Gene Expression Profiling of Multiple Histone Deacetylases (HDAC) and Its Correlation with NRF2-Mediated Redox Regulation in the Pathogenesis of Diabetic Foot Ulcers

 , ,
, ,
Abstract
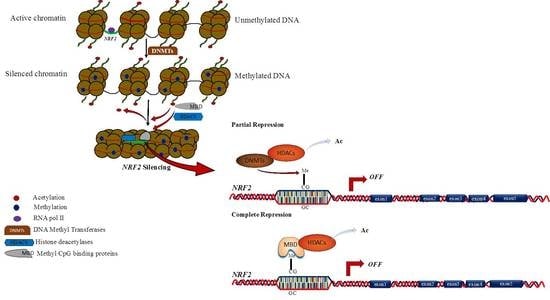
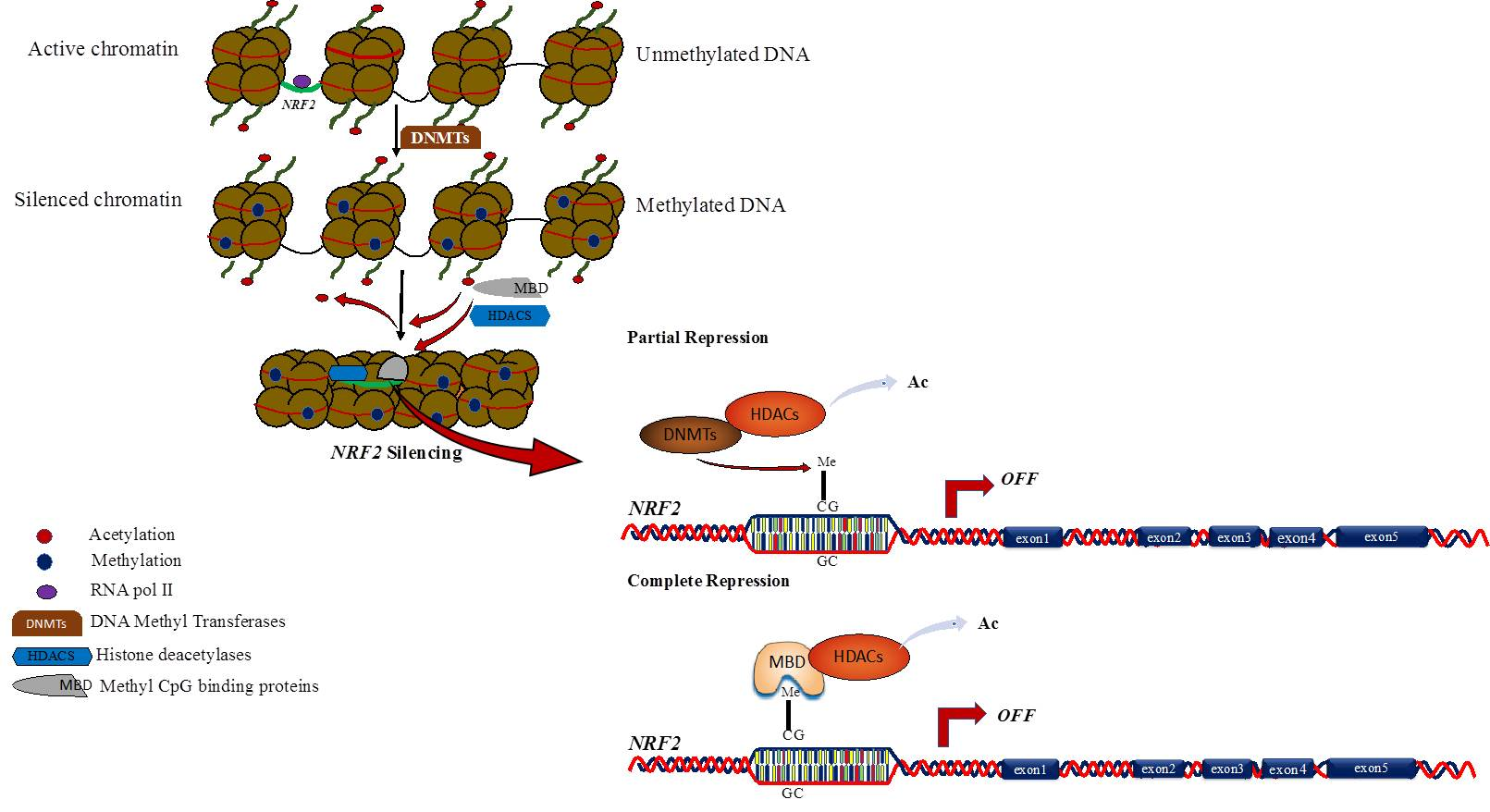
1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Basic Clinical and Biochemical Characteristics of the Study Participants
2.3. Sample Size Calculation and Power of the Study
2.4. Isolation of Peripheral Blood Mononuclear Cells (PBMCs) from Blood
2.5. Quantitative RT-PCR Analysis
2.6. Statistical Analysis
3. Results
3.1. Clinical and Biochemical Characteristics of the Study Subjects
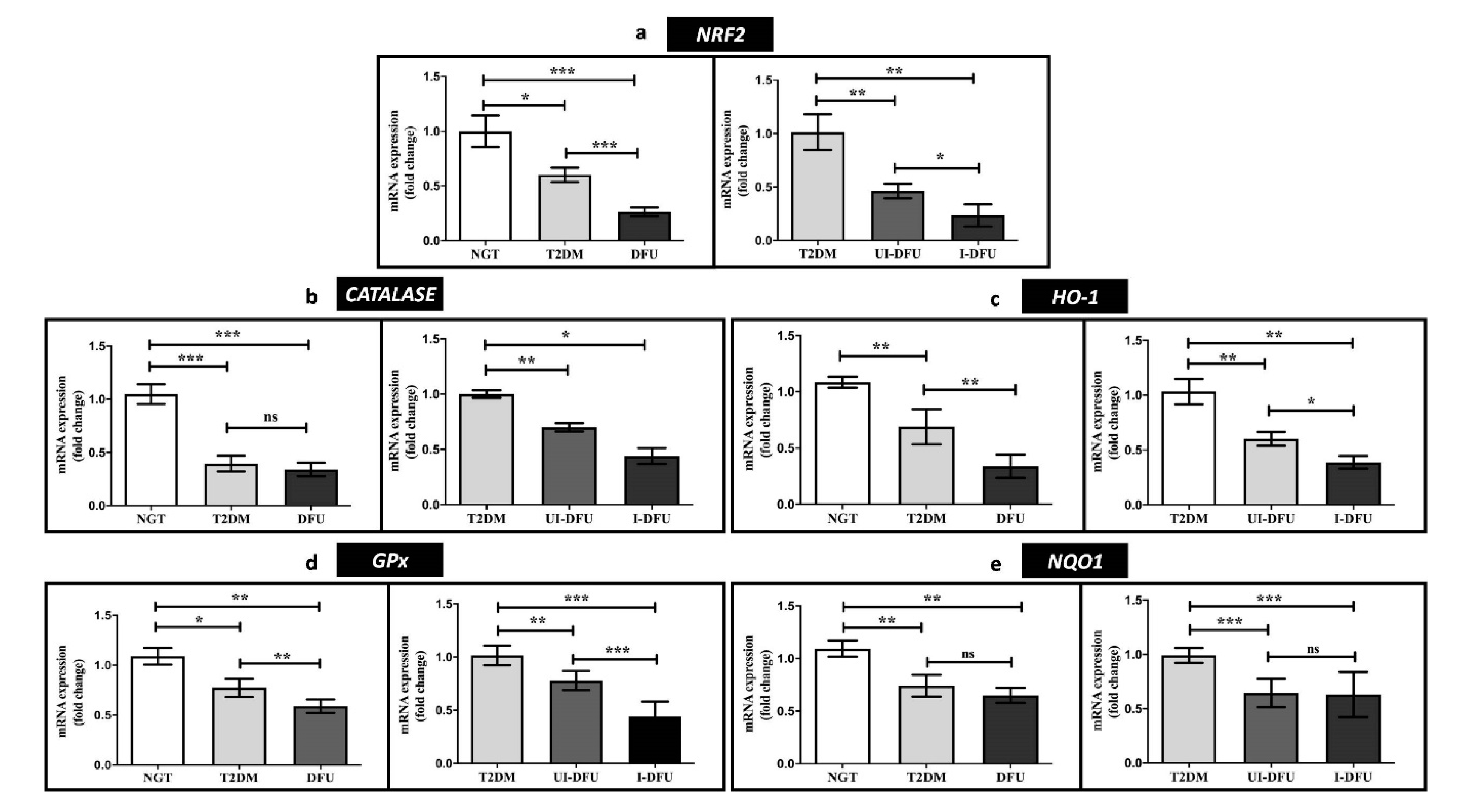
3.2. Expression of NRF2 and Downstream Targets in PBMCs
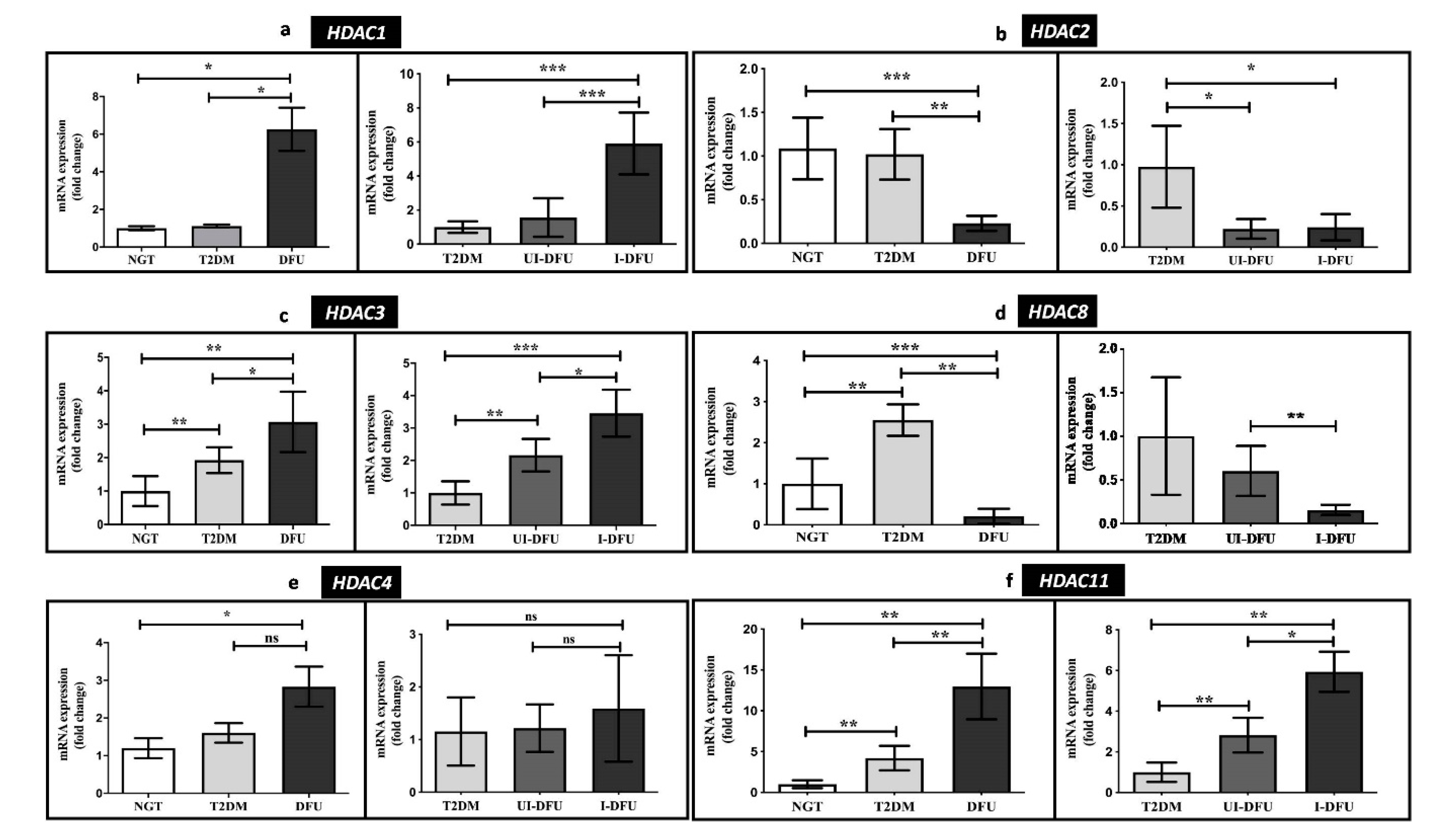
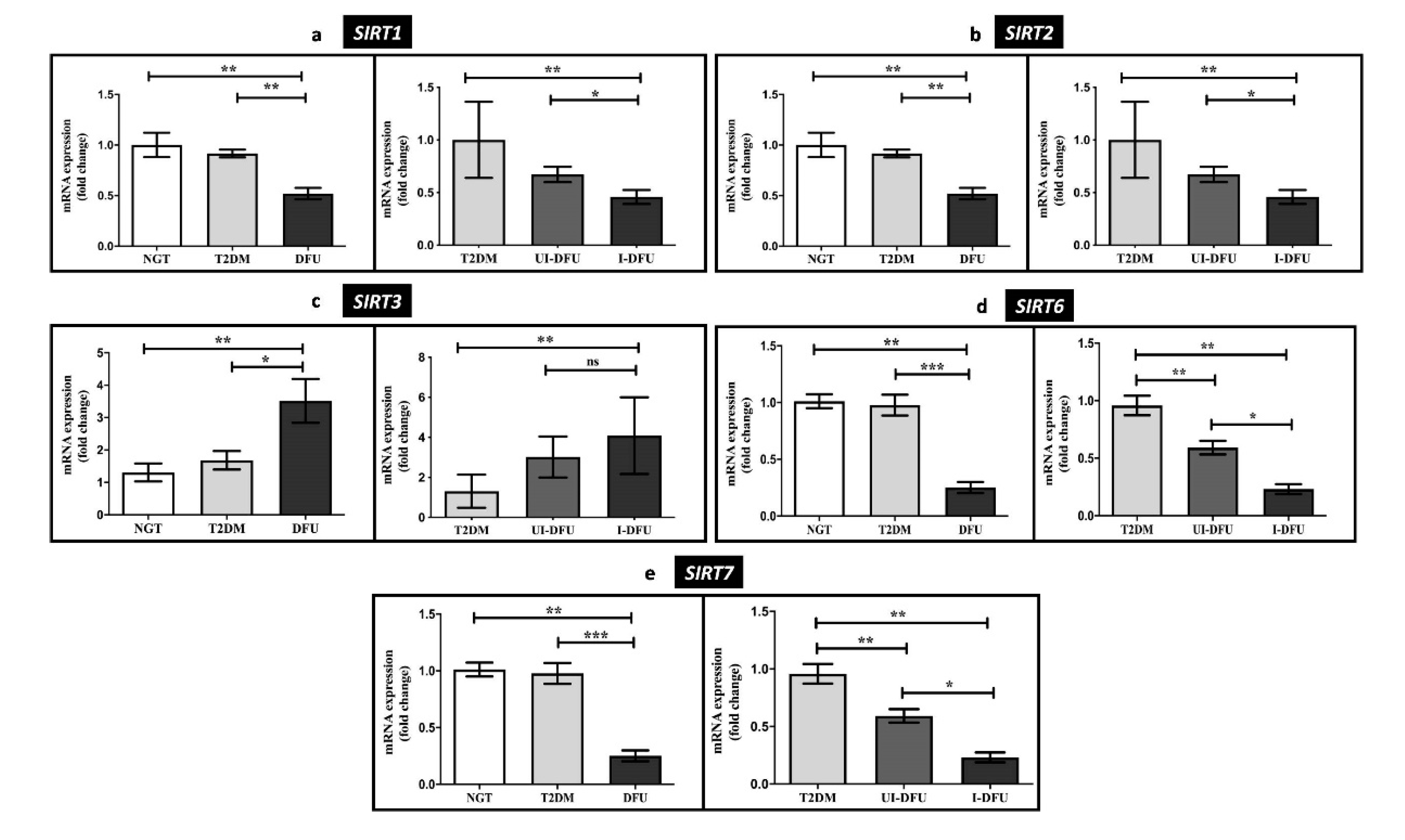
3.3. Quantitative RT-PCR Analysis of HDACs
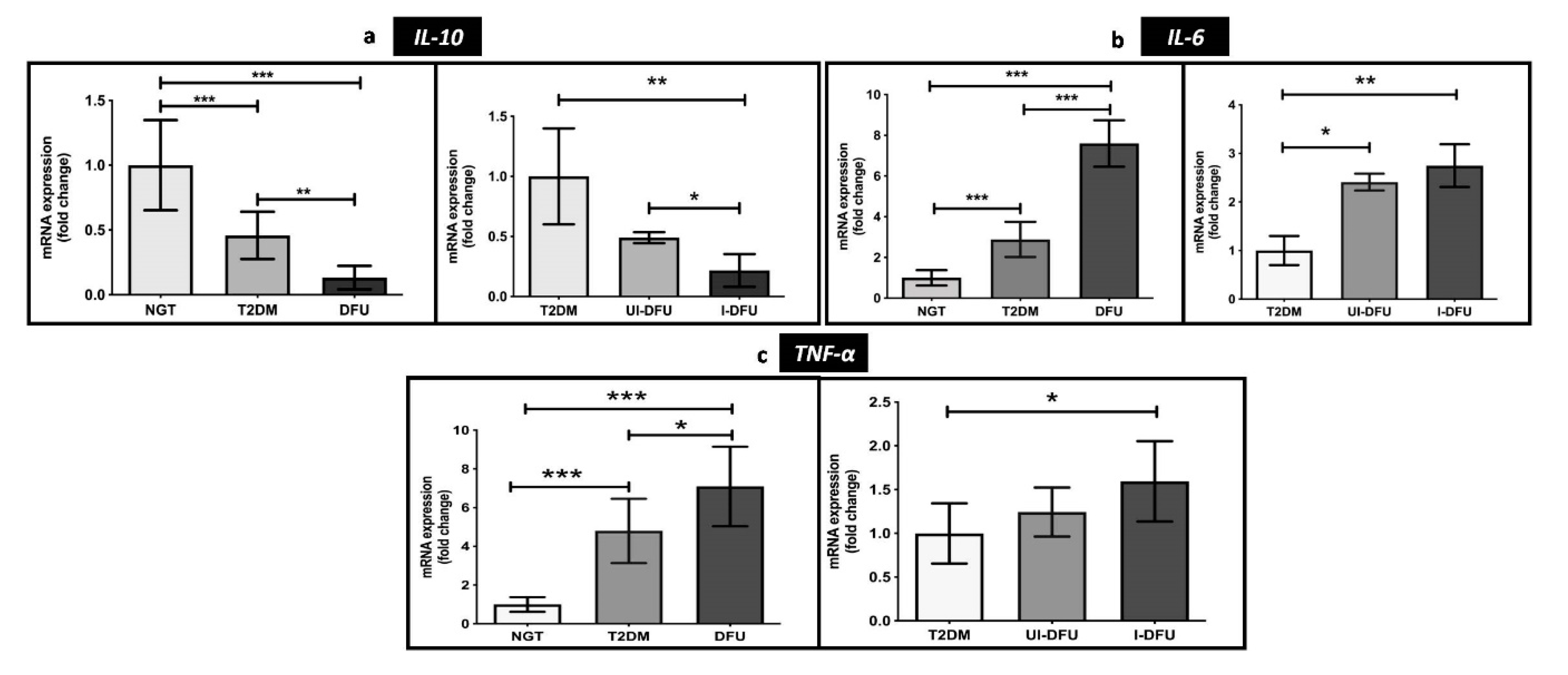
3.4. Transcriptional Levels of Pro-Inflammatory and Anti-Inflammatory Markers
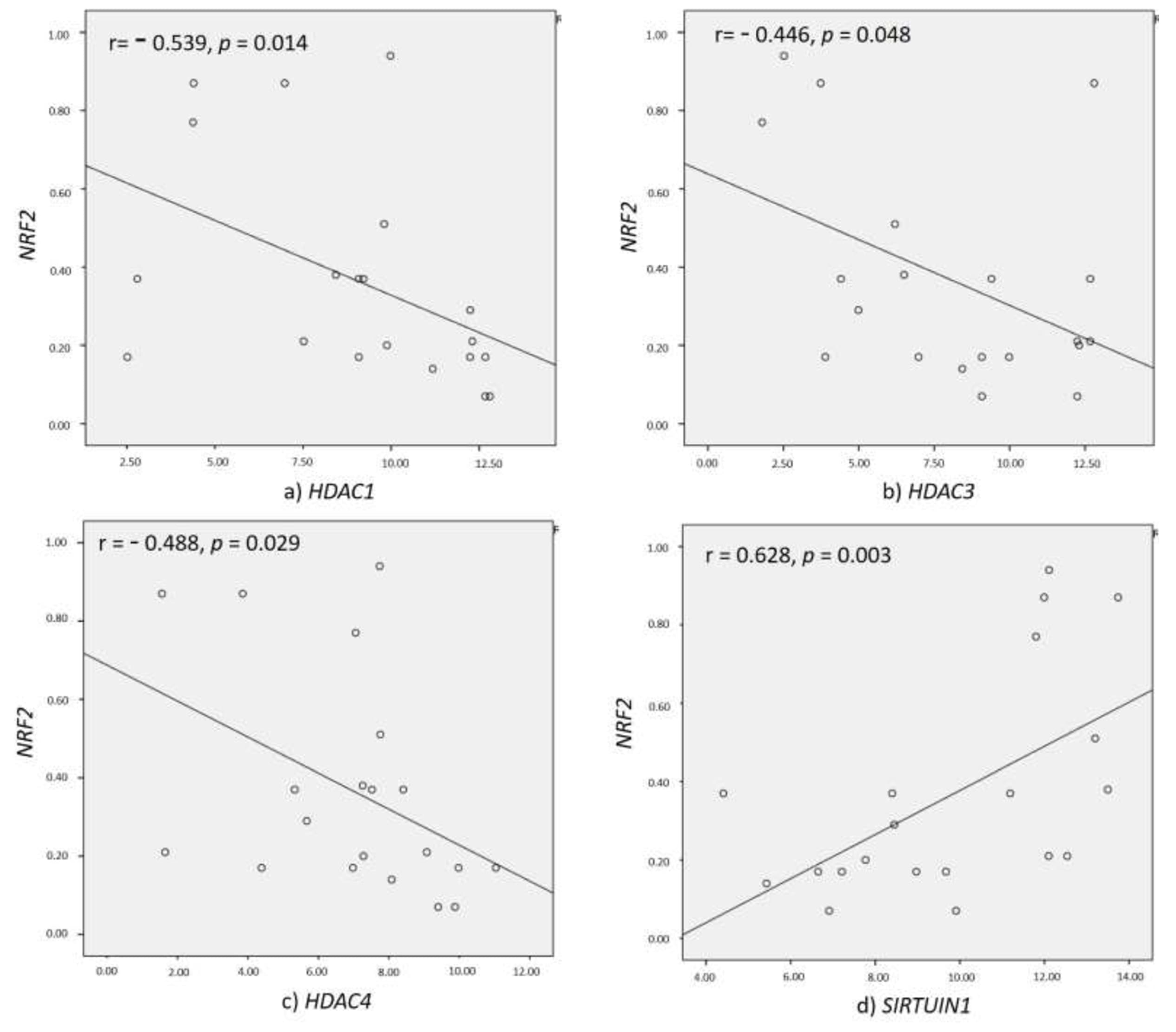
3.5. Correlation of HDACs with NRF2 and Inflammatory Markers
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pendsey, S.P. Understanding diabetic foot. Int. J. Diabetes Dev. Ctries 2010, 30, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Everett, E.; Mathioudakis, N. Update on management of diabetic foot ulcers. Ann. N. Y. Acad. Sci. 2018, 1411, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.; Doupis, J. Diabetic foot disease: From the evaluation of the "foot at risk" to the novel diabetic ulcer treatment modalities. World J. Diabetes 2016, 7, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Shih, A.Y.; Murphy, T.H.; Johnson, J.A. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J. Biol. Chem. 2003, 278, 37948–37956. [Google Scholar] [CrossRef] [PubMed]
- Dhamodharan, U.; Karan, A.; Sireesh, D.; Vaishnavi, A.; Somasundar, A.; Rajesh, K.; Ramkumar, K.M. Tissue-specific role of Nrf2 in the treatment of diabetic foot ulcers during hyperbaric oxygen therapy. Free Radic. Biol. Med. 2019, 138, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Teena, R.; Dhamodharan, U.; Ali, D.; Rajesh, K.; Ramkumar, K.M. Genetic Polymorphism of the Nrf2 Promoter Region (rs35652124) Is Associated with the Risk of Diabetic Foot Ulcers. Oxid. Med. Cell. Longev. 2020, 2020, 9825028. [Google Scholar] [CrossRef]
- Cheng, D.; Wu, R.; Guo, Y.; Kong, A.N. Regulation of Keap1-Nrf2 signaling: The role of epigenetics. Curr. Opin. Toxicol. 2016, 1, 134–138. [Google Scholar] [CrossRef]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef]
- Kagohara, L.T.; Stein-O’Brien, G.L.; Kelley, D.; Flam, E.; Wick, H.C.; Danilova, L.V.; Easwaran, H.; Favorov, A.V.; Qian, J.; Gaykalova, D.A.; et al. Epigenetic regulation of gene expression in cancer: Techniques, resources and analysis. Brief. Funct. Genom. 2017, 17, 49–63. [Google Scholar] [CrossRef]
- Moosavi, A.; Motevalizadeh Ardekani, A. Role of Epigenetics in Biology and Human Diseases. Iran Biomed. J. 2016, 20, 246–258. [Google Scholar] [CrossRef]
- Sarkar, S.; Abujamra, A.L.; Loew, J.E.; Forman, L.W.; Perrine, S.P.; Faller, D.V. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res. 2011, 31, 2723–2732. [Google Scholar] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Kim, K.C.; Kang, H.K.; Chang, W.Y.; Park, I.C.; Keum, Y.S.; Surh, Y.J.; Hyun, J.W. Epigenetic modification of Nrf2 in 5-fluorouracil-resistant colon cancer cells: Involvement of TET-dependent DNA demethylation. Cell Death Dis. 2014, 5, e1183. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, I.; Cao, M.; Su, Z.Y.; Wu, R.; Guo, Y.; Fang, M.; Kong, A.N. Fucoxanthin Elicits Epigenetic Modifications, Nrf2 Activation and Blocking Transformation in Mouse Skin JB6 P+ Cells. AAPS J. 2018, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.W.; Wang, M.; Sun, N.X.; Qing, Y.; Yin, T.F.; Li, C.; Wu, D. Sulforaphane-induced epigenetic regulation of Nrf2 expression by DNA methyltransferase in human Caco-2 cells. Oncol. Lett. 2019, 18, 2639–2647. [Google Scholar] [CrossRef] [PubMed]
- Lucio-Eterovic, A.K.; Cortez, M.A.; Valera, E.T.; Motta, F.J.; Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Neder, L.; Scrideli, C.A.; Tone, L.G. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: Class II and IV are hypoexpressed in glioblastomas. BMC Cancer 2008, 8, 243. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Zhen, J.; Zhang, C.; Wan, Q.; Liu, G.; Wei, X.; Zhang, Y.; Wang, Z.; Han, H.; et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014, 86, 712–725. [Google Scholar] [CrossRef]
- Kao, G.D.; McKenna, W.G.; Guenther, M.G.; Muschel, R.J.; Lazar, M.A.; Yen, T.J. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J. Cell Biol. 2003, 160, 1017–1027. [Google Scholar] [CrossRef]
- Imbriano, C.; Gurtner, A.; Cocchiarella, F.; Di Agostino, S.; Basile, V.; Gostissa, M.; Dobbelstein, M.; Del Sal, G.; Piaggio, G.; Mantovani, R. Direct p53 transcriptional repression: In vivo analysis of CCAAT-containing G2/M promoters. Mol. Cell. Biol. 2005, 25, 3737–3751. [Google Scholar] [CrossRef]
- Basile, V.; Mantovani, R.; Imbriano, C. DNA damage promotes histone deacetylase 4 nuclear localization and repression of G2/M promoters, via p53 C-terminal lysines. J. Biol. Chem. 2006, 281, 2347–2357. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Jun, J.A.; Jeong, K.J.; Heo, H.J.; Sohn, J.S.; Lee, H.Y.; Park, C.G.; Kang, J. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol. Rep. 2011, 25, 1677–1681. [Google Scholar] [CrossRef]
- American Diabetes, A. Diagnosis and classification of diabetes mellitus. Diabetes Care 2009, 32 (Suppl. S1), S62–S67. [Google Scholar] [CrossRef] [PubMed]
- Lipsky, B.A.; Aragon-Sanchez, J.; Diggle, M.; Embil, J.; Kono, S.; Lavery, L.; Senneville, E.; Urbancic-Rovan, V.; Van Asten, S.; International Working Group on the Diabetic Foot; et al. IWGDF guidance on the diagnosis and management of foot infections in persons with diabetes. Diabetes Metab. Res. Rev. 2016, 32 (Suppl. S1), 45–74. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Young, M.J.; Breddy, J.L.; Veves, A.; Boulton, A.J. The prediction of diabetic neuropathic foot ulceration using vibration perception thresholds. A prospective study. Diabetes Care 1994, 17, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Rooke, T.W.; Hirsch, A.T.; Misra, S.; Sidawy, A.N.; Beckman, J.A.; Findeiss, L.K.; Golzarian, J.; Gornik, H.L.; Halperin, J.L.; Jaff, M.R.; et al. 2011 ACCF/AHA Focused Update of the Guideline for the Management of Patients With Peripheral Artery Disease (updating the 2005 guideline): A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2011, 58, 2020–2045. [Google Scholar] [CrossRef]
- Rabbani, P.S.; Soares, M.A.; Hameedi, S.G.; Kadle, R.L.; Mubasher, A.; Kowzun, M.; Ceradini, D.J. Dysregulation of Nrf2/Keap1 Redox Pathway in Diabetes Affects Multipotency of Stromal Cells. Diabetes 2019, 68, 141–155. [Google Scholar] [CrossRef]
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and Its Activators in Respiratory Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 7090534. [Google Scholar] [CrossRef]
- Sireesh, D.; Dhamodharan, U.; Ezhilarasi, K.; Vijay, V.; Ramkumar, K.M. Association of NF-E2 Related Factor 2 (Nrf2) and inflammatory cytokines in recent onset Type 2 Diabetes Mellitus. Sci. Rep. 2018, 8, 5126. [Google Scholar] [CrossRef]
- Jayasuriya, R.; Dhamodharan, U.; Amin, K.N.; Anandharaj, A.; Rajesh, K.; Ramkumar, K.M. Role of Nrf2 in MALAT1/ HIF-1alpha loop on the regulation of angiogenesis in diabetic foot ulcer. Free Radic. Biol. Med. 2020, 156, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Florczyk, U.; Jazwa, A.; Maleszewska, M.; Mendel, M.; Szade, K.; Kozakowska, M.; Grochot-Przeczek, A.; Viscardi, M.; Czauderna, S.; Bukowska-Strakova, K.; et al. Nrf2 regulates angiogenesis: Effect on endothelial cells, bone marrow-derived proangiogenic cells and hind limb ischemia. Antioxid. Redox Signal. 2014, 20, 1693–1708. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Tak Park, J.; Natarajan, R. Epigenetic modifications in the pathogenesis of diabetic nephropathy. Semin. Nephrol. 2013, 33, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Chen, X.F.; Chen, H.Z.; Liu, D.P. Mitochondrial Sirtuins in cardiometabolic diseases. Clin. Sci. 2017, 131, 2063–2078. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Gerhart-Hines, Z.; Puigserver, P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008, 582, 46–53. [Google Scholar] [CrossRef]
- Johnson, E.; Marsh, S. Anti-diabetic effects of class 1 histone deacetylase inhibition in a rodent model of type 2 diabetes mellitus. FASEB J. 2016, 30, 1273–1276. [Google Scholar]
- Lkhagva, B.; Kao, Y.H.; Lee, T.I.; Lee, T.W.; Cheng, W.L.; Chen, Y.J. Activation of Class I histone deacetylases contributes to mitochondrial dysfunction in cardiomyocytes with altered complex activities. Epigenetics 2018, 13, 376–385. [Google Scholar] [CrossRef]
- Jeong, Y.; Du, R.; Zhu, X.; Yin, S.; Wang, J.; Cui, H.; Cao, W.; Lowenstein, C.J. Histone deacetylase isoforms regulate innate immune responses by deacetylating mitogen-activated protein kinase phosphatase-1. J. Leukoc. Biol. 2014, 95, 651–659. [Google Scholar] [CrossRef]
- Mercado, N.; Thimmulappa, R.; Thomas, C.M.; Fenwick, P.S.; Chana, K.K.; Donnelly, L.E.; Biswal, S.; Ito, K.; Barnes, P.J. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem. Biophys. Res. Commun. 2011, 406, 292–298. [Google Scholar] [CrossRef]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.M.; Hogg, J.C.; et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Sathishkumar, C.; Prabu, P.; Balakumar, M.; Lenin, R.; Prabhu, D.; Anjana, R.M.; Mohan, V.; Balasubramanyam, M. Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin. Epigenet. 2016, 8, 125. [Google Scholar] [CrossRef]
- Park, D.; Park, H.; Kim, Y.; Kim, H.; Jeoung, D. HDAC3 acts as a negative regulator of angiogenesis. BMB Rep. 2014, 47, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, Z.; Gu, J.; Jiang, S.; Liu, Q.; Zheng, Y.; Freedman, J.H.; Sun, J.; Cai, L. HDAC3 inhibition in diabetic mice may activate Nrf2 preventing diabetes-induced liver damage and FGF21 synthesis and secretion leading to aortic protection. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E150–E162. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhang, F.; Yu, Z.; Guo, S.; Liu, N.; Jiang, Y.; Lo, E.H.; Xu, Y.; Wang, X. HDAC3 inhibition prevents blood-brain barrier permeability through Nrf2 activation in type 2 diabetes male mice. J. Neuroinflamm. 2019, 16, 103. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wong, V.W.; Wong, G.L.; Yang, W.; Sun, H.; Shen, J.; Tong, J.H.; Go, M.Y.; Cheung, Y.S.; Lai, P.B.; et al. Histone Deacetylase HDAC8 Promotes Insulin Resistance and beta-Catenin Activation in NAFLD-Associated Hepatocellular Carcinoma. Cancer Res. 2015, 75, 4803–4816. [Google Scholar] [CrossRef]
- Yan, W.; Liu, S.; Xu, E.; Zhang, J.; Zhang, Y.; Chen, X.; Chen, X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene 2013, 32, 599–609. [Google Scholar] [CrossRef]
- Qian, Y.; Zhang, J.; Jung, Y.S.; Chen, X. DEC1 coordinates with HDAC8 to differentially regulate TAp73 and DeltaNp73 expression. PLoS ONE 2014, 9, e84015. [Google Scholar] [CrossRef]
- Zhong, Q.; Kowluru, R.A. Role of histone acetylation in the development of diabetic retinopathy and the metabolic memory phenomenon. J. Cell. Biochem. 2010, 110, 1306–1313. [Google Scholar] [CrossRef]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Okada, M.; Mizuno, W.; Oda, M.; Ide, N.; Morita, T.; Hara, Y.; Yamawaki, H. HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1894–H1904. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; Verdin, E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008, 7, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Laemmle, A.; Lechleiter, A.; Roh, V.; Schwarz, C.; Portmann, S.; Furer, C.; Keogh, A.; Tschan, M.P.; Candinas, D.; Vorburger, S.A.; et al. Inhibition of SIRT1 impairs the accumulation and transcriptional activity of HIF-1alpha protein under hypoxic conditions. PLoS ONE 2012, 7, e33433. [Google Scholar] [CrossRef]
- Huang, K.; Gao, X.; Wei, W. The crosstalk between Sirt1 and Keap1/Nrf2/ARE anti-oxidative pathway forms a positive feedback loop to inhibit FN and TGF-beta1 expressions in rat glomerular mesangial cells. Exp. Cell Res. 2017, 361, 63–72. [Google Scholar] [CrossRef]
- Elkhwanky, M.S.; Hakkola, J. Extranuclear Sirtuins and Metabolic Stress. Antioxid. Redox Signal. 2018, 28, 662–676. [Google Scholar] [CrossRef]
- Zeng, H.; Li, L.; Chen, J.X. Loss of Sirt3 limits bone marrow cell-mediated angiogenesis and cardiac repair in post-myocardial infarction. PLoS ONE 2014, 9, e107011. [Google Scholar] [CrossRef]
- Wei, T.; Huang, G.; Gao, J.; Huang, C.; Sun, M.; Wu, J.; Bu, J.; Shen, W. Sirtuin 3 Deficiency Accelerates Hypertensive Cardiac Remodeling by Impairing Angiogenesis. J. Am. Heart Assoc. 2017, 6, e006114. [Google Scholar] [CrossRef]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef]
- Sun, L.; Marin de Evsikova, C.; Bian, K.; Achille, A.; Telles, E.; Pei, H.; Seto, E. Programming and Regulation of Metabolic Homeostasis by HDAC11. EBioMedicine 2018, 33, 157–168. [Google Scholar] [CrossRef]
- Bagchi, R.A.; Ferguson, B.S.; Stratton, M.S.; Hu, T.; Cavasin, M.A.; Sun, L.; Lin, Y.H.; Liu, D.; Londono, P.; Song, K.; et al. HDAC11 suppresses the thermogenic program of adipose tissue via BRD2. JCI Insight 2018, 3, e120159. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Names | Gene | Forward Primer | Reverse Primer |
|---|---|---|---|
| NRF2 and downstream target genes | NRF2 | TGTAGATGACAATGAGGTTTC | ACTGAGCCTGATTAGTAGCAA |
| CAT | ATCCGTGTAACCCGCTCATC | ACCTTCATTTTCCCCTGGGG | |
| HO-1 | GGGAATTCTCTTGGCTGGCT | AACTGAGGATGCTGAAGGGC | |
| GPx | TATCGAGAATGTGGCGTCCC | CAAACTGGTTGCACGGGAAG | |
| NQO1 | AGTCATCTCATTCCACTGTTGG | GCTGTCTCCCATTTTTCAGG | |
| HDACs | HDAC1 | GGCTGGCAAAGGCAAGTAT | CGCACTAGGCTGGAACATCT |
| HDAC2 | ATTGGGGAACAGGTGGTG | GGGGCGAGGGATAAAAGA | |
| HDAC3 | GTATGAAGTCGGGGCAGAGA | CGTGGGTTGGTAGAAGTCC | |
| HDAC8 | GTGGGAATTGGCAAGTGTCT | CCAGCACATAATCAGGACCA | |
| HDAC4 | GCACAGTCCTTGGTTGGTG | AGAAACTGCTGATGCTGCTG | |
| SIRT1 | GCCGACAACTTGTACGACGA | CACCGAACAGAAGGTTATCTCG | |
| SIRT2 | CCCCCTCTTAACCAGCAGTT | GATGCCTGTTTAAGCCTTGG | |
| SIRT3 | CTCAGCCTCTCCTCCAGAAA | TAATGCCTTCCCTGTCTCAG | |
| SIRT6 | AGGGTGGGGCTTTTTGTA | CTCTGGGGTGTGGCTTCTT | |
| SIRT7 | AGCAGAGCAGACACCATCCT | CAGCCCAGTCATCCTTCG | |
| HDAC11 | GGGGGAGGGCAGAAGAAG | CCGCCTCACCAGTGTCTG | |
| Inflammatory markers | IL-10 | ACATCAGGGTGGCGACTCTA | AAGGTTTCTCAAGGGGCTGG |
| IL-6 | GTCCAGTTGCCTTCTCCCTG | AGCACGACCACGACCTTG | |
| TNF-α | TCTGGGCAGGTCTACTTTGG | GGTTGAGGGTGTCTGAAGGA | |
| Housekeeping gene | GAPDH | AAGAAGGTGGTGAAGCAGGC | GTCAAAGGTGGAGGAGTGGG |
| Clinical Parameters | NGT (n = 20) | T2DM (n = 20) a | UI-DFU (n = 10) b | I-DFU (n = 10) c |
|---|---|---|---|---|
| Gender (M/F) | 11M/9F | 12M/8F | 6M/4F | 5M/5F |
| Age (years) | 51.6 ± 1.3 | 51.5 ± 1.2 | 51.7 ± 1.2 | 51.5 ± 1.3 |
| BMI (kg/m2) | 26.1 ± 1.7 | 26.5 ± 1.1 ns | 27.9 ± 1.7 * | 28.1± 1ns |
| SBP (mm Hg) | 115.8 ± 4.2 | 125.30 ± 2.5 **** | 131.7 ± 3.1 **** | 135.4 ± 2.1 ** |
| DBP (mm Hg) | 74.6 ± 4.1 | 83.10 ± 1.8 **** | 85.1 ± 2.77 ns | 88.6 ± 1.8 ** |
| FPG (mg/dL) | 90.5 ± 4.9 | 143.3 ± 11.6 **** | 167.8 ± 14.5 **** | 241.1 ± 14.5 **** |
| PPG (mg/dL) | 108 ± 6.1 | 227.7 ± 4.7 **** | 242.6 ± 5.3 **** | 293.6 ± 27.6 **** |
| HbA1c (%) | 5 ± 0.3 | 8.01 ± 0.49 **** | 9.3 ± 0.5 **** | 10.3 ± 0.5 ** |
| TSC (mg/dL) | 175.4 ± 10.1 | 182.5 ± 3.4 ** | 184.8 ± 7.8 ns | 187.1 ± 3.1 ns |
| HDL-c (mg/dL) | 49.1 ± 6.9 | 46.2 ± 5 ns | 44.7 ± 11.7 ns | 40.9 ± 8.7 ns |
| LDL-c (mg/dL) | 91.6 ± 6.6 | 104 ± 15 ** | 123.3 ± 3.9 *** | 129 ± 6 * |
| Urea (mg/dL) | 24.2 ± 3.2 | 29.1 ± 4.4 *** | 32.6 ± 2.6 * | 37.6 ± 1.4 *** |
| Creatinine (mg/dL) | 0.7 ± 0.1 | 0.8 ± 0.2 * | 0.9 ± 0.1 * | 1 ± 0.1 ns |
| HOMA-IR | 1.0 ± 0.3 | 3.5 ± 0.9 **** | 5.1 ± 0.7 *** | 8 ± 0.6 **** |
| Inflammation markers | ||||
| CRP (mg/L) | 2 ± 0.6 | 4 ± 0.8 **** | 15.6 ± 2.5 **** | 37.1 ± 6.2 **** |
| ESR (mm/hour) | 2.9 ± 1.1 | 21 ± 2.1 **** | 45.6 ± 3.3 **** | 56.4 ± 3.6 **** |
| WBC (109/L) | 4.7 ± 1.1 | 6.8 ± 1.0 **** | 8.1 ± 0.5 ** | 12.1 ± 0.6 **** |
| NRF2 vs. HDACs | r Value | p Value |
|---|---|---|
| HDAC1 | −0.539 | 0.014 |
| HDAC2 | 0.584 | 0.128 |
| HDAC3 | −0.446 | 0.048 |
| HDAC8 | 0.036 | 0.933 |
| HDAC4 | −0.488 | 0.029 |
| HDAC11 | −0.302 | 0.114 |
| SIRT1 | 0.628 | 0.003 |
| SIRT2 | 0.155 | 0.598 |
| SIRT3 | −0.147 | 0.09 |
| SIRT6 | 0.144 | 0.758 |
| SIRT7 | 0.348 | 0.499 |
| Variables | IL-6 | TNF-ɑ | IL-10 | |||
|---|---|---|---|---|---|---|
| r Value | p Value | r Value | p Value | r Value | p Value | |
| SIRTUIN1 | −0.683 | 0.042 | −0.2 | 0.606 | 0.536 | 0.137 |
| HDAC1 | 0.477 | 0.194 | 0.259 | 0.5 | −0.424 | 0.255 |
| HDAC3 | 0.383 | 0.308 | 0.6 | 0.088 | −0.167 | 0.667 |
| HDAC4 | 0.733 | 0.025 | 0.483 | 0.187 | −0.561 | 0.116 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teena, R.; Dhamodharan, U.; Ali, D.; Rajesh, K.; Ramkumar, K.M. Gene Expression Profiling of Multiple Histone Deacetylases (HDAC) and Its Correlation with NRF2-Mediated Redox Regulation in the Pathogenesis of Diabetic Foot Ulcers. Biomolecules 2020, 10, 1466. https://doi.org/10.3390/biom10101466
Teena R, Dhamodharan U, Ali D, Rajesh K, Ramkumar KM. Gene Expression Profiling of Multiple Histone Deacetylases (HDAC) and Its Correlation with NRF2-Mediated Redox Regulation in the Pathogenesis of Diabetic Foot Ulcers. Biomolecules. 2020; 10(10):1466. https://doi.org/10.3390/biom10101466
Chicago/Turabian StyleTeena, Rajan, Umapathy Dhamodharan, Daoud Ali, Kesavan Rajesh, and Kunka Mohanram Ramkumar. 2020. "Gene Expression Profiling of Multiple Histone Deacetylases (HDAC) and Its Correlation with NRF2-Mediated Redox Regulation in the Pathogenesis of Diabetic Foot Ulcers" Biomolecules 10, no. 10: 1466. https://doi.org/10.3390/biom10101466
APA StyleTeena, R., Dhamodharan, U., Ali, D., Rajesh, K., & Ramkumar, K. M. (2020). Gene Expression Profiling of Multiple Histone Deacetylases (HDAC) and Its Correlation with NRF2-Mediated Redox Regulation in the Pathogenesis of Diabetic Foot Ulcers. Biomolecules, 10(10), 1466. https://doi.org/10.3390/biom10101466