi.org/10.3390/biom10111565
Chicago/Turabian StyleWijerathne, Charith U. B., Susara Madduma Hewage, Yaw L. Siow, and Karmin O. 2020. "Kidney Ischemia-Reperfusion Decreases Hydrogen Sulfide and Increases Oxidative Stress in the Heart" Biomolecules 10, no. 11: 1565. https://doi.org/10.3390/biom10111565
APA StyleWijerathne, C. U. B., Madduma Hewage, S., Siow, Y. L., & O, K. (2020). Kidney Ischemia-Reperfusion Decreases Hydrogen Sulfide and Increases Oxidative Stress in the Heart. Biomolecules, 10(11), 1565. https://doi.org/10.3390/biom10111565