Alpha-Synuclein in Alcohol Use Disorder, Connections with Parkinson’s Disease and Potential Therapeutic Role of 5’ Untranslated Region-Directed Small Molecules


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Alpha Synuclein Expression is Associated with Alcohol Use Disorder (AUD)
3. Parkinson’s Disease, Parkinsonism and Alcohol Use Disorder
4. Alcohol Affects the Cholinergic and Dopaminergic Systems in the Brain
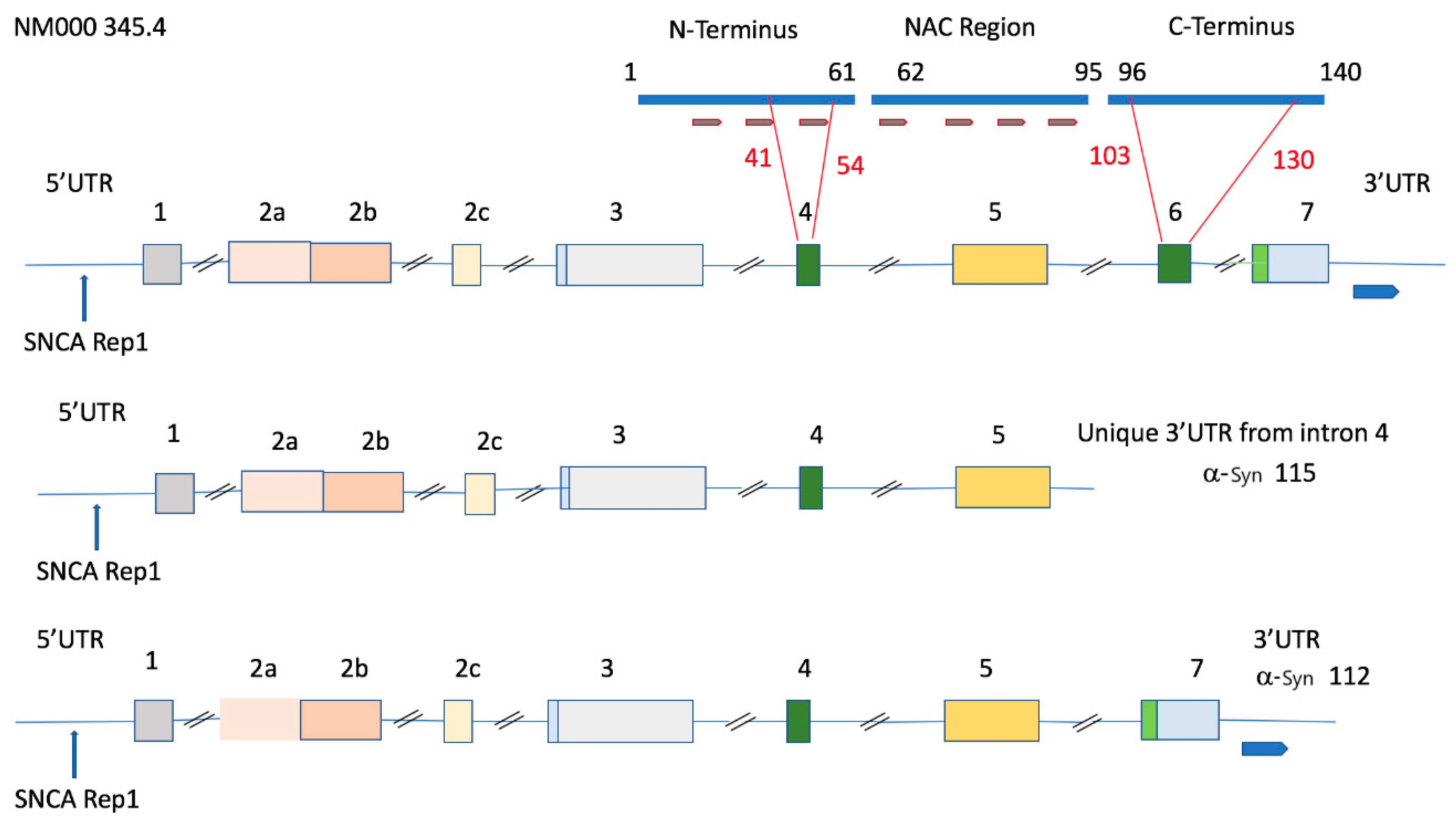
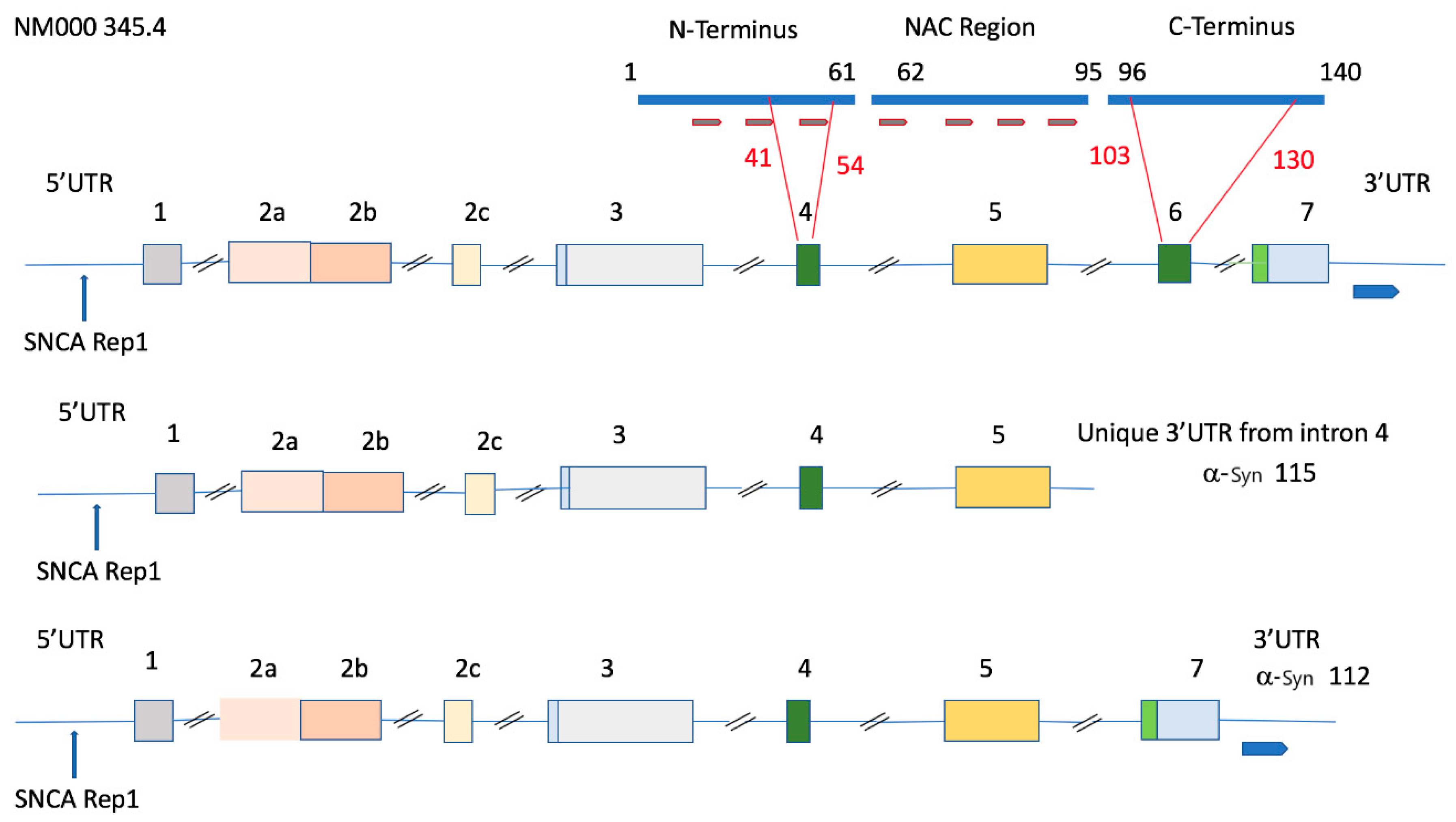
5. Genomic Organization of the SNCA Gene and Association of the 115 SNCA Variant with Alcohol Use Disorder
6. REP1 Alleles and Variations in the 5’ and 3’UTRs of α-Syn; Associations with Parkinson’s Disease and Alcohol Use Disorder
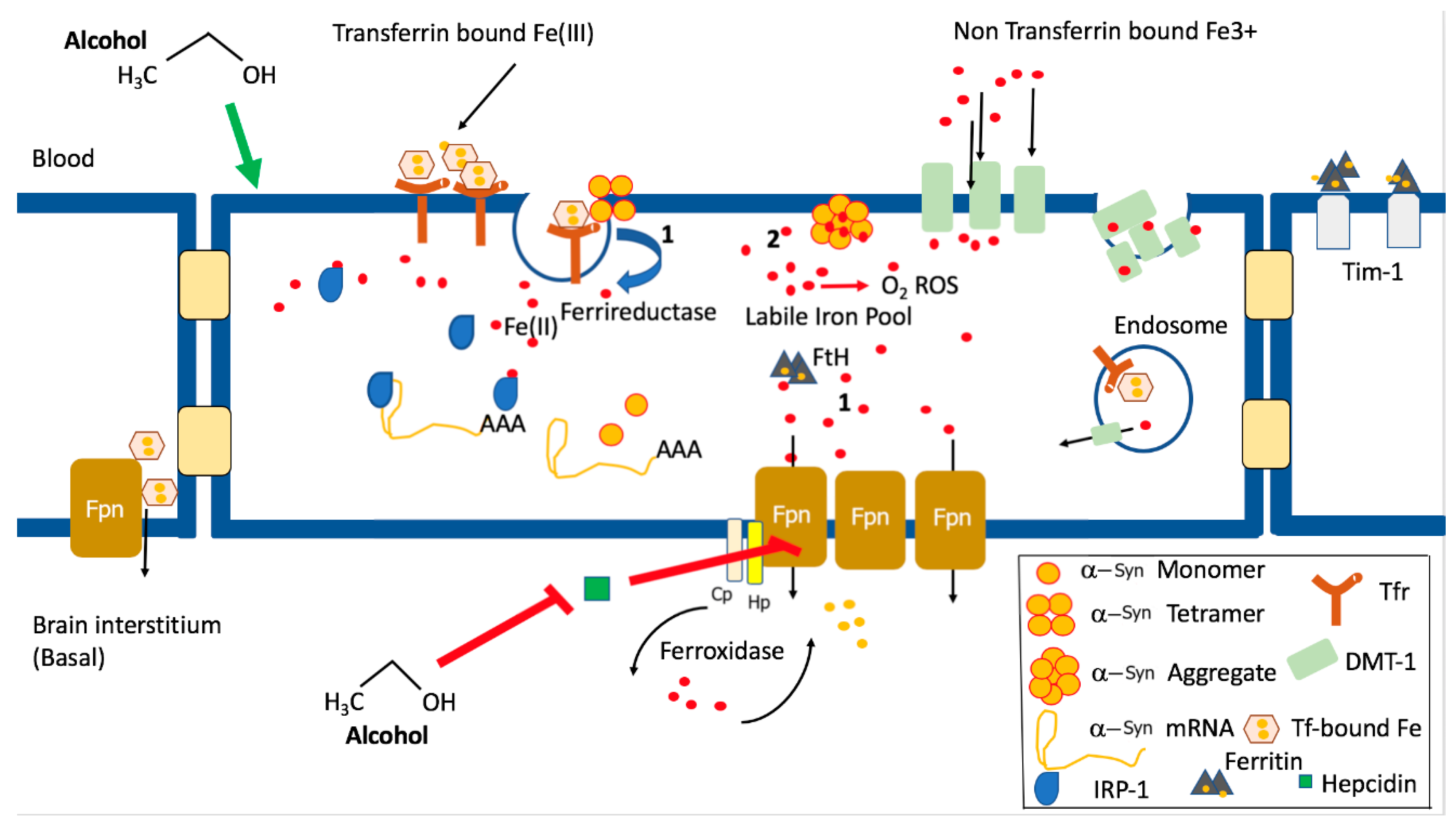
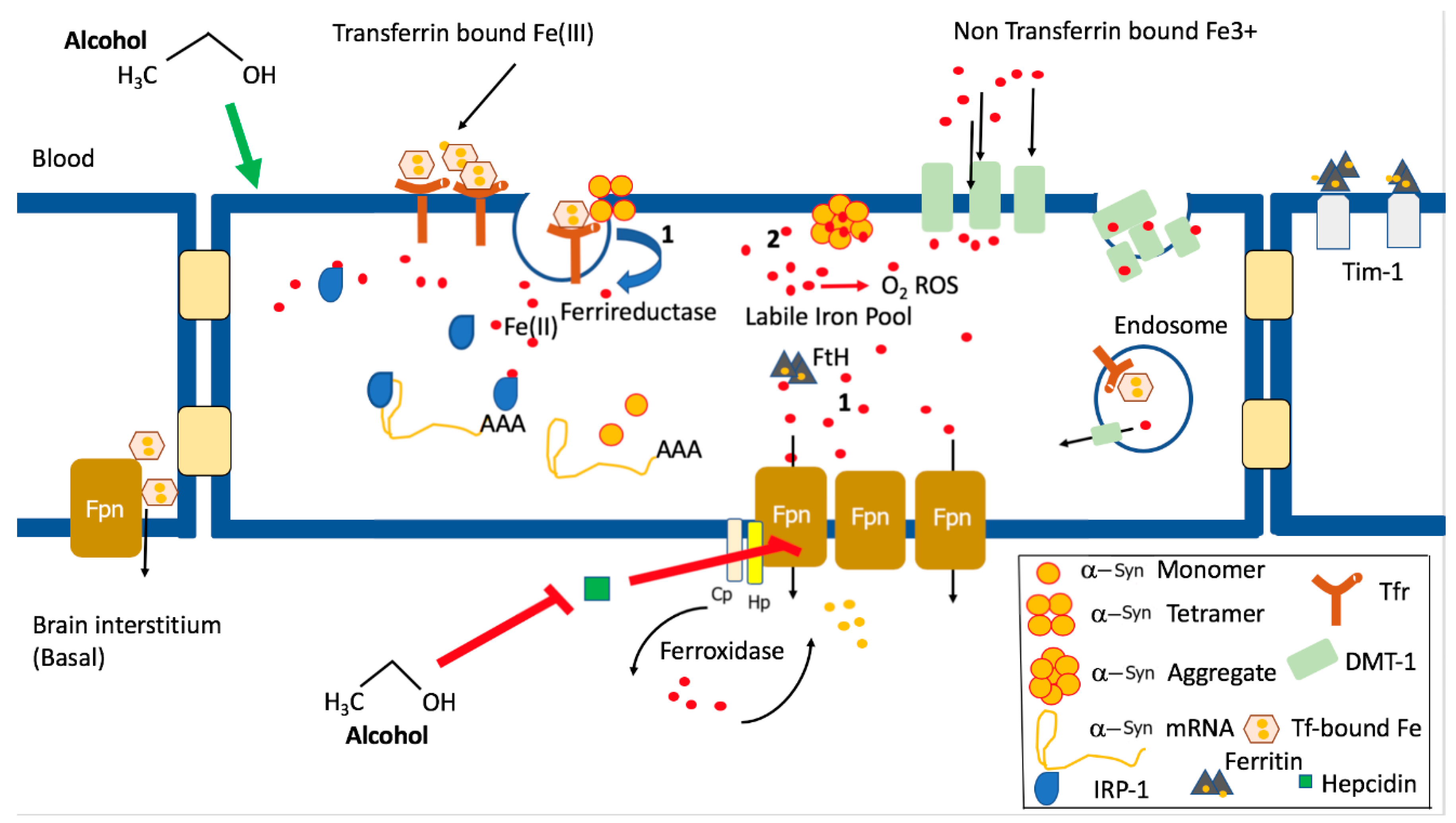
7. Alpha Synuclein and Iron Homeostasis
8. Iron Dyshomeostasis and Alcohol Use Disorder
9. α-Synuclein 5’UTR-Directed Small Molecules as Potential Therapy for Alcohol Use Disorder and Parkinson’s Disease
10. Stem Cells, α-Synuclein, and Alcohol Use Disorder
11. Looking to the Future
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Atsmon-Raz, Y.; Miller, Y. Non-Amyloid-beta Component of Human alpha-Synuclein Oligomers Induces Formation of New Abeta Oligomers: Insight into the Mechanisms That Link Parkinson’s and Alzheimer’s Diseases. ACS Chem. Neurosci. 2016, 7, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Biere, A.L.; Wood, S.J.; Wypych, J.; Steavenson, S.; Jiang, Y.; Anafi, D.; Jacobsen, F.W.; Jarosinski, M.A.; Wu, G.M.; Louis, J.C.; et al. Parkinson’s disease-associated alpha-synuclein is more fibrillogenic than beta- and gamma-synuclein and cannot cross-seed its homologs. J. Biol. Chem. 2000, 275, 34574–34579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Attoub, S.; Singh, M.N.; Martin, F.L.; El-Agnaf, O.M.A. γ-Synuclein and the progression of cancer. FASEB J. 2007, 21, 3419–3430. [Google Scholar] [CrossRef] [Green Version]
- Burre, J.; Sharma, M.; Sudhof, T.C. Cell Biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Butler, B.; Sambo, D.; Khoshbouei, H. Alpha-synuclein modulates dopamine neurotransmission. J. Chem. Neuroanat. 2017, 83–84, 41–49. [Google Scholar] [CrossRef]
- Butler, B.; Saha, K.; Rana, T.; Becker, J.P.; Sambo, D.; Davari, P.; Goodwin, J.S.; Khoshbouei, H. Dopamine Transporter Activity Is Modulated by alpha-Synuclein. J. Biol. Chem. 2015, 290, 29542–29554. [Google Scholar] [CrossRef] [Green Version]
- Abdelkarim, H.; Marshall, M.S.; Scesa, G.; Smith, R.A.; Rue, E.; Marshall, J.; Elackattu, V.; Stoskute, M.; Issa, Y.; Santos, M.; et al. Alpha-Synuclein interacts directly but reversibly with psychosine: Implications for alpha-synucleinopathies. Sci. Rep. 2018, 8, 12462. [Google Scholar] [CrossRef] [Green Version]
- Trubetckaia, O.; Lane, A.E.; Qian, L.; Zhou, P.; Lane, D.A. Alpha-synuclein is strategically positioned for afferent modulation of midbrain dopamine neurons and is essential for cocaine preference. Commun. Biol. 2019, 2, 418. [Google Scholar] [CrossRef]
- Mash, D.C.; Ouyang, Q.; Pablo, J.; Basile, M.; Izenwasser, S.; Lieberman, A.; Perrin, R.J. Cocaine abusers have an overexpression of alpha-synuclein in dopamine neurons. J. Neurosci. 2003, 23, 2564–2571. [Google Scholar] [CrossRef] [Green Version]
- Chiba-Falek, O. Structural variants in SNCA gene and the implication to synucleinopathies. Curr. Opin. Genet. Dev. 2017, 44, 110–116. [Google Scholar] [CrossRef]
- Rotter, A.; Lenz, B.; Pitsch, R.; Richter-Schmidinger, T.; Kornhuber, J.; Rhein, C. Alpha-Synuclein RNA Expression is Increased in Major Depression. Int. J. Mol. Sci. 2019, 20, 2029. [Google Scholar] [CrossRef] [Green Version]
- Soldner, F.; Jaenisch, R. Dissecting risk haplotypes in sporadic Alzheimer’s disease. Cell Stem Cell 2015, 16, 341–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persyn, W.; Houchi, H.; Papillon, C.A.; Martinetti, M.; Antol, J.; Guillaumont, C.; Dervaux, A.; Naassila, M. Ethanol (EtOH)-Related Behaviors in alpha-Synuclein Mutant Mice and Association of SNCA SNPs with Anxiety in EtOH-Dependent Patients. Alcohol. Clin. Exp. Res. 2018, 42, 2172–2185. [Google Scholar] [CrossRef] [PubMed]
- Foroud, T.; Wetherill, L.F.; Liang, T.; Dick, D.M.; Hesselbrock, V.; Kramer, J.; Nurnberger, J.; Schuckit, M.; Carr, L.; Porjesz, B.; et al. Association of alcohol craving with alpha-synuclein (SNCA). Alcohol. Clin. Exp. Res. 2007, 31, 537–545. [Google Scholar] [PubMed]
- Levey, D.F.; Le-Niculescu, H.; Frank, J.; Ayalew, M.; Jain, N.; Kirlin, B.; Learman, R.; Winiger, E.; Rodd, Z.; Shekhar, A.; et al. Genetic risk prediction and neurobiological understanding of alcoholism. Transl. Psychiatry 2014, 4, e391. [Google Scholar] [CrossRef] [Green Version]
- Bonsch, D.; Reulbach, U.; Bayerlein, K.; Hillemacher, T.; Kornhuber, J.; Bleich, S. Elevated alpha synuclein mRNA levels are associated with craving in patients with alcoholism. Biol. Psychiatry 2004, 56, 984–986. [Google Scholar] [CrossRef]
- Bonsch, D.; Greifenberg, V.; Bayerlein, K.; Biermann, T.; Reulbach, U.; Hillemacher, T.; Kornhuber, J.; Bleich, S. Alpha-synuclein protein levels are increased in alcoholic patients and are linked to craving. Alcohol. Clin. Exp. Res. 2005, 29, 763–765. [Google Scholar] [CrossRef]
- Bonsch, D.; Lederer, T.; Reulbach, U.; Hothorn, T.; Kornhuber, J.; Bleich, S. Joint analysis of the NACP-REP1 marker within the alpha synuclein gene concludes association with alcohol dependence. Hum. Mol. Genet. 2005, 14, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Walker, S.J.; Grant, K.A. Peripheral blood alpha-synuclein mRNA levels are elevated in cynomolgus monkeys that chronically self-administer ethanol. Alcohol 2006, 38, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Carr, L.G. Regulation of alpha-synuclein expression in alcohol-preferring and -non preferring rats. J. Neurochem. 2006, 99, 470–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, W.; Hyytia, P.; Kiianmaa, K. The alcohol-preferring AA and alcohol-avoiding ANA rats: Neurobiology of the regulation of alcohol drinking. Addict. Biol. 2006, 11, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowska, B.; Gieryk, A.; Wawrzczak-Bargiela, A.; Krowka, T.; Kaminska, D.; Korkosz, A.; Bienkowski, P.; Przewlocki, R. Alpha-Synuclein expression in the brain and blood during abstinence from chronic alcohol drinking in mice. Neuropharmacology 2008, 54, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, C.; Reolon, G.K.; Leixner, S.; Boden, C.; Bilbao, A.; Kahle, P.J. Enhanced motivation to alcohol in transgenic mice expressing human alpha-synuclein. J. Neurochem. 2017, 143, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Mayfield, R.D.; Lewohl, J.M.; Dodd, P.R.; Herlihy, A.; Liu, J.; Harris, R.A. Patterns of gene expression are altered in the frontal and motor cortices of human alcoholics. J. Neurochem. 2002, 81, 802–813. [Google Scholar] [CrossRef]
- Fan, L.; Bellinger, F.; Ge, Y.L.; Wilce, P. Genetic study of alcoholism and novel gene expression in the alcoholic brain. Addict. Biol. 2004, 9, 11–18. [Google Scholar] [CrossRef]
- Hoffman, P.L.; Miles, M.; Edenberg, H.J.; Sommer, W.; Tabakoff, B.; Wehner, J.M.; Lewohl, J. Gene expression in brain: A window on ethanol dependence, neuroadaptation, and preference. Alcohol. Clin. Exp. Res. 2003, 27, 155–168. [Google Scholar] [CrossRef]
- Liu, J.; Lewohl, J.M.; Harris, R.A.; Iyer, V.R.; Dodd, P.R.; Randall, P.K.; Mayfield, R.D. Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology 2006, 31, 1574–1582. [Google Scholar] [CrossRef] [Green Version]
- Janeczek, P.; MacKay, R.K.; Lea, R.A.; Dodd, P.R.; Lewohl, J.M. Reduced expression of alpha-synuclein in alcoholic brain: Influence of SNCA-Rep1 genotype. Addict. Biol. 2014, 19, 509–515. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.; Kimpel, M.W.; McClintick, J.N.; Skillman, A.R.; McCall, K.; Edenberg, H.J.; Carr, L.G. Candidate genes for alcohol preference identified by expression profiling in alcohol-preferring and -nonpreferring reciprocal congenic rats. Genome Biol. 2010, 11, R11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, T.; Spence, J.; Liu, L.; Strother, W.N.; Chang, H.W.; Ellison, J.A.; Lumeng, L.; Li, T.K.; Foroud, T.; Carr, L.G. Alpha-Synuclein maps to a quantitative trait locus for alcohol preference and is differentially expressed in alcohol-preferring and -nonpreferring rats. Proc. Natl. Acad. Sci. USA 2003, 100, 4690–4695. [Google Scholar] [CrossRef] [Green Version]
- Pelkonen, A.; Hiltunen, M.; Kiianmaa, K.; Yavich, L. Stimulated dopamine overflow and alpha-synuclein expression in the nucleus accumbens core distinguish rats bred for differential ethanol preference. J. Neurochem. 2010, 114, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Edenberg, H.J.; Gelernter, J.; Agrawal, A. Genetics of Alcoholism. Curr. Psychiatry Rep. 2019, 21, 26. [Google Scholar] [CrossRef] [Green Version]
- Edenberg, H.J.; Foroud, T. The genetics of alcoholism: Identifying specific genes through family studies. Addict. Biol. 2006, 11, 386–396. [Google Scholar] [CrossRef]
- Rosborough, K.; Patel, N.; Kalia, L.V. Alpha-Synuclein and Parkinsonism: Updates and Future Perspectives. Curr. Neurol. Neurosci. Rep. 2017, 17, 31. [Google Scholar] [CrossRef]
- Pang, S.Y.; Ho, P.W.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef]
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2, a009258. [Google Scholar] [CrossRef] [Green Version]
- Skogar, O.; Nilsson, M. Distribution of non-motor symptoms in idiopathic Parkinson’s disease and secondary Parkinsonism. J. Multidiscip. Healthc. 2018, 11, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, A.K.; Lofving, S.; Callaghan, R.C.; Allebeck, P. Alcohol use disorders and risk of Parkinson’s disease: Findings from a Swedish national cohort study 1972–2008. BMC Neurol. 2013, 13, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Guo, X.; Park, Y.; Wang, J.; Huang, X.; Hollenbeck, A.; Blair, A.; Chen, H. Alcohol Consumption, Types of Alcohol, and Parkinson’s Disease. PLoS ONE 2013, 8, e66452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Commenges, D.; Scotet, V.; Renaud, S.; Jacqmin-Gadda, H.; Barberger-Gateau, P.; Dartigues, J.F. Intake of flavonoids and risk of dementia. Eur. J. Epidemiol. 2000, 16, 357–363. [Google Scholar] [CrossRef] [PubMed]
- de Gaetano, G.; Costanzo, S.; Di Castelnuovo, A.; Badimon, L.; Bejko, D.; Alkerwi, A.; Chiva-Blanch, G.; Estruch, R.; La Vecchia, C.; Panico, S.; et al. Effects of moderate beer consumption on health and disease: A consensus document. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 443–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neiman, J.; Lang, A.E.; Fornazzari, L.; Carlen, P.L. Movement disorders in alcoholism: A review. Neurology 1990, 40, 741–746. [Google Scholar] [CrossRef]
- Carlen, P.L.; Wilkinson, D.A. Reversibility of alcohol-related brain damage: Clinical and experimental observations. Acta Med. Scand. Suppl. 1987, 717, 19–26. [Google Scholar] [CrossRef]
- Shandling, M.; Carlen, P.L.; Lang, A.E. Parkinsonism in alcohol withdrawal: A follow-up study. Mov. Disord. 1990, 5, 36–39. [Google Scholar] [CrossRef]
- Carlen, P.L.; Lee, M.A.; Jacob, M.; Livshits, O. Parkinsonism provoked by alcoholism. Ann. Neurol. 1981, 9, 84–86. [Google Scholar] [CrossRef]
- Lang, A.E.; Marsden, C.D.; Obeso, J.A.; Parkes, J.D. Alcohol and Parkinson disease. Ann. Neurol. 1982, 12, 254–256. [Google Scholar] [CrossRef]
- Buervenich, S.; Sydow, O.; Carmine, A.; Zhang, Z.; Anvret, M.; Olson, L. Alcohol dehydrogenase alleles in Parkinson’s disease. Mov. Disord. 2000, 15, 813–818. [Google Scholar] [CrossRef]
- Buervenich, S.; Carmine, A.; Galter, D.; Shahabi, H.N.; Johnels, B.; Holmberg, B.; Ahlberg, J.; Nissbrandt, H.; Eerola, J.; Hellstrom, O.; et al. A rare truncating mutation in ADH1C (G78Stop) shows significant association with Parkinson disease in a large international sample. Arch. Neurol. 2005, 62, 74–78. [Google Scholar] [CrossRef] [Green Version]
- Hafer, G.; Agarwal, D.P.; Goedde, H.W. Human brain aldehyde dehydrogenase: Activity with DOPAL and isozyme distribution. Alcohol 1987, 4, 413–418. [Google Scholar] [CrossRef]
- Jinsmaa, Y.; Sullivan, P.; Gross, D.; Cooney, A.; Sharabi, Y.; Goldstein, D.S. Divalent metal ions enhance DOPAL-induced oligomerization of alpha-synuclein. Neurosci. Lett. 2014, 569, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisaglia, M.; Tessari, I.; Mammi, S.; Bubacco, L. Interaction between alpha-synuclein and metal ions, still looking for a role in the pathogenesis of Parkinson’s disease. Neuromol. Med. 2009, 11, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Sullivan, P.; Holmes, C.; Kopin, I.J.; Sharabi, Y.; Mash, D.C. Decreased vesicular storage and aldehyde dehydrogenase activity in multiple system atrophy. Parkinsonism Relat. Disord. 2015, 21, 567–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunblatt, E.; Riederer, P. Aldehyde dehydrogenase (ALDH) in Alzheimer’s and Parkinson’s disease. J. Neural Transm. 2016, 123, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yu, J.; Ding, J.; Xie, C.; Sun, L.; Rudenko, I.; Zheng, W.; Sastry, N.; Luo, J.; Rudow, G.; et al. Aldehyde dehydrogenase 1 defines and protects a nigrostriatal dopaminergic neuron subpopulation. J. Clin. Investig. 2014, 124, 3032–3046. [Google Scholar] [CrossRef]
- Liljequist, S. Changes in the sensitivity of dopamine receptors in the nucleus accumbens and in the striatum induced by chronic ethanol administration. Acta Pharmacol. Toxicol. 1978, 43, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Balldin, J.; Alling, C.; Gottfries, C.G.; Lindstedt, G.; Langstrom, G. Changes in dopamine receptor sensitivity in humans after heavy alcohol intake. Psychopharmacology 1985, 86, 142–146. [Google Scholar] [CrossRef]
- Engelhard, B.; Finkelstein, J.; Cox, J.; Fleming, W.; Jang, H.J.; Ornelas, S.; Koay, S.A.; Thiberge, S.Y.; Daw, N.D.; Tank, D.W.; et al. Specialized coding of sensory, motor and cognitive variables in VTA dopamine neurons. Nature 2019, 570, 509–513. [Google Scholar] [CrossRef]
- Boileau, I.; Assaad, J.M.; Pihl, R.O.; Benkelfat, C.; Leyton, M.; Diksic, M.; Tremblay, R.E.; Dagher, A. Alcohol promotes dopamine release in the human nucleus accumbens. Synapse 2003, 49, 226–231. [Google Scholar] [CrossRef]
- Weiss, F.; Lorang, M.T.; Bloom, F.E.; Koob, G.F. Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: Genetic and motivational determinants. J. Pharmacol. Exp. Ther. 1993, 267, 250–258. [Google Scholar] [PubMed]
- Wu, G.; Tonner, P.H.; Miller, K.W. Ethanol stabilizes the open channel state of the Torpedo nicotinic acetylcholine receptor. Mol. Pharmacol. 1994, 45, 102–108. [Google Scholar]
- Larsson, A.; Edstrom, L.; Svensson, L.; Soderpalm, B.; Engel, J.A. Voluntary ethanol intake increases extracellular acetylcholine levels in the ventral tegmental area in the rat. Alcohol Alcohol. 2005, 40, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Brodie, M.S.; Appel, S.B. Ethanol inhibition of m-current and ethanol-induced direct excitation of ventral tegmental area dopamine neurons. J. Neurophysiol. 2007, 97, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Bacopoulos, N.G.; Bhatnagar, R.K.; Van Orden, L.S. The effects of subhypnotic doses of ethanol on regional catecholamine turnover. J. Pharmacol. Exp. Ther. 1978, 204, 1–10. [Google Scholar] [PubMed]
- Bacopoulos, N.G.; Bize, I.; Levine, J.; Van Orden, L.S., 3rd. Modification of ethanol intoxication by dopamine agonists and antagonists. Psychopharmacology 1979, 60, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Darden, J.H.; Hunt, W.A. Reduction of striatal dopamine release during an ethanol withdrawal syndrome. J. Neurochem. 1977, 29, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Neiman, J.; Borg, S.; Wahlund, L.O. Parkinsonism and dyskinesias during ethanol withdrawal. Br. J. Addict. 1988, 83, 437–439. [Google Scholar] [CrossRef]
- Gepshtein, S.; Li, X.; Snider, J.; Plank, M.; Lee, D.; Poizner, H. Dopamine function and the efficiency of human movement. J. Cogn. Neurosci. 2014, 26, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Beyer, K.; Ariza, A. Alpha-Synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Mol. Neurobiol. 2013, 47, 509–524. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. Alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitan, K.; Chereau, D.; Cohen, S.I.; Knowles, T.P.; Dobson, C.M.; Fink, A.L.; Anderson, J.P.; Goldstein, J.M.; Millhauser, G.L. Conserved C-terminal charge exerts a profound influence on the aggregation rate of alpha-synuclein. J. Mol. Biol. 2011, 411, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Beyer, K.; Humbert, J.; Ferrer, A.; Lao, J.I.; Carrato, C.; Lopez, D.; Ferrer, I.; Ariza, A. Low alpha-synuclein 126 mRNA levels in dementia with Lewy bodies and Alzheimer disease. Neuroreport 2006, 17, 1327–1330. [Google Scholar] [CrossRef]
- Beyer, K.; Domingo-Sabat, M.; Lao, J.I.; Carrato, C.; Ferrer, I.; Ariza, A. Identification and characterization of a new alpha-synuclein isoform and its role in Lewy body diseases. Neurogenetics 2008, 9, 15–23. [Google Scholar] [CrossRef]
- Murray, I.V.; Giasson, B.I.; Quinn, S.M.; Koppaka, V.; Axelsen, P.H.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 2003, 42, 8530–8540. [Google Scholar] [CrossRef]
- Janeczek, P.; Brooker, C.; Dodd, P.R.; Lewohl, J.M. Differential expression of alpha-synuclein splice variants in the brain of alcohol misusers: Influence of genotype. Drug Alcohol Depend. 2015, 155, 284–292. [Google Scholar] [CrossRef]
- Brighina, L.; Schneider, N.K.; Lesnick, T.G.; de Andrade, M.; Cunningham, J.M.; Mrazek, D.; Rocca, W.A.; Maraganore, D.M. Alpha-synuclein, alcohol use disorders, and Parkinson disease: A case-control study. Parkinsonism Relat. Disord. 2009, 15, 430–434. [Google Scholar] [CrossRef] [Green Version]
- Chiba-Falek, O.; Nussbaum, R.L. Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum. Mol. Genet. 2001, 10, 3101–3109. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Zhang, Y.; Sun, Q.; Pan, H.; Guo, J.; Tang, B. SNCA REP1 and Parkinson’s disease. Neurosci. Lett. 2018, 682, 79–84. [Google Scholar] [CrossRef]
- Janeczek, P.; Lewohl, J.M. The role of alpha-synuclein in the pathophysiology of alcoholism. Neurochem. Int. 2013, 63, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Bonsch, D.; Lenz, B.; Kornhuber, J.; Bleich, S. DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism. Neuroreport 2005, 16, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Marchese, D.; Botta-Orfila, T.; Cirillo, D.; Rodriguez, J.; Livi, C.; Fernández-Santiago, R.; Ezquerra, M.; Martí, M.; Bechara, E.; Tartaglia, G.; et al. Discovering the 3′ UTR-mediated regulation of alpha-synuclein. Nucleic Acids Res. 2017, 45, 12888–12903. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Saitoh, T.; Ueda, K.; Tanaka, S.; Chen, X.; Hashimoto, M.; Hsu, L.; Conrad, C.; Sundsmo, M.; Yoshimoto, M.; et al. Characterization of the human alpha-synuclein gene: Genomic structure, transcription start site, promoter region and polymorphisms. J. Alzheimers Dis. 2001, 3, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Friedlich, A.L.; Tanzi, R.E.; Rogers, J.T. The 5′-untranslated region of Parkinson’s disease alpha-synuclein messengerRNA contains a predicted iron responsive element. Mol. Psychiatry 2007, 12, 222–223. [Google Scholar] [CrossRef] [PubMed]
- Koukouraki, P.; Doxakis, E. Constitutive translation of human alpha-synuclein is mediated by the 5′-untranslated region. Open Biol. 2016, 6, 160022. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Wetherill, L.; Bucholz, K.K.; Kramer, J.; Kuperman, S.; Lynskey, M.T.; Nurnberger, J.I., Jr.; Schuckit, M.; Tischfield, J.A.; Edenberg, H.J.; et al. Genetic influences on craving for alcohol. Addict. Behav. 2013, 38, 1501–1508. [Google Scholar] [CrossRef] [Green Version]
- Soldner, F.; Stelzer, Y.; Shivalila, C.S.; Abraham, B.J.; Latourelle, J.C.; Barrasa, M.I.; Goldmann, J.; Myers, R.H.; Young, R.A.; Jaenisch, R. Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 2016, 533, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Kariks, J. Extensive damage to substantia nigra in chronic alcoholics. Med. J. Aust. 1978, 2, 628–629. [Google Scholar] [CrossRef]
- Crews, F.T.; Collins, M.A.; Dlugos, C.; Littleton, J.; Wilkins, L.; Neafsey, E.J.; Pentney, R.; Snell, L.D.; Tabakoff, B.; Zou, J.; et al. Alcohol-induced neurodegeneration: When, where and why? Alcohol. Clin. Exp. Res. 2004, 28, 350–364. [Google Scholar] [CrossRef]
- Pfefferbaum, A.; Sullivan, E.V.; Rosenbloom, M.J.; Mathalon, D.H.; Lim, K.O. A controlled study of cortical gray matter and ventricular changes in alcoholic men over a 5-year interval. Arch. Gen. Psychiatry 1998, 55, 905–912. [Google Scholar] [CrossRef]
- Brown, D.R. α-Synuclein as a ferrireductase. Biochem. Soc. Trans. 2013, 41, 1513–1517. [Google Scholar] [CrossRef] [Green Version]
- McDowall, J.S.; Ntai, I.; Hake, J.; Whitley, P.R.; Mason, J.M.; Pudney, C.R.; Brown, D.R. Steady-State Kinetics of alpha-Synuclein Ferrireductase Activity Identifies the Catalytically Competent Species. Biochemistry 2017, 56, 2497–2505. [Google Scholar] [CrossRef]
- Ortega, R.; Carmona, A.; Roudeau, S.; Perrin, L.; Ducic, T.; Carboni, E.; Bohic, S.; Cloetens, P.; Lingor, P. Alpha-Synuclein Over-Expression Induces Increased Iron Accumulation and Redistribution in Iron-Exposed Neurons. Mol. Neurobiol. 2016, 53, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Munro, H.N. Translation of ferritin light and heavy subunit mRNAs is regulated by intracellular chelatable iron levels in rat hepatoma cells. Proc. Natl. Acad. Sci. USA 1987, 84, 2277–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, J. APP and Ferritin Translation, Metals and Alzheimer’s Disease. J. Am. Chem. Soc. 2006, 13, 215–251. [Google Scholar]
- Zhou, Z.D.; Tan, E.K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Bush, A.I.; Cho, H.H.; Smith, D.H.; Thomson, A.M.; Friedlich, A.L.; Lahiri, D.K.; Leedman, P.J.; Huang, X.; Cahill, C.M. Iron and the translation of the amyloid precursor protein (APP) and ferritin mRNAs: Riboregulation against neural oxidative damage in Alzheimer’s disease. Biochem. Soc. Trans. 2008, 36 Pt 6, 1282–1287. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.H.; Cahill, C.M.; Vanderburg, C.R.; Scherzer, C.R.; Wang, B.; Huang, X.; Rogers, J.T. Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J. Biol. Chem. 2010, 285, 31217–31232. [Google Scholar] [CrossRef] [Green Version]
- Ross, N.T.; Metkar, S.R.; Le, H.; Burbank, J.; Cahill, C.; Germain, A.; MacPherson, L.; Bittker, J.; Palmer, M.; Rogers, J.; et al. Identification of a small molecule that selectively inhibits alpha-synuclein translational expression. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Harrison-Findik, D.D. Role of alcohol in the regulation of iron metabolism. World J. Gastroenterol. 2007, 13, 4925–4930. [Google Scholar] [CrossRef]
- Harrison-Findik, D.D.; Klein, E.; Crist, C.; Evans, J.; Timchenko, N.; Gollan, J. Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology 2007, 46, 1979–1985. [Google Scholar] [CrossRef]
- Cui, J.; Guo, X.; Li, Q.; Song, N.; Xie, J. Hepcidin-to-Ferritin Ratio Is Decreased in Astrocytes with Extracellular Alpha-Synuclein and Iron Exposure. Front. Cell. Neurosci. 2020, 14, 47. [Google Scholar] [PubMed]
- Klos, K.J.; Ahlskog, J.E.; Kumar, N.; Cambern, S.; Butz, J.; Burritt, M.; Fealey, R.D.; Cowl, C.T.; Parisi, J.E.; Josephs, K.A. Brain metal concentrations in chronic liver failure patients with pallidal T1 MRI hyperintensity. Neurology 2006, 67, 1984–1989. [Google Scholar] [CrossRef] [PubMed]
- Chiou, B.; Lucassen, E.; Sather, M.; Kallianpur, A.; Connor, J. Semaphorin4A and H-ferritin utilize Tim-1 on human oligodendrocytes: A novel neuro-immune axis. Glia 2018, 66, 1317–1330. [Google Scholar]
- Kim, Y.; Bowler, R.M.; Abdelouahab, N.; Harris, M.; Gocheva, V.; Roels, H.A. Motor function in adults of an Ohio community with environmental manganese exposure. Neurotoxicology 2011, 32, 606–614. [Google Scholar] [CrossRef]
- Kim, Y.; Jeong, K.S.; Song, H.J.; Lee, J.J.; Seo, J.H.; Kim, G.C.; Lee, H.J.; Kim, H.J.; Ahn, J.H.; Park, S.J.; et al. Altered white matter microstructural integrity revealed by voxel-wise analysis of diffusion tensor imaging in welders with manganese exposure. Neurotoxicology 2011, 32, 100–109. [Google Scholar] [CrossRef]
- Butterworth, R.F. Parkinsonism in cirrhosis: Pathogenesis and current therapeutic options. Metab. Brain Dis. 2013, 28, 261–267. [Google Scholar]
- Tryc, A.B.; Goldbecker, A.; Berding, G.; Rumke, S.; Afshar, K.; Shahrezaei, G.H.; Pflugrad, H.; Barg-Hock, H.; Strassburg, C.P.; Hecker, H.; et al. Cirrhosis-related Parkinsonism: Prevalence, mechanisms and response to treatments. J. Hepatol. 2013, 58, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Doeppner, T.R.; Willkommen, D.; Cahill, C.M.; Xin, Y.; Ye, G.; Liu, Y.; Southon, A.; Aron, A.; Au-Yeung, H.Y.; et al. Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H-Ferritin. J. Neurochem. 2018, 147, 831–848. [Google Scholar]
- Wang, T.Y.; Ma, Z.; Wang, C.; Liu, C.; Yan, D.Y.; Deng, Y.; Liu, W.; Xu, Z.F.; Xu, B. Manganese-induced alpha-synuclein overexpression impairs synaptic vesicle fusion by disrupting the Rab3 cycle in primary cultured neurons. Toxicol. Lett. 2018, 285, 34–42. [Google Scholar] [CrossRef]
- Steimle, B.L.; Smith, F.M.; Kosman, D.J. The solute carriers ZIP8 and ZIP14 regulate manganese accumulation in brain microvascular endothelial cells and control brain manganese levels. J. Biol. Chem. 2019, 294, 19197–19208. [Google Scholar] [CrossRef]
- Rodd, Z.A.; Bertsch, B.A.; Strother, W.N.; Le-Niculescu, H.; Balaraman, Y.; Hayden, E.; Jerome, R.E.; Lumeng, L.; Nurnberger, J.I., Jr.; Edenberg, H.J.; et al. Candidate genes, pathways and mechanisms for alcoholism: An expanded convergent functional genomics approach. Pharm. J. 2007, 7, 222–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golka, K.; Sondermann, R.; Reich, S.E.; Wiese, A. Carbohydrate-deficient transferrin (CDT) as a biomarker in persons suspected of alcohol abuse. Toxicol. Lett. 2004, 151, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Golka, K.; Wiese, A. Carbohydrate-deficient transferrin (CDT)—A biomarker for long-term alcohol consumption. J. Toxicol. Environ. Health B Crit. Rev. 2004, 7, 319–337. [Google Scholar] [CrossRef]
- Baksi, S.; Singh, N. Alpha-Synuclein impairs ferritinophagy in the retinal pigment epithelium: Implications for retinal iron dyshomeostasis in Parkinson’s disease. Sci. Rep. 2017, 7, 12843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baksi, S.; Tripathi, A.K.; Singh, N. Alpha-synuclein modulates retinal iron homeostasis by facilitating the uptake of transferrin-bound iron: Implications for visual manifestations of Parkinson’s disease. Free Radic. Biol. Med. 2016, 97, 292–306. [Google Scholar] [CrossRef] [Green Version]
- Juhas, M.; Sun, H.; Brown, M.R.G.; MacKay, M.B.; Mann, K.F.; Sommer, W.H.; Wilman, A.H.; Dursun, S.M.; Greenshaw, A.J. Deep grey matter iron accumulation in alcohol use disorder. Neuroimage 2017, 148, 115–122. [Google Scholar] [CrossRef]
- Harper, C.G.; Kril, J.J. Corpus callosal thickness in alcoholics. Br. J. Addict. 1988, 83, 577–580. [Google Scholar] [CrossRef]
- Harper, C.G.; Kril, J.J.; Daly, J.M. Brain shrinkage in alcoholics is not caused by changes in hydration: A pathological study. J. Neurol. Neurosurg. Psychiatry 1988, 51, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Cahill, C.M.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim. Biophys. Acta 2009, 1790, 615–628. [Google Scholar] [CrossRef] [Green Version]
- Rogers, J.T.; Mikkilineni, S.; Cantuti-Castelvetri, I.; Smith, D.H.; Huang, X.; Bandyopadhyay, S.; Cahill, C.M.; Maccecchini, M.L.; Lahiri, D.K.; Greig, N.H. The alpha-synuclein 5′untranslated region targeted translation blockers: Anti-alpha synuclein efficacy of cardiac glycosides and Posiphen. J. Neural Transm. (Vienna) 2011, 118, 493–507. [Google Scholar] [CrossRef]
- Zhang, P.; Park, H.J.; Zhang, J.; Junn, E.; Andrews, R.J.; Velagapudi, S.P.; Abegg, D.; Vishnu, K.; Costales, M.G.; Childs-Disney, J.L.; et al. Translation of the intrinsically disordered protein alpha-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl. Acad. Sci. USA 2020, 117, 1457–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkilineni, S.; Cantuti-Castelvetri, I.; Cahill, C.M.; Balliedier, A.; Greig, N.H.; Rogers, J.T. The anticholinesterase phenserine and its enantiomer posiphen as 5′ untranslated-region-directed translation blockers of the Parkinson’s alpha synuclein expression. Parkinson’s Dis. 2012, 2012, 142372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, N.T.; Metkar, S.R.; Le, H.; Burbank, J.; Cahill, C.; Germain, A.; MacPherson, L.; Bittker, J.; Palmer, M.; Rogers, J.; et al. Identification of a small molecule that selectively activates alpha-synuclein translational expression. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Kim, T.W.; Koo, S.Y.; Studer, L. Pluripotent Stem Cell Therapies for Parkinson Disease: Present Challenges and Future Opportunities. Front. Cell Dev. Biol. 2020, 8, 729. [Google Scholar] [CrossRef]
- Barker, R.A.; Carpenter, M.K.; Forbes, S.; Goldman, S.A.; Jamieson, C.; Murry, C.E.; Takahashi, J.; Weir, G. The Challenges of First-in-Human Stem Cell Clinical Trials: What Does This Mean for Ethics and Institutional Review Boards? Stem Cell Rep. 2018, 10, 1429–1431. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human Trials of Stem Cell-Derived Dopamine Neurons for Parkinson’s Disease: Dawn of a New Era. Cell Stem Cell 2017, 21, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.H.; Kim, H.N.; Park, H.J.; Shin, J.Y.; Kim, D.Y.; Lee, P.H. The Cleavage Effect of Mesenchymal Stem Cell and Its Derived Matrix Metalloproteinase-2 on Extracellular alpha-Synuclein Aggregates in Parkinsonian Models. Stem Cells Transl. Med. 2017, 6, 949–961. [Google Scholar] [CrossRef]
- Oh, S.H.; Lee, S.C.; Kim, D.Y.; Kim, H.N.; Shin, J.Y.; Ye, B.S.; Lee, P.H. Mesenchymal Stem Cells Stabilize Axonal Transports for Autophagic Clearance of alpha-Synuclein in Parkinsonian Models. Stem Cells 2017, 35, 1934–1947. [Google Scholar] [CrossRef] [Green Version]
- Ezquer, F.; Quintanilla, M.E.; Morales, P.; Ezquer, M.; Lespay-Rebolledo, C.; Herrera-Marschitz, M.; Israel, Y. Activated mesenchymal stem cell administration inhibits chronic alcohol drinking and suppresses relapse-like drinking in high-alcohol drinker rats. Addict. Biol. 2019, 24, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Ezquer, F.; Morales, P.; Quintanilla, M.E.; Santapau, D.; Lespay-Rebolledo, C.; Ezquer, M.; Herrera-Marschitz, M.; Israel, Y. Intravenous administration of anti-inflammatory mesenchymal stem cell spheroids reduces chronic alcohol intake and abolishes binge-drinking. Sci. Rep. 2018, 8, 4325. [Google Scholar] [CrossRef] [Green Version]
- Perez-Villalba, A.; Sirerol-Piquer, M.S.; Belenguer, G.; Soriano-Canton, R.; Munoz-Manchado, A.B.; Villadiego, J.; Alarcon-Aris, D.; Soria, F.N.; Dehay, B.; Bezard, E.; et al. Synaptic Regulator alpha-Synuclein in Dopaminergic Fibers Is Essentially Required for the Maintenance of Subependymal Neural Stem Cells. J. Neurosci. 2018, 38, 814–825. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Vivacqua, G.; Yu, S. The role of alpha-synuclein in neurotransmission and synaptic plasticity. J. Chem. Neuroanat. 2011, 42, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Regensburger, M.; Schreglmann, S.; Boyer, L.; Prots, I.; Rockenstein, E.; Mante, M.; Zhao, C.; Winkler, J.; Masliah, E.; et al. Role of alpha-synuclein in adult neurogenesis and neuronal maturation in the dentate gyrus. J. Neurosci. 2012, 32, 16906–16916. [Google Scholar] [CrossRef]
- Schlachetzki, J.C.; Grimm, T.; Schlachetzki, Z.; Ben Abdallah, N.M.; Ettle, B.; Vohringer, P.; Ferger, B.; Winner, B.; Nuber, S.; Winkler, J. Dopaminergic lesioning impairs adult hippocampal neurogenesis by distinct modification of alpha-synuclein. J. Neurosci. Res. 2016, 94, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Moualla, D.; Brown, D.R. Alpha-synuclein is a cellular ferrireductase. PLoS ONE 2011, 6, e15814. [Google Scholar] [CrossRef]
- Hidese, S.; Saito, K.; Asano, S.; Kunugi, H. Association between iron-deficiency anemia and depression: A web-based Japanese investigation. Psychiatry Clin. Neurosci. 2018, 72, 513–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zeng, Y.N.; Yang, P.; Jin, L.Q.; Xiong, W.C.; Zhu, M.Z.; Zhang, J.Z.; He, X.; Zhu, X.H. Axonal iron transport in the brain modulates anxiety-related behaviors. Nat. Chem. Biol. 2019, 15, 1214–1222. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cahill, C.M.; Aleyadeh, R.; Gao, J.; Wang, C.; Rogers, J.T. Alpha-Synuclein in Alcohol Use Disorder, Connections with Parkinson’s Disease and Potential Therapeutic Role of 5’ Untranslated Region-Directed Small Molecules. Biomolecules 2020, 10, 1465. https://doi.org/10.3390/biom10101465
Cahill CM, Aleyadeh R, Gao J, Wang C, Rogers JT. Alpha-Synuclein in Alcohol Use Disorder, Connections with Parkinson’s Disease and Potential Therapeutic Role of 5’ Untranslated Region-Directed Small Molecules. Biomolecules. 2020; 10(10):1465. https://doi.org/10.3390/biom10101465
Chicago/Turabian StyleCahill, Catherine M., Rozaleen Aleyadeh, Jin Gao, Changning Wang, and Jack T. Rogers. 2020. "Alpha-Synuclein in Alcohol Use Disorder, Connections with Parkinson’s Disease and Potential Therapeutic Role of 5’ Untranslated Region-Directed Small Molecules" Biomolecules 10, no. 10: 1465. https://doi.org/10.3390/biom10101465
APA StyleCahill, C. M., Aleyadeh, R., Gao, J., Wang, C., & Rogers, J. T. (2020). Alpha-Synuclein in Alcohol Use Disorder, Connections with Parkinson’s Disease and Potential Therapeutic Role of 5’ Untranslated Region-Directed Small Molecules. Biomolecules, 10(10), 1465. https://doi.org/10.3390/biom10101465