1. Introduction
Methyl-CpG binding protein 2 (MeCP2) is an intrinsically disordered protein (IDP) involved in early stages of neuronal development, differentiation, maturation, and synaptic plasticity control [
1]. Although it was identified as a methyl-dependent chromatin binding protein and an epigenetic methylation reader, and, therefore, associated to gene silencing, recent evidences suggest it could be considered as a transcriptional regulator whose primary role is recruiting co-repressor complexes to methylated sites and contributing to decreasing transcriptional noise [
2].
MeCP2 exhibits a promoter-specific dsDNA interaction required for finely tuning gene transcription, but it also binds massively to heterochromatin when acting as a chromatin architecture remodeling factor. From the initial embryonic development stages, MeCP2 gradually replaces histone 1 as a sort of nucleosomal linker [
3,
4,
5]. The possibility to establish different types of interaction with DNA together with its ability to interact with other many biological partners (RNA, structural and transcriptional proteins, nucleosomal elements) and its central role as an important network interaction hub within gene transcription regulation networks, as well as the additional regulatory level of MeCP2 activity through post-translational modifications are made possible thanks to its modular, dynamic and adaptive structure [
6,
7].
Abnormal MeCP2 activity leads to disease [
2,
8,
9]. MeCP2 point mutations or deletions causing activity loss are associated with Rett syndrome (RTT). RTT is the main cause of mental retardation in females (1:10,000 births), exhibiting a clinically broad expression phenotype gradation. RTT shares features with other neurological diseases from the autistic spectrum. Importantly, duplication of mecp2 gene results in overexpression of MeCP2 and leads to MeCP2 duplication syndrome (MDS), another much rarer disorder affecting males and, strikingly, sharing phenotypic features with RTT, such as severe intellectual disability and impaired motor function.
Each of the six domains MeCP2 is either completely or partially disordered: N-terminal domain (NTD), methyl binding domain (MBD), intervening domain (ID), transcriptional repression domain (TRD), C-terminal domain α (CTDα), and C-terminal domain β (CTDβ) (
Supplemental Figure S1) [
3,
10]. Because of the importance of the interaction of MeCP2 with the nuclear co-receptor co-repressor (NCoR), an additional NCoR/SMRT interaction domain (NID) is often considered between TRD and CTDα [
11]. Most of MeCP2 polypeptide chain (≥60%) lacks well-defined secondary/tertiary structure. Flexible, disordered regions facilitate structural rearrangements necessary for exposing different interaction motifs and adapting to the many interacting partners, as well as the giving rise to the allosteric regulation through which the protein conformational landscape is modulated by ligand binding.
The most important domains are MBD, initially associated with methylated CpG (mCpG) DNA binding, and TRD, associated with transcription repression activities [
12,
13]. Most RTT-associated mutations are concentrated within these two domains, including missense and nonsense mutations, insertions, duplications, and deletions [
14]. Nevertheless, only eight missense and nonsense mutations (R106W, R133C, T158M, R168X, R255X, R270X, R294X and R306C) account for approximately 70% of all mutations in RTT [
15]. In particular, R133C, T158M, and R106W (in increasing order for phenotype severity and disease burden) represent 5%, 12%, and 3% of RTT cases [
16,
17].
MBD is the best characterized domain in MeCP2. MBD structure basically consists of a wedge-shaped structured core containing a 3-stranded anti-parallel β-sheet with an α-helix on the C-terminal side, with two unstructured regions flanking this core [
16,
17]. MBD is considered to be directly involved in maintaining the global organization of the protein through interactions with other domains through inter-domain coupling [
5,
18,
19]. Mutations in this domain would have an impact on the local and the global stability in MeCP2 [
3,
18].
In a previous biophysical study of three MeCP2 variants (MBD, and NTD-MBD, and NTD-MBD-ID), we established that the isolated MBD might not be the appropriate construct to study and assay its dsDNA binding features, because the presence of NTD and ID increased considerably the dsDNA binding affinity and the structural stability, besides adding a second, functionally independent dsDNA binding site [
20]. Here we report a biophysical study of the structural stability and the dsDNA interaction of mutant variants containing the substitutions R106W and R133C, two main RTT-mutations. These mutations were selected because they consist in an arginine substitution by a bulkier or a smaller residue, they are located in different positions regarding the dsDNA binding interface, and they correspond to different disease severity and burden levels. According to the results presented here, the inclusion of those substitutions into different protein constructions (MBD and NTD-MBD-ID) results in different structural and functional effects, highlighting the importance of selecting an appropriate molecular context (i.e., protein construction) when evaluation mutational effects, and emphasizing, in particular for MeCP2, the potential interdomain interaction in intrinsically disordered proteins [
18,
19].
2. Materials and Methods
2.1. Plasmid Construction
MeCP2 variants from isoform were expressed in
E. coli using a pET30b plasmid. The different protein variants were obtained by inserting appropriate substitutions: MBD, MBD R106W, MBD R133C, NTD-MBD-ID, NTD-MBD-ID R106W, and NTD-MBD-ID R133C (
Supplemental Figure S1). An N-terminal polyhistidine-tag was inserted for quick purification, and it was removed through an inserted PreScission Protease cleavage site. Appropriate expression was assessed by sequencing analysis: Sanger sequencing using a BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies, Carlsbad, CA, USA) in an Applied Biosystems 3730/DNA Analyzer (Thermo Fisher Scientific, Waltham, MA, USA).
2.2. Protein Expression and Purification
Protein variants (MBD, MBD R106W, MBD R133C, NTD-MBD-ID, NTD-MBD-ID R106W, NTD-MBD-ID R133C) were expressed and purified following identical procedures. Plasmids were transformed into BL21 (DE3) Star E. coli strain. Cultures were grown in 150 mL of LB/kanamycin (50 µg/mL) media at 37 °C overnight. Then, 4 L of LB/kanamycin (25 µg/mL) were inoculated (1:100 dilution) and incubated under the same conditions until reaching an OD (λ = 600 nm) of 0.6. Protein expression was induced with 1 mM isopropyl 1-thio-β-D-galactopyranoside (IPTG) at 18 °C overnight. Cells were sonicated in ice and benzonase (Merck-Millipore, Madrid, Spain) was added (20 U/mL) to remove nucleic acids. Proteins were purified using metal affinity chromatography employing a HiTrap TALON column (GE-Healthcare Life Sciences, Barcelona, Spain) with two washing steps: buffer sodium phosphate 50 mM, pH 7, NaCl 300 mM, and buffer sodium phosphate 50 mM, pH 7, NaCl 800 mM. Elution was performed applying an imidazole 10–150 mM elution gradient. Protein purity was evaluated by SDS-PAGE.
The polyhistidine-tag was removed by processing with GST-tagged PreScission Protease in protease buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.5) at 4 °C for 4 h. Progress of the proteolytic processing was monitored by SDS-PAGE. In the final step the protein was further purified with a combination of two affinity chromatographic steps to remove the polyhistidine-tag (HiTrap TALON column) and the GST-tagged PreScission Protease (GST TALON column, from GE-Healthcare Life Sciences, Barcelona, Spain). Purity and homogeneity were evaluated by SDS-PAGE and size-exclusion chromatography. Storage buffer consisted of Tris 50 mM pH 7.0 and pooled samples were kept at −80 °C. The identity of all proteins was checked by mass spectrometry (4800plus MALDI-TOF/MS, from Applied Biosystems-Thermo Fisher Scientific, Waltham, MA, USA). Potential DNA contamination was always estimated by UV absorption 260/280 ratio. Because a single tryptophan is located in MBD, an extinction coefficient of 11,460 M−1 cm−1 at 280 nm was employed for all variants, except for the R106W mutants for which a value of 16,960 M−1 cm−1 was applied.
Stability and binding assays were performed at different pH and buffer conditions (Tris 50 mM pH 7–9, NaCl 0–150 mM; Pipes 50 mM, pH 7; Phosphate 50 mM, pH 7). When needed, buffer exchange was done employing a 3 or 10 kDa-pore size ultrafiltration device (Amicon centrifugal filter, Merck-Millipore, Madrid, Spain) at 4000 rpm and 4 °C.
2.3. Double-Stranded DNA
HPLC-purified methylated and unmethylated 45-bp single-stranded DNA (ssDNA) oligomers corresponding to the promoter IV of the mouse brain-derived neurotrophic factor (BDNF) gene [
18,
19], were purchased from Integrated DNA Technologies. Two complementary pairs of DNA were used for DNA binding assays: forward unmethylated: 5’- GCCATGCCCTGGAACGGAACTCTCCTAATAAAAG-ATGTATCATTT-3’; reverse unmethylated: 5’- AAATGATACATCTTTTATTAGGAGAGTTCCGTTCC-AGGGCATGGC-3’; forward mCpG: 5’- GCCATGCCCTGGAA(5-Me)CGGAACTCTCCTAATAAA-AGATGTATCATTT-3’; reverse mCpG: 5’- AAATGATACATCTTTTATTAGGAGAGTTC(5-Me)CGTT-CCAGGGCATGGC-3’.
The ssDNA oligonucleotides were dissolved at a concentration of 0.5 mM, mixed at equimolar ratio, and annealed to obtain 45-bp double-stranded DNA (dsDNA) using a Stratagene Mx3005P qPCR real-time thermal cycler (Agilent Technologies, Santa Clara, CA, USA). The thermal annealing profile consisted of: (1) equilibration at 25 °C for 30 s; (2) heating ramp up to 99 °C; (3) equilibration at 99 °C for 1 min; and (4) 3-h cooling process down to 25 °C at a rate of 1 °C/3 min.
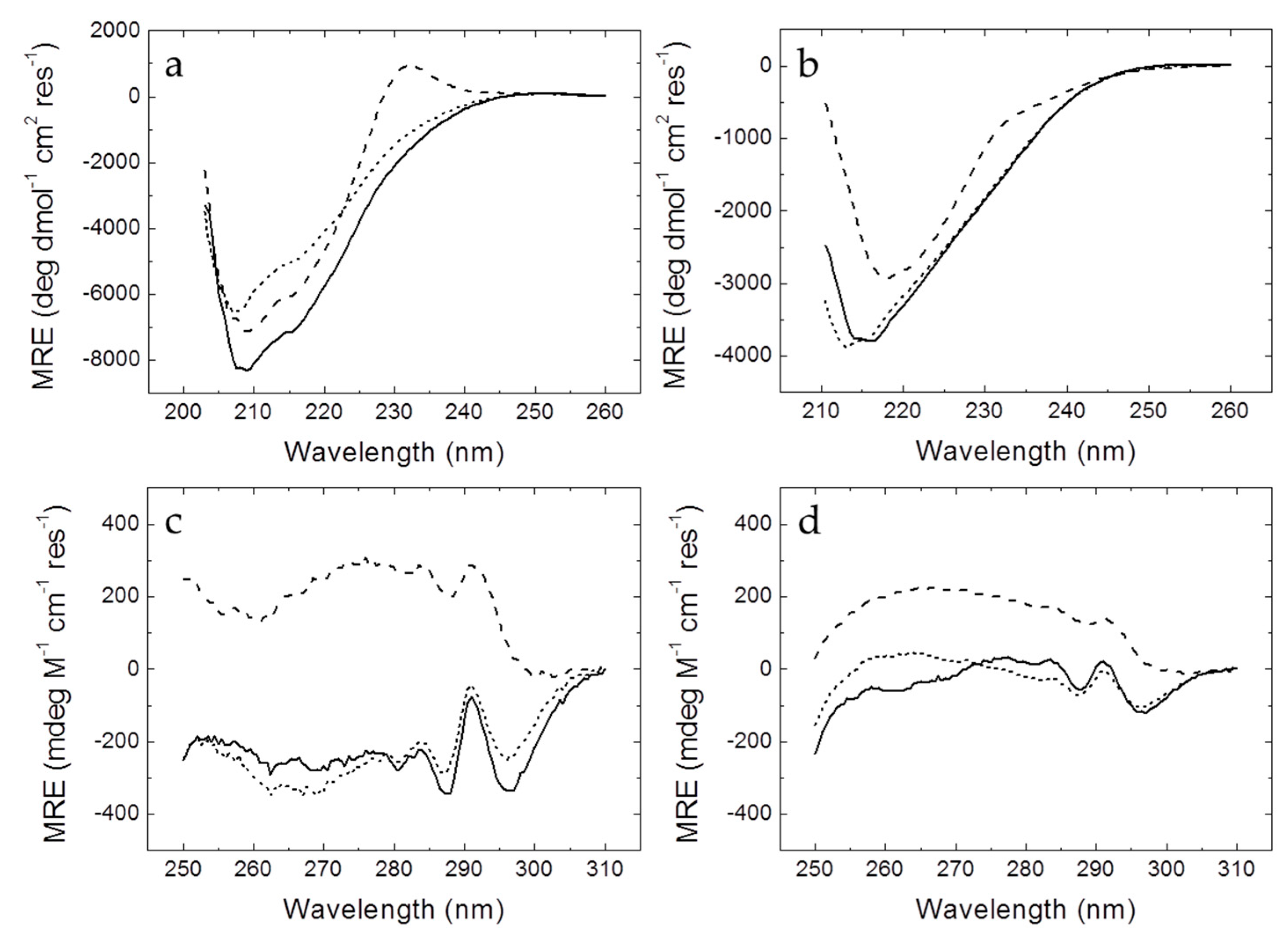
2.4. Circular Dichroism
Circular dichroism spectra were recorded in a thermostated Chirascan spectrometer (Applied Photophysics, Leatherhead, UK) using a 0.1 cm (far-UV) or 0.4 cm (near-UV) path-length quartz cuvette (Hellma Analytics, Müllheim, Germany) with a bandwidth of 1 nm, a spectral resolution of 0.5 nm, and a response time of 5 s. Temperature was controlled by a Peltier unit and monitored using a temperature probe. The assays were performed in the far-UV (200–260 nm) and the near-UV (250–310 nm) ranges. Protein concentration was set at 10–50 µM, depending on the signal-to-noise ratio.
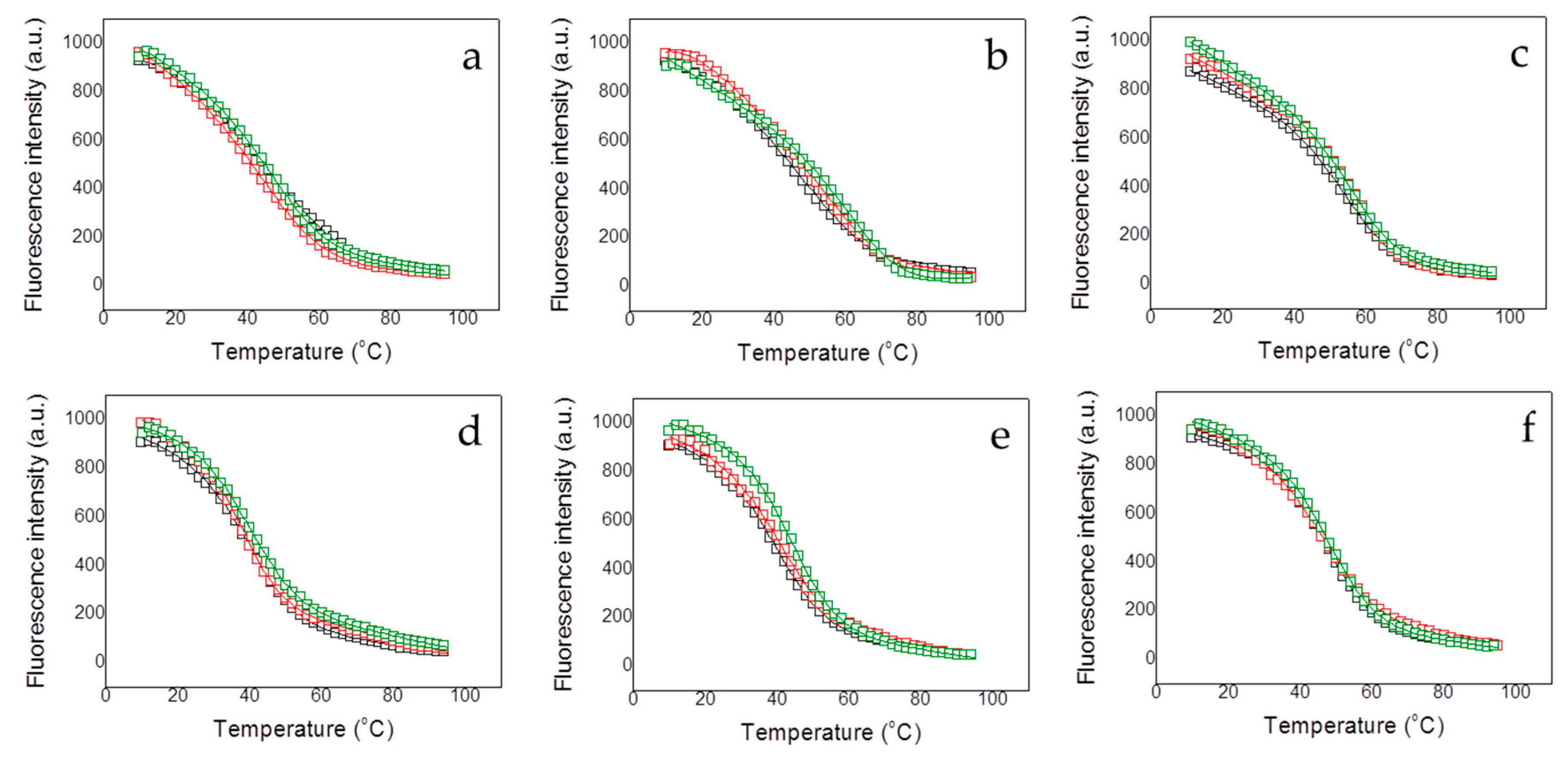
2.5. Fluorescence Spectroscopy
Protein thermal unfolding studies were performed in a Cary Eclipse fluorescence spectrophotometer (Varian—Agilent, Santa Clara, CA, USA) using a protein concentration of 5 µM and a 1 cm path-length quartz cuvette (Hellma Analytics, Müllheim, Germany). The temperature was controlled by a Peltier unit and monitored using a temperature probe, at a heating rate of 1 °C/min. Fluorescence emission spectra were recorded from 300 to 400 nm using an excitation wavelength of 290 nm and a bandwidth of 5 nm. Assays were performed and at the emission wavelength of 330 nm (maximal protein spectral change along the unfolding). A simple two-state unfolding model was considered for analyzing the assays:
where
F(
T) is the fluorescence signal at a given absolute temperature
T,
Tm is the unfolding temperature, Δ
H(
Tm) is the unfolding enthalpy (at the
Tm), Δ
CP is the unfolding heat capacity, and Δ
G(
T) is the stabilization Gibbs energy (which is a temperature function). The adjustable parameters
AN,
BN,
AU, and
BU are instrumental parameters defining the pre- (native) and post-transition (unfolded) regions in the unfolding trace. The stabilizing effect upon dsDNA interaction was assessed performing thermal denaturations of the different proteins (at 5 µM) in the presence of methylated and unmethylated DNA (at 10 µM) under the same conditions.
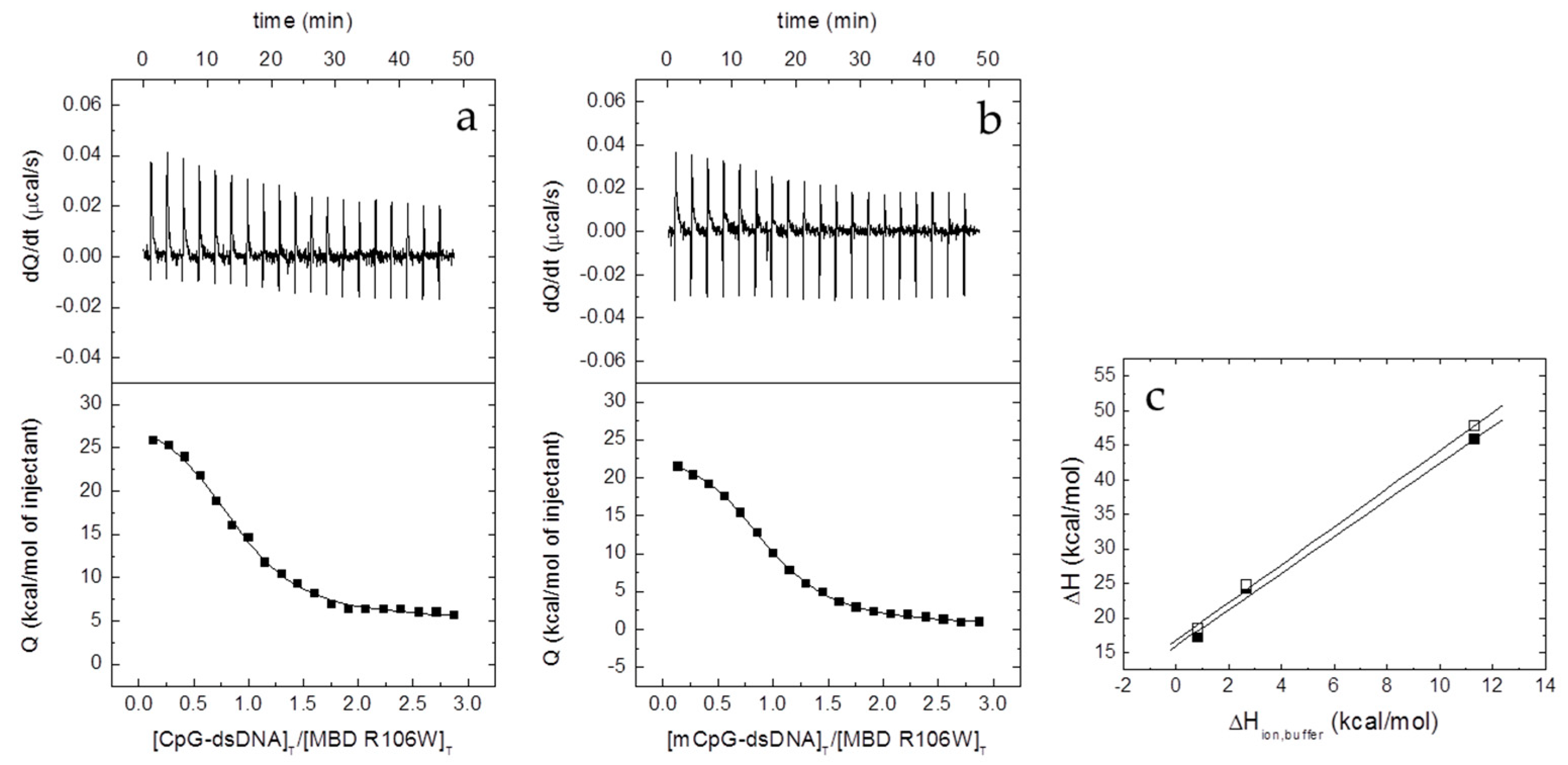
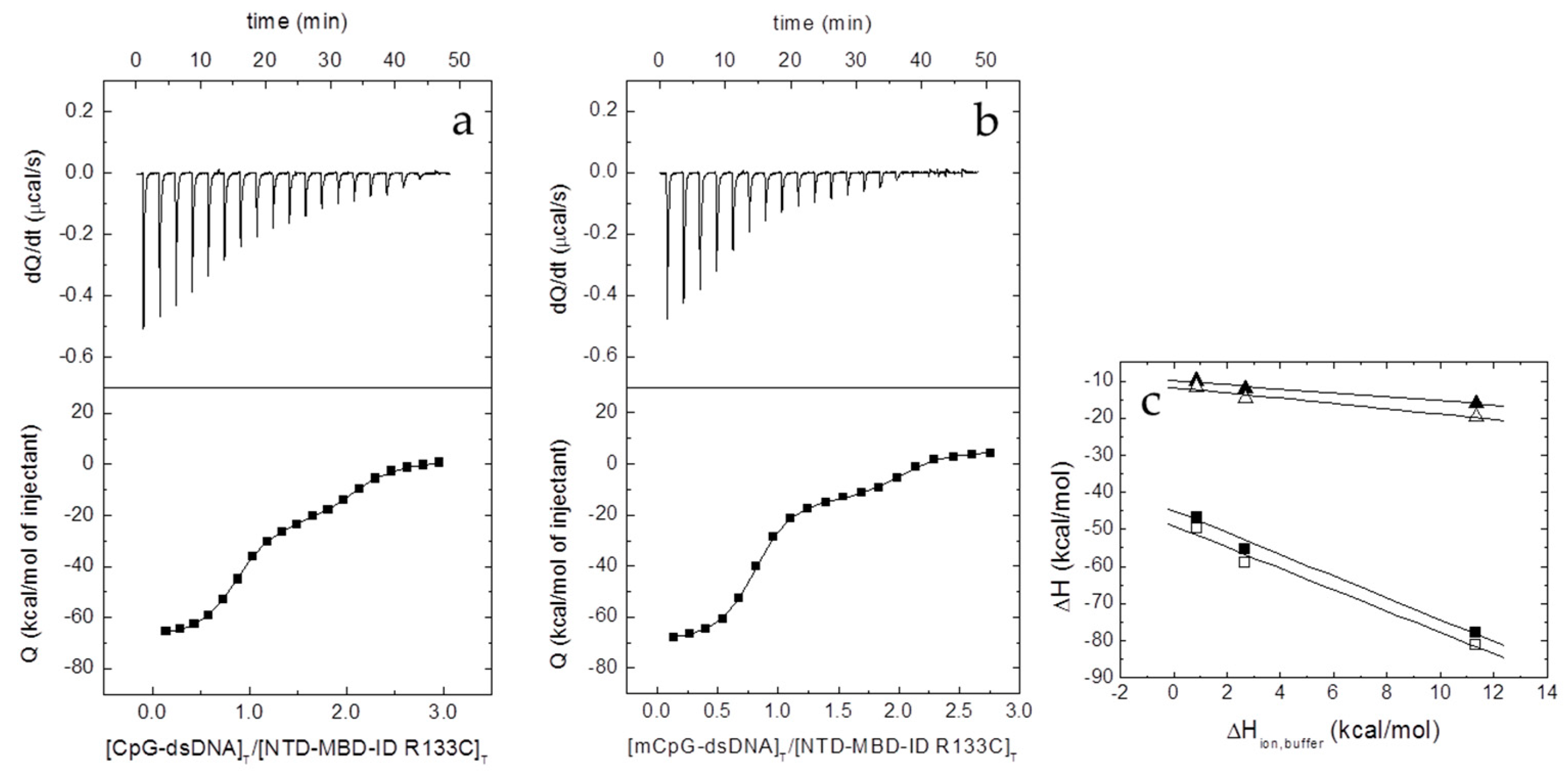
2.6. Isothermal Titration Calorimetry (ITC)
The interaction between the different proteins and dsDNA was studied in an Auto-iTC200 (MicroCal, Malvern-Panalytical, Malvern, UK). dsDNA (50 µM) in the injecting syringe was titrated into protein in the calorimetric cell (3–5 µM). Series of 2 µL-injections of titrant with a time-spacing of 150 s were programmed, maintaining a stirring speed of 750 rpm, and a reference power of 10 μcal/s. The association constant,
KB, and the observed enthalpy of binding, Δ
HB,obs, were estimated through non-linear regression of the experimental data employing a single ligand binding site model (1:1 protein:dsDNA stoichiometry) or a two ligand binding sites model (1:2 protein:dsDNA stoichiometry) implemented in Origin (OriginLab, Northampton, MA, USA) [
21,
22]. The dissociation constant
Kd was calculated as the inverse of
KB,obs, and the binding Gibbs energy and entropy were calculated applying standard well-known relationships: Δ
G = −
RT ln
KB, Δ
G = Δ
H −
TΔ
S.
The number of protons released from or uptaken by the protein-dsDNA complex upon dsDNA binding, Δ
nH, was determined, according to [
23,
24,
25]:
where Δ
H is the buffer-independent binding enthalpy, and Δ
Hbuffer is the ionization enthalpy of the buffer. Titrations were performed in buffers with different ionization enthalpies (Tris, 11.35 kcal/mol; Pipes, 2.67 kcal/mol; and phosphate, 0.86 kcal/mol) [
26] in order to estimate the buffer-independent thermodynamic parameters (Δ
H and Δ
nH) from linear regression using Equation (2). From Δ
G and Δ
H, the buffer-independent binding entropy can be readily calculated. The parameter Δ
nH may be non-zero if ligand binding results in changes in the proton dissociation constant of certain ionizable residues (either in the protein or the ligand) as a consequence of changes in their microenvironment upon complex formation. The association binding constant
KB will be not affected by the buffer ionization as long as the
pKa of the buffer is close to the experimental pH. However, the observed binding enthalpy (and, therefore, the observed entropic contribution) will contain an additional contribution from buffer ionization as indicated above. The experimental strategy allows removing the extrinsic contribution from buffer ionization. Noticeably, Δ
nH has practical utility since it reports the change in binding affinity as a result of a (moderate) change in pH, according to Wyman’s linkage relationships [
27]:
4. Discussion
Disordered regions in proteins are characterized by a biased amino acid composition, where residues exhibiting considerable propensity to be exposed to the solvent (polar and charged amino acids) predominate [
29]. They may influence protein conformation and function through steric effects, or exerting long-distance attractive or repulsive electrostatic interactions due to their highly polar/charged character, or making contacts with other structured regions affecting the global stability and the dynamics of the protein, as well as modulating the interaction with a binding partner. Thus, even lacking a well-defined structure, disordered regions may contribute to the overall stability of the protein, as it happens in MeCP2. Related to that, we have recently reported that: (1) NTD and ID, the two completely disordered MBD-flanking domains significantly increase the thermal stability of MBD [
20]; and (2) the two differentially expressed MeCP2 isoforms (E1 and E2) as a result of differential splicing and differing in just a few amino acids at the N-terminal part of the completely disordered NTD, differ in their thermal stability and functional capabilities [
30]. Thus, it may be possible that the conformation and/or the dynamics of MBD is altered by presence of the two disordered flanking domains, resulting in a different stability and different affinity toward binding partners.
While it is reasonable to expect that the structural and functional impact of point mutations located on structured regions may be predicted with certain reliability, the impact of those located on or close to disordered regions may be more difficult to assess. The two mutations studied in this work, R106W and R133C, are some of the most relevant clinically associated with RTT. They are not located in disordered regions, but in the structured region of the MeCP2 MBD. However, the MBD is very dynamic and susceptible to many environmental factors (pH, temperature, solutes, ligands …), being considered as a key element able to interact with or allosterically regulate the other functional domains [
5,
18,
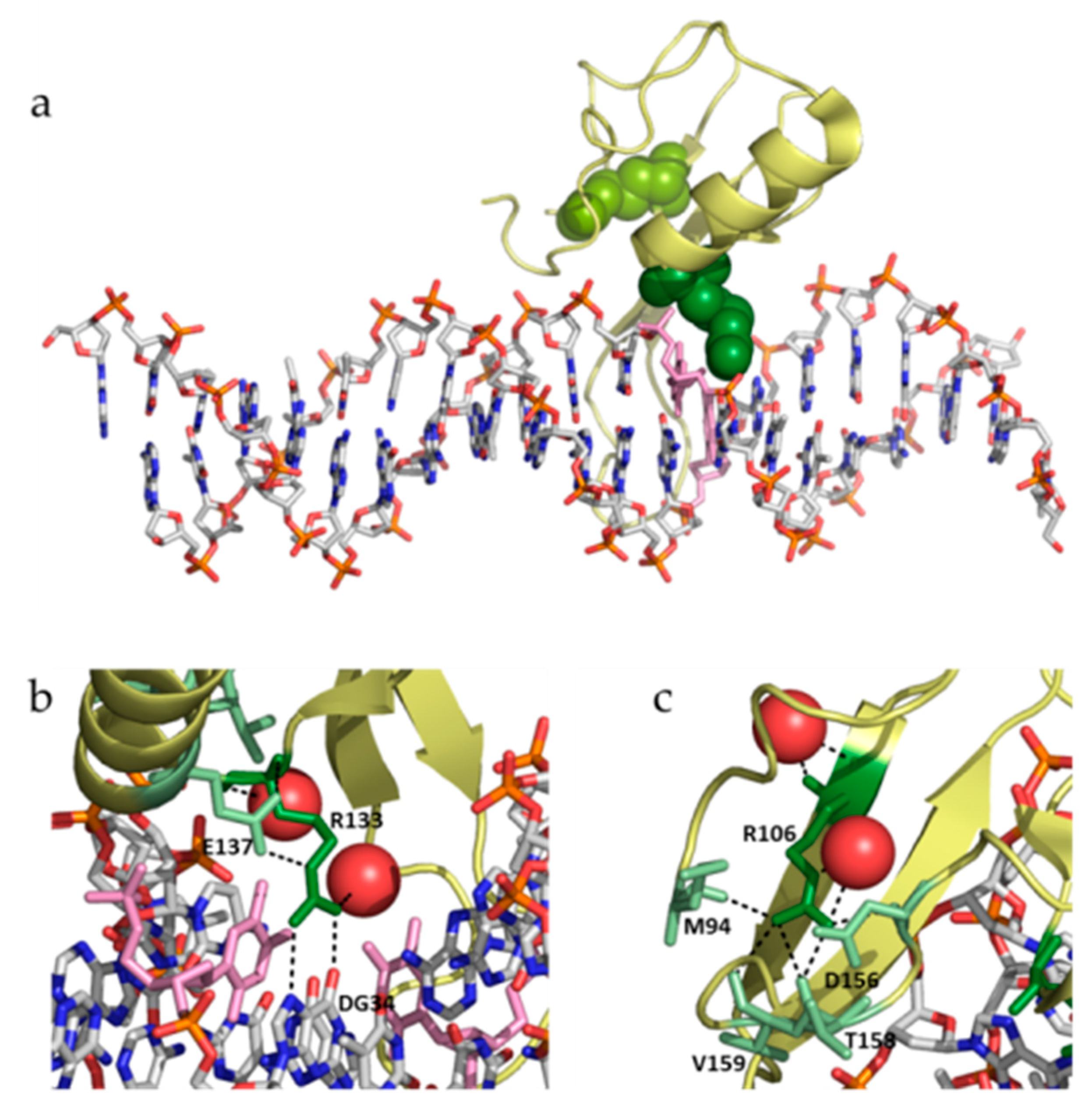
19]. Both mutations show some similarities and many dissimilarities: (1) both involve the substitution of an arginine residue, but R106W involves the substitution by a bulkier aromatic hydrophobic residue, while R133C involves the substitution by a smaller polar aliphatic residue; (2) R106 is located far from the DNA binding interface, while R133 is located in the DNA binding interface (
Figure 7); (3) R106 establishes many interactions with many surrounding residues (in particular, four hydrogen bonding residues: M94, D156, T158, and V159), while R133 interacts with fewer residues (only one hydrogen bonding residue: E137) (
Figure 7); and (4) R106 does not interact with DNA, while R133 interacts with DNA through hydrogen bonds and van der Waals contacts (
Figure 7). Therefore, the impact of both substitutions is expected to be structurally and functionally different. In fact, if the main rotamers for tryptophan and cysteine are introduced in positions 106 and 133, respectively, all W106 rotamers clash with neighboring residues, while C133 shows no clashes at all, indicating that R106W substitution would result in considerable structural distortion in the vicinity of that position to accommodate such substitution.
As indicated above, the purpose of this work was to gain insight into the relationship between the phenotypic effect and the molecular effect of RTT-associated mutations by assessing the impact of two clinically relevant substitutions in MeCP2 MBD. MBD variants containing the two R106W and R133C mutations were studied regarding their structural stability and dsDNA interaction. In addition, because we have reported before an allosteric coupling between MBD and ID, in which the presence of ID dramatically increased the MBD dsDNA binding affinity and contributed an additional dsDNA binding site, we wanted to address whether the location of those mutations on different scaffolds (MBD or NTD-MBD-ID) would result in different structural and/or functional properties. The experimental strategy consisted of a combination of spectroscopic (CD and fluorescence) and calorimetric (ITC), taking advantage of their strengths and overcoming their limitations. Thus, CD and fluorescence are suitable for gathering coarse-grained structural information, and ITC is the gold-standard for determining binding affinity and providing a complete thermodynamic description of biomolecular interactions. In addition, contrary to other techniques, ITC is appropriate for studying biological interactions with more than one binding site, where the interplay between binding affinity and enthalpy makes easy to observe different binding processes occurring at different locations in a macromolecule.
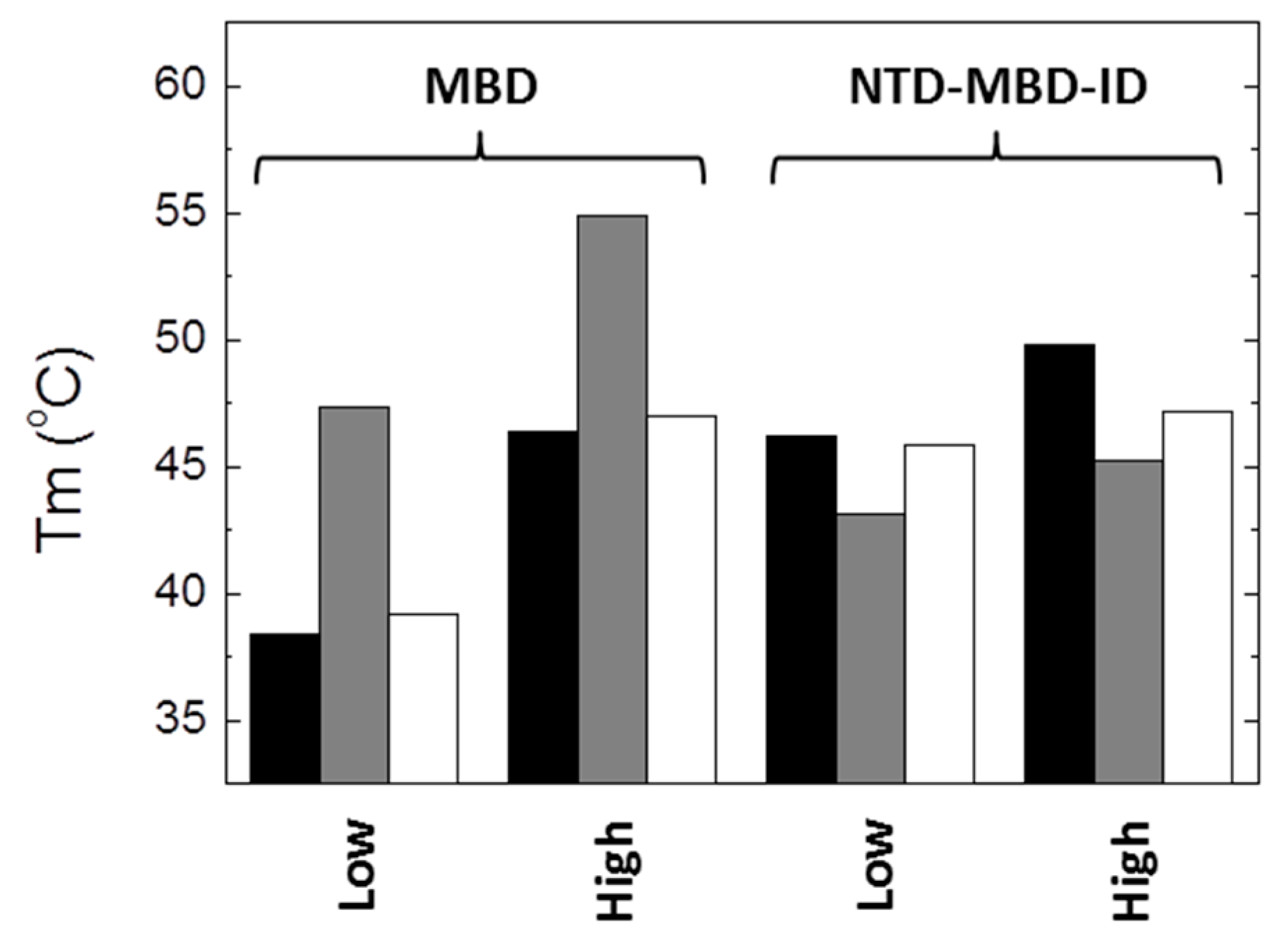
From the results presented here, it is apparent that the impact of R106W and R133C substitutions on the structural stability and the dsDNA binding capability depends on the molecular context, i.e., the scaffold (MBD or NTD-MBD-ID) in which the substitutions are introduced. Thus, to highlight some of the most important findings: (1) R106W and R133C substitutions increase the thermal stability of MBD, but decrease the thermal stability of NTD-MBD-ID (
Figure 8); (2) high ionic strength induces a large stabilization in MBD wild-type and mutants R106W and R133C, maintaining the same stability ranking, but a minor stabilization could be observed for NTD-MBD-ID variants (
Figure 8); (3) R106W induces an increase in dsDNA binding affinity in MBD, but a decrease in dsDNA binding affinity in NTD-MBD-ID, compared to their respective wild-type variants; and 4) R133C abolishes dsDNA binding in MBD, but behaves similar to the wild-type variant in NTD-MBD-ID in terms of dsDNA affinity and methyl-dependent discrimination.
The large stabilization observed in NTD-MBD-ID R106W when bound to dsDNA compared to the small stabilization observed for NTD-MBD-ID R133C, taking wild-type NTD-MBD-ID as a reference, may be considered a largely unexpected result (
Table 2 and
Table 3). The higher dsDNA affinity for the R133C mutant should have induced a larger stabilization extent for that mutant when bound to dsDNA, because the stabilization energy provided by dsDNA binding is equal to +
RTln(1+[dsDNA]/
Kd). Thus, the extent of the stabilization effect caused by the presence of dsDNA on protein conformation (quantified as increase in stability energy or increase in
Tm) depends on the dsDNA binding affinity, the binding stoichiometry, and the concentration of dsDNA. But the binding affinity is dependent on temperature, and, as a consequence, it will change along the thermal denaturation process. The temperature dependency of the binding affinity will be determined by the dsDNA binding enthalpy (and the binding heat capacity), which might not be the same for each interaction (as it occurs for R106W and R133C variants) and will further modulate the overall extent of the ligand-induced stabilization effect. The R106W variant exhibits a strongly endothermic dsDNA binding to the high-affinity site, indicating that, as the temperature starts increasing during the thermal denaturation process, initially the binding affinity and the strength of the complex would increase (according to the van’t Hoff equation) until a temperature in which the binding enthalpy becomes zero (the binding heat capacity is expected to be negative, as found for MBD and NTD-MBD-ID), and then the binding affinity decreases from that temperature. On the contrary, the R133C variant exhibits a strongly exothermic dsDNA binding, and the binding affinity and the strength of the complex would continuously decrease from the beginning of the thermal denaturation process. Therefore, the temperature evolution of
Kd would be different for the two mutants and the stabilization extent would also be different. This is a nice example of two molecules (R106W and R133C variants) binding to a common molecule with different binding affinities, but exerting different stabilization effects: the higher affinity interaction is associated with a smaller stabilization effect.
There are several intriguing facts derived from the experimental results previously shown here. First, how R106W substitution, which is far from the dsDNA binding interface, could affect dsDNA binding and increase its affinity? Of course, even located far from the dsDNA binding interface, the large structural rearrangements resulting from R106W substitution could very likely propagated to distal regions in MBD thanks to its intrinsic structural plasticity. Second, how R133C substitution of a main player in the dsDNA interaction would result in abolishment of interaction for isolated MBD, but almost no effect in NTD-MBD-ID. And third, how these molecular findings can be related to the phenotypic outcomes associated with those mutations: What is the final consequence of R106W substitution at molecular level? Is R106W substitution interfering with the interaction of MeCP2 with other biological partners through the surface residues around R106? What is the final consequence of R133C substitution at molecular level? Is R133C substitution causing an overlooked rearrangement that interferes with other interactions? According to the current classification of RTT mutations, R106W is associated to a severe phenotype, whereas R133C is associated to a mild phenotype [
31]. Interestingly, from the evidence gathered in this work, we expect larger functional alterations due to R106W substitution.



 and
and 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}