Robust Cell-Free Expression of Sub-Pathological and Pathological Huntingtin Exon-1 for NMR Studies. General Approaches for the Isotopic Labeling of Low-Complexity Proteins

 ,
,  ,
, 
Abstract
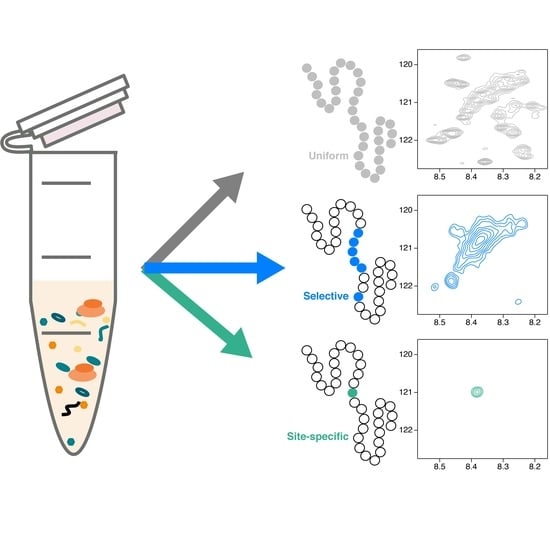
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. DNA Constructs
2.2. Expression, Purification and Activation of EF-Tu
2.3. Lysate Preparation and CF Expression Conditions
2.3.1. Lysate Preparation
2.3.2. Standard Batch Mode CF Expression Conditions
2.3.3. Optimization of CF Suppression Conditions
2.3.4. Uniform and Selective Labeling in CF Expression Systems
2.3.5. Site-Specific Isotopic Labeling in CF Expression Systems
2.3.6. Addition of EF-Tu to CF Expression and CF Suppression Reactions
2.4. EF-Tu:aa-tRNACUA Stability Assay
2.5. NMR Experiments
3. Results
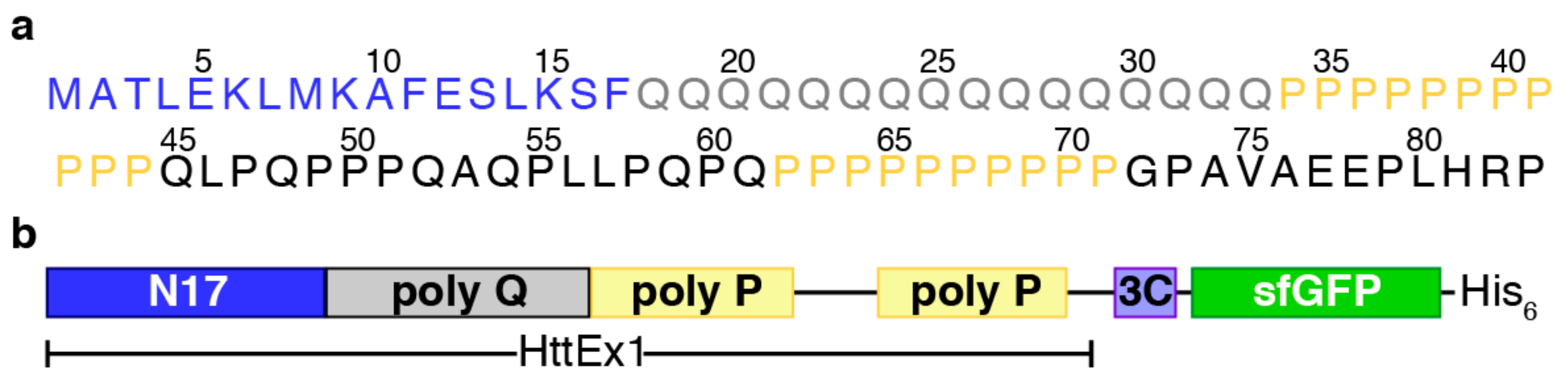
3.1. An Optimal Huntingtin Exon-1 Construct for CF Expression
3.2. Optimization of CF Expression Conditions
3.3. Optimization of the Glutamine Concentration
3.4. Uniform Isotopic Labeling of Huntingtin
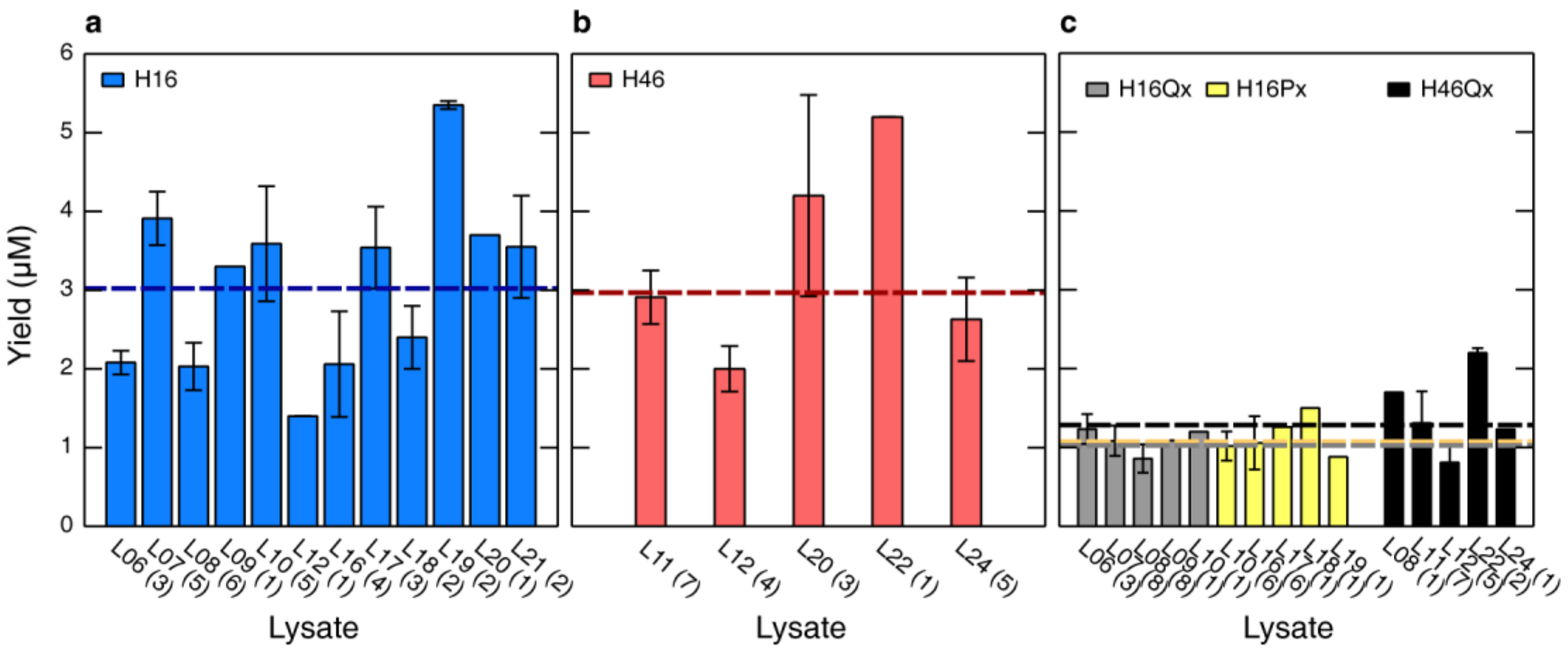
3.5. HttEx1 Production Variability Across Different Lysates
3.6. CF Synthesis as a Flexible Platform for Tailored Isotopically Labeled HttEx1 Samples
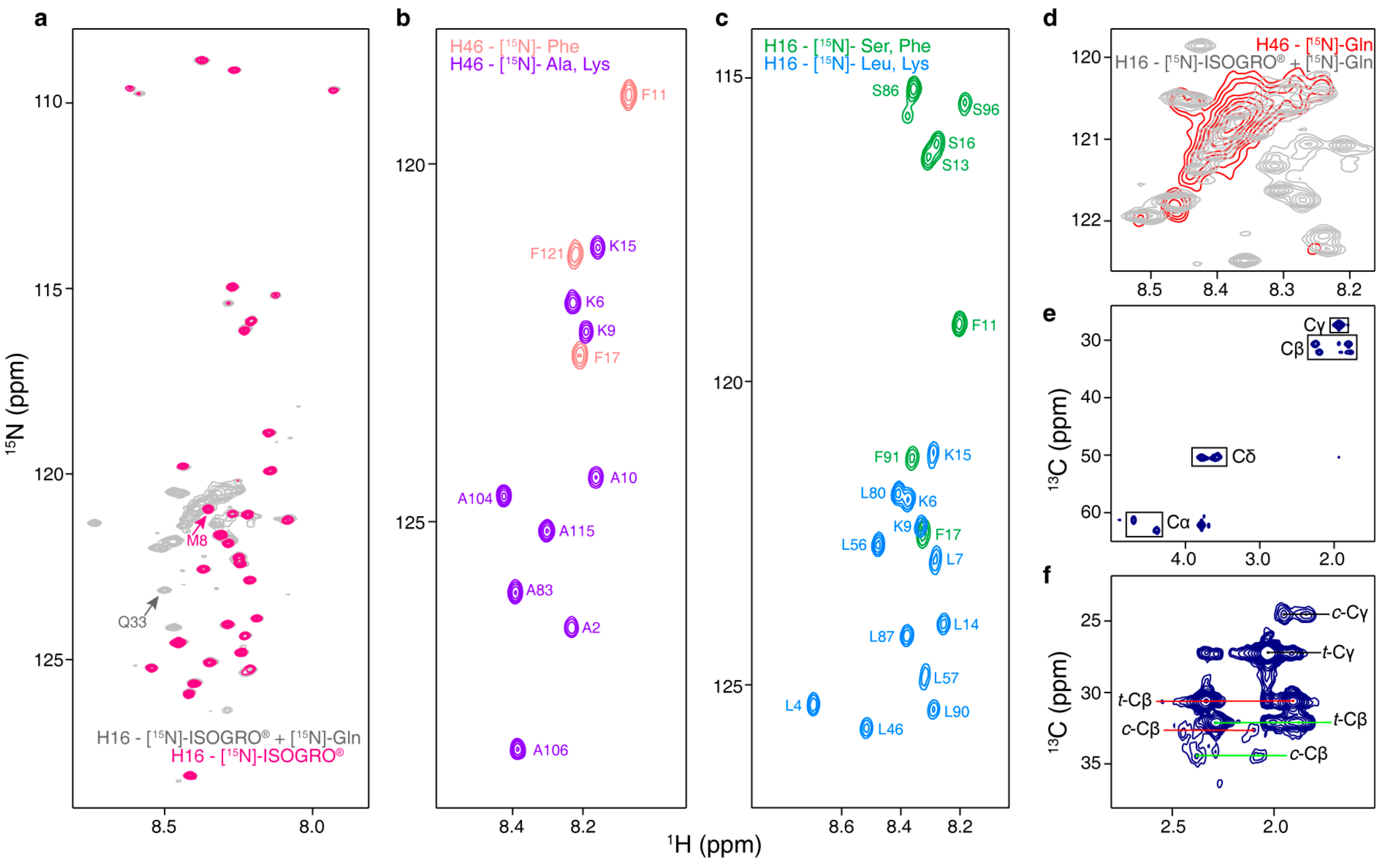
3.7. Glutamine and Proline Site-Specific Isotopic Labeling
3.8. EF-Tu as an Additive for CF Protein Production
3.9. Reproducibility of SSIL in CF Reactions
3.10. NMR on Site-Specific Isotopically Labeled Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benn, C.L.; Landles, C.; Li, H.; Strand, A.D.; Woodman, B.; Sathasivam, K.; Li, S.-H.; Ghazi-Noori, S.; Hockly, E.; Faruque, S.M.N.N.; et al. Contribution of nuclear and extranuclear polyQ to neurological phenotypes in mouse models of Huntington’s disease. Hum. Mol. Genet. 2005, 14, 3065–3078. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.K.; Deng, Y.; Slow, E.J.; Haigh, B.; Bissada, N.; Lu, G.; Pearson, J.; Shehadeh, J.; Bertram, L.; Murphy, Z.; et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006, 125, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon I of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Masino, L.; Kelly, G.; Leonard, K.; Trottier, Y.; Pastore, A. Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett. 2002, 513, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Monsellier, E.; Redeker, V.; Ruiz-Arlandis, G.; Bousset, L.; Melki, R. Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation. J. Biol. Chem. 2015, 290, 2560–2576. [Google Scholar] [CrossRef] [Green Version]
- Isas, J.M.; Langen, R.; Siemer, A.B. Solid-state nuclear magnetic resonance on the static and dynamic domains of huntingtin exon-1 fibrils. Biochemistry 2015, 54, 3942–3949. [Google Scholar] [CrossRef] [Green Version]
- Baias, M.; Smith, P.E.S.; Shen, K.; Joachimiak, L.A.; Żerko, S.; Koźmiński, W.; Frydman, J.; Frydman, L. Structure and dynamics of the huntingtin exon-1 N-terminus: A solution NMR perspective. J. Am. Chem. Soc. 2017, 139, 1168–1176. [Google Scholar] [CrossRef]
- Newcombe, E.A.; Ruff, K.M.; Sethi, A.; Ormsby, A.R.; Ramdzan, Y.M.; Fox, A.; Purcell, A.W.; Gooley, P.R.; Pappu, R.V.; Hatters, D.M. Tadpole-like conformations of huntingtin exon 1 are characterized by conformational heterogeneity that persists regardless of polyglutamine length. J. Mol. Biol. 2018, 430, 1442–1458. [Google Scholar] [CrossRef]
- Peskett, T.R.; Rau, F.; O’Driscoll, J.; Patani, R.; Lowe, A.R.; Saibil, H.R. A Liquid to Solid Phase Transition Underlying Pathological Huntingtin Exon1 Aggregation. Mol. Cell 2018, 70, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Calamini, B.; Fauerbach, J.A.; Ma, B.; Shahmoradian, S.H.; Serrano Lachapel, I.L.; Chiu, W.; Lo, D.C.; Frydman, J. Control of the structural landscape and neuronal proteotoxicity of mutant huntingtin by domains flanking the polyQ tract. Elife 2016, 5, e18065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherzinger, E.; Lurz, R.; Turmaine, M.; Mangiarini, L.; Hollenbach, B.; Hasenbank, R.; Bates, G.P.; Davies, S.W.; Lehrach, H.; Wanker, E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997, 90, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Thakur, A.K.; Jayaraman, M.; Mishra, R.; Thakur, M.; Chellgren, V.M.; Byeon, I.-J.L.; Anjum, D.H.; Kodali, R.; Creamer, T.P.; Conway, J.F.; et al. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 2009, 16, 380–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, A.; Thakur, A.K.; Chellgren, V.M.; Thiagarajan, G.; Williams, A.D.; Chellgren, B.W.; Creamer, T.P.; Wetzel, R. Oligoproline effects on polyglutamine conformation and aggregation. J. Mol. Biol. 2006, 355, 524–535. [Google Scholar] [CrossRef]
- Dehay, B.; Bertolotti, A. Critical role of the proline-rich region in huntingtin for aggregation and cytotoxicity in yeast. J. Biol. Chem. 2006, 281, 35608–35615. [Google Scholar] [CrossRef] [Green Version]
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Slepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.-Z.; Cattaneo, E.; et al. SUMO modification of huntingtin and huntington’s disease pathology. Science 2004, 304, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Duennwald, M.L.; Jagadish, S.; Muchowski, P.J.; Lindquist, S. Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc. Natl. Acad. Sci. USA 2006, 103, 11045–11050. [Google Scholar] [CrossRef] [Green Version]
- Urbanek, A.; Popovic, M.; Morató, A.; Estaña, A.; Elena-Real, C.A.; Mier, P.; Fournet, A.; Allemand, F.; Delbecq, S.; Andrade-Navarro, M.A.; et al. Flanking Regions Determine the Structure of the Poly-Glutamine in Huntingtin through Mechanisms Common among Glutamine-Rich Human Proteins. Structure 2020, 28, 733–746.e5. [Google Scholar] [CrossRef]
- Kim, M.W.; Chelliah, Y.; Kim, S.W.; Otwinowski, Z.; Bezprozvanny, I. Secondary structure of huntingtin amino-terminal region. Structure 2009, 17, 1205–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, B.; Arduini, I.; Drombosky, K.W.; Kodali, R.; Sanders, L.H.; Greenamyre, J.T.; Wetzel, R. Folding Landscape of Mutant Huntingtin Exon1: Diffusible Multimers, Oligomers and Fibrils, and No Detectable Monomer. PLoS ONE 2016, 11, e0155747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.J.; Huey-Tubman, K.E.; Herr, A.B.; West, A.P.; Ross, S.A.; Bjorkman, P.J. A linear lattice model for polyglutamine in CAG-expansion diseases. Proc. Natl. Acad. Sci. USA 2002, 99, 11634–11639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Huang, B.; Cheng, J.; Seefelder, M.; Engler, T.; Pfeifer, G.; Oeckl, P.; Otto, M.; Moser, F.; Maurer, M.; et al. The cryo-electron microscopy structure of huntingtin. Nature 2018, 555, 117–120. [Google Scholar] [CrossRef]
- Urbanek, A.; Morató, A.; Allemand, F.; Delaforge, E.; Fournet, A.; Popovic, M.; Delbecq, S.; Sibille, N.; Bernadó, P. A general strategy to access structural information at atomic resolution in polyglutamine homorepeats. Angew. Chem. Int. Ed. Engl. 2018, 57, 3598–3601. [Google Scholar] [CrossRef] [Green Version]
- Escobedo, A.; Chiesa, G.; Salvatella, X. Recombinant Production of Monomeric Isotope-Enriched Aggregation-Prone Peptides: Polyglutamine Tracts and Beyond. In Intrinsically Disordered Proteins. Methods in Molecular Biology; Kragelund, B., Skriver, K., Eds.; Humana Press: New York, NY, USA, 2020; Volume 2141, pp. 211–231. ISBN 9781071605240. [Google Scholar]
- Urbanek, A.; Elena-Real, C.A.; Popovic, M.; Morató, A.; Fournet, A.; Allemand, F.; Delbecq, S.; Sibille, N.; Bernadó, P. Site-specific isotopic labeling (SSIL): Access to high-resolution structural and dynamic information in low-complexity proteins. Chembiochem 2020, 21, 769–775. [Google Scholar] [CrossRef]
- Kigawa, T.; Yabuki, T.; Yoshida, Y.; Tsutsui, M.; Ito, Y.; Shibata, T.; Yokoyama, S. Cell-free production and stable-isotope labeling of milligram quantities of proteins. FEBS Lett. 1999, 442, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, K.; Headlam, M.J.; Schaeffer, P.M.; Henderson, B.R.; Dixon, N.E.; Otting, G. Optimization of an Escherichia coli system for cell-free synthesis of selectively 15N-labelled proteins for rapid analysis by NMR spectroscopy. Eur. J. Biochem. 2004, 271, 4084–4093. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Z.; Qiao, X.; Li, J.; Shu, X.; Qi, H. Emerging Methods for Efficient and Extensive Incorporation of Non-canonical Amino Acids Using Cell-Free Systems. Front. Bioeng. Biotechnol. 2020, 8, 863. [Google Scholar] [CrossRef]
- Wu, P.S.C.; Ozawa, K.; Lim, S.P.; Vasudevan, S.G.; Dixon, N.E.; Otting, G. Cell-free transcription/translation from PCR-amplified DNA for high-throughput NMR studies. Angew. Chem. Int. Ed. Engl. 2007, 46, 3356–3358. [Google Scholar] [CrossRef]
- Dopp, J.L.; Rothstein, S.M.; Mansell, T.J.; Reuel, N.F. Rapid prototyping of proteins: Mail order gene fragments to assayable proteins within 24 h. Biotechnol. Bioeng. 2019, 116, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, G.; Goto, M.; Saeki, M.; Ito, K.; Hori, T.; Kigawa, T.; Shirouzu, M.; Yokoyama, S. Expression of G protein coupled receptors in a cell-free translational system using detergents and thioredoxin-fusion vectors. Protein Expr. Purif. 2005, 41, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Klammt, C.; Schwarz, D.; Fendler, K.; Haase, W.; Dötsch, V.; Bernhard, F. Evaluation of detergents for the soluble expression of alpha-helical and beta-barrel-type integral membrane proteins by a preparative scale individual cell-free expression system. FEBS J. 2005, 272, 6024–6038. [Google Scholar] [CrossRef] [PubMed]
- Kai, L.; Dötsch, V.; Kaldenhoff, R.; Bernhard, F. Artificial environments for the co-translational stabilization of cell-free expressed proteins. PLoS ONE 2013, 8, e56637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kainosho, M.; Torizawa, T.; Iwashita, Y.; Terauchi, T.; Mei Ono, A.; Güntert, P. Optimal isotope labelling for NMR protein structure determinations. Nature 2006, 440, 52–57. [Google Scholar] [CrossRef]
- Torizawa, T.; Shimizu, M.; Taoka, M.; Miyano, H.; Kainosho, M. Efficient production of isotopically labeled proteins by cell-free synthesis: A practical protocol. J. Biomol. NMR 2004, 30, 311–325. [Google Scholar] [CrossRef]
- Linser, R.; Gelev, V.; Hagn, F.; Arthanari, H.; Hyberts, S.G.; Wagner, G. Selective methyl labeling of eukaryotic membrane proteins using cell-free expression. J. Am. Chem. Soc. 2014, 136, 11308–11310. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, J.; Matsuda, T.; Koshiba, S.; Kigawa, T. An economical method for producing stable-isotope labeled proteins by the E. coli cell-free system. J. Biomol. NMR 2010, 48, 193–201. [Google Scholar] [CrossRef]
- Loscha, K.V.; Herlt, A.J.; Qi, R.; Huber, T.; Ozawa, K.; Otting, G. Multiple-site labeling of proteins with unnatural amino acids. Angew. Chem. Int. Ed. Engl. 2012, 51, 2243–2246. [Google Scholar] [CrossRef]
- Ozawa, K.; Dixon, N.E.; Otting, G. Cell-free synthesis of 15N-labeled proteins for NMR studies. IUBMB Life 2005, 57, 615–622. [Google Scholar] [CrossRef]
- Noren, C.J.; Anthony-Cahill, S.J.; Griffith, M.C.; Schultz, P.G. A general method for site-specific incorporation of unnatural amino acids into proteins. Science 1989, 244, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Schultz, P.G. At the Interface of Chemical and Biological Synthesis: An Expanded Genetic Code. Cold Spring Harb. Perspect. Biol. 2016, 8, a023945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mier, P.; Paladin, L.; Tamana, S.; Petrosian, S.; Hajdu-Soltész, B.; Urbanek, A.; Gruca, A.; Plewczynski, D.; Grynberg, M.; Bernadó, P.; et al. Disentangling the complexity of low complexity proteins. Brief. Bioinform. 2019, 72, 137–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, B.; Fernandes, S.; Abreu, I.A.; Macedo-Ribeiro, S. Trinucleotide repeats: A structural perspective. Front. Neurol. 2013, 4, 76. [Google Scholar] [CrossRef] [Green Version]
- Pak, C.W.; Kosno, M.; Holehouse, A.S.; Padrick, S.B.; Mittal, A.; Ali, R.; Yunus, A.A.; Liu, D.R.; Pappu, R.V.; Rosen, M.K. Sequence Determinants of Intracellular Phase Separation by Complex Coacervation of a Disordered Protein. Mol. Cell 2016, 63, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Choi, J.M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699.e16. [Google Scholar] [CrossRef] [Green Version]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Nazarenko, I.A.; Harrington, K.M.; Uhlenbeck, O.C. Many of the conserved nucleotides of tRNA(Phe) are not essential for ternary complex formation and peptide elongation. EMBO J. 1994, 13, 2464–2471. [Google Scholar] [CrossRef]
- Pleiss, J.A.; Uhlenbeck, O.C. Identification of thermodynamically relevant interactions between EF-Tu and backbone elements of tRNA. J. Mol. Biol. 2001, 308, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Apponyi, M.A.; Ozawa, K.; Dixon, N.E.; Otting, G. Cell-free protein synthesis for analysis by NMR spectroscopy. In Structural Proteomics. Methods in Molecular Biology™; Kobe, B., Guss, M., Huber, T., Eds.; Humana Press: New York, NY, USA, 2008; Volume 426, pp. 257–268. ISBN 978-1-60327-058-8. [Google Scholar]
- Urbanek, A.; Popovic, M.; Elena-Real, C.A.; Morató, A.; Estaña, A.; Fournet, A.; Allemand, F.; Gil, A.M.; Cativiela, C.; Cortés, J.; et al. Evidence of the Reduced Abundance of Proline cis Conformation in Protein Poly Proline Tracts. J. Am. Chem. Soc. 2020, 142, 7976–7986. [Google Scholar] [CrossRef] [Green Version]
- Kigawa, T.; Yabuki, T.; Matsuda, N.; Matsuda, T.; Nakajima, R.; Tanaka, A.; Yokoyama, S. Preparation of Escherichia coli cell extract for highly productive cell-free protein expression. J. Struct. Funct. Genom. 2004, 5, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Kigawa, T.; Muto, Y.; Yokoyama, S. Cell-free synthesis and amino acid-selective stable isotope labeling of proteins for NMR analysis. J. Biomol. NMR 1995, 6, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Wu, P.S.C.; Dixon, N.E.; Otting, G. N-Labelled proteins by cell-free protein synthesis. Strategies for high-throughput NMR studies of proteins and protein-ligand complexes. FEBS J. 2006, 273, 4154–4159. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.E.; Fredrick, K. Preparation and evaluation of acylated tRNAs. Methods 2008, 44, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef]
- Markley, J.L.; Bax, A.; Arata, Y.; Hilbers, C.W.; Kaptein, R.; Sykes, B.D.; Wright, P.E.; Wüthrich, K. Recommendations for the presentation of NMR structures of proteins and nucleic acids. J. Mol. Biol. 1998, 280, 933–952. [Google Scholar] [CrossRef]
- Pédelacq, J.-D.; Cabantous, S.; Tran, T.; Terwilliger, T.C.; Waldo, G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006, 24, 79–88. [Google Scholar] [CrossRef]
- Borkowski, O.; Koch, M.; Zettor, A.; Pandi, A.; Batista, A.C.; Soudier, P.; Faulon, J.L. Large scale active-learning-guided exploration for in vitro protein production optimization. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Dopp, B.J.L.; Tamiev, D.D.; Reuel, N.F. Cell-free supplement mixtures: Elucidating the history and biochemical utility of additives used to support in vitro protein synthesis in E. coli extract. Biotechnol. Adv. 2019, 37, 246–258. [Google Scholar] [CrossRef]
- Yokoyama, J.; Matsuda, T.; Koshiba, S.; Tochio, N.; Kigawa, T. A practical method for cell-free protein synthesis to avoid stable isotope scrambling and dilution. Anal. Biochem. 2011, 411, 223–229. [Google Scholar] [CrossRef]
- Hoffmann, B.; Löhr, F.; Laguerre, A.; Bernhard, F.; Dötsch, V. Protein labeling strategies for liquid-state NMR spectroscopy using cell-free synthesis. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 105, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.D.; Beabout, K.; Turner, K.B.; Smith, Z.K.; Funk, V.L.; Harbaugh, S.V.; Liem, A.T.; Roth, P.A.; Geier, B.A.; Emanuel, P.A.; et al. Quantification of Interlaboratory Cell-Free Protein Synthesis Variability. ACS Synth. Biol. 2019, 8, 2080–2091. [Google Scholar] [CrossRef] [PubMed]
- Dopp, J.L.; Reuel, N.F. Process optimization for scalable E. coli extract preparation for cell-free protein synthesis. Biochem. Eng. J. 2018, 138, 21–28. [Google Scholar] [CrossRef]
- Kwon, Y.-C.; Jewett, M.C. High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Sci. Rep. 2015, 5, 8663. [Google Scholar] [CrossRef] [Green Version]
- Alderson, T.R.; Lee, J.H.; Charlier, C.; Ying, J.; Bax, A. Propensity for cis-proline formation in unfolded proteins. Chembiochem 2018, 19, 37–42. [Google Scholar] [CrossRef]
- Murrali, M.G.; Piai, A.; Bermel, W.; Felli, I.C.; Pierattelli, R. Proline fingerprint in intrinsically disordered proteins. Chembiochem 2018, 19, 1625–1629. [Google Scholar] [CrossRef]
- Mateos, B.; Conrad-Billroth, C.; Schiavina, M.; Beier, A.; Kontaxis, G.; Konrat, R.; Felli, I.C.; Pierattelli, R. The ambivalent role of proline residues in an intrinsically disordered protein: From disorder promoters to compaction facilitators. J. Mol. Biol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Schuler, B.; Lipman, E.A.; Steinbach, P.J.; Kumke, M.; Eaton, W.A. Polyproline and the “spectroscopic ruler” revisited with single-molecule fluorescence. Proc. Natl. Acad. Sci. USA 2005, 102, 2754–2759. [Google Scholar] [CrossRef] [Green Version]
- Kremer, B.; Goldberg, P.; Andrew, S.E.; Theilmann, J.; Telenius, H.; Zeisler, J.; Squitieri, F.; Lin, B.; Bassett, A.; Almqvist, E. A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 1994, 330, 1401–1406. [Google Scholar] [CrossRef]
- Darnell, G.; Orgel, J.P.R.O.; Pahl, R.; Meredith, S.C. Flanking polyproline sequences inhibit beta-sheet structure in polyglutamine segments by inducing PPII-like helix structure. J. Mol. Biol. 2007, 374, 688–704. [Google Scholar] [CrossRef]
- Darnell, G.D.; Derryberry, J.; Kurutz, J.W.; Meredith, S.C. Mechanism of cis-inhibition of polyQ fibrillation by polyP: PPII oligomers and the hydrophobic effect. Biophys. J. 2009, 97, 2295–2305. [Google Scholar] [CrossRef] [Green Version]
- Fei, J.; Wang, J.; Sternberg, S.H.; MacDougall, D.D.; Elvekrog, M.M.; Pulukkunat, D.K.; Englander, M.T.; Gonzalez, R.L. A highly purified, fluorescently labeled in vitro translation system for single-molecule studies of protein synthesis. Methods Enzymol. 2010, 472, 221–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freischmidt, A.; Meysing, M.; Liss, M.; Wagner, R.; Kalbitzer, H.R.; Horn, G. Limiting factors of the translation machinery. J. Biotechnol. 2010, 150, 44–50. [Google Scholar] [CrossRef]
- Foshag, D.; Henrich, E.; Hiller, E.; Schäfer, M.; Kerger, C.; Burger-Kentischer, A.; Diaz-Moreno, I.; García-Mauriño, S.M.; Dötsch, V.; Rupp, S.; et al. The E. coli S30 lysate proteome: A prototype for cell-free protein production. New Biotechnol. 2018, 40, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Bremer, H.; Dennis, P.P. Modulation of Chemical Composition and Other Parameters of the Cell at Different Exponential Growth Rates. EcoSal Plus 2008, 3. [Google Scholar] [CrossRef]
- Yabuki, T.; Kigawa, T.; Dohmae, N.; Takio, K.; Terada, T.; Ito, Y.; Laue, E.D.; Cooper, J.A.; Kainosho, M.; Yokoyama, S. Dual amino acid-selective and site-directed stable-isotope labeling of the human c-Ha-Ras protein by cell-free synthesis. J. Biomol. NMR 1998, 11, 295–306. [Google Scholar] [CrossRef]
- Ellman, J.A.; Volkman, B.F.; Mendel, D.; Schultz, P.G.; Wemmer, D.E. Site-specific isotopic labeling of proteins for NMR studies. J. Am. Chem. Soc. 1992, 114, 7959–7961. [Google Scholar] [CrossRef]
- Peuker, S.; Andersson, H.; Gustavsson, E.; Maiti, K.S.; Kania, R.; Karim, A.; Niebling, S.; Pedersen, A.; Erdelyi, M.; Westenhoff, S. Efficient isotope editing of proteins for site-directed vibrational spectroscopy. J. Am. Chem. Soc. 2016, 138, 2312–2318. [Google Scholar] [CrossRef]
- Escobedo, A.; Topal, B.; Kunze, M.B.A.; Aranda, J.; Chiesa, G.; Mungianu, D.; Bernardo-Seisdedos, G.; Eftekharzadeh, B.; Gairí, M.; Pierattelli, R.; et al. Side chain to main chain hydrogen bonds stabilize a polyglutamine helix in a transcription factor. Nat. Commun. 2019, 10, 2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorda, J.; Kajava, A. V Protein homorepeats. Adv. Protein Chem. Struct. Biol. 2010, 79, 59–88. [Google Scholar] [CrossRef]
- Chavali, S.; Chavali, P.L.; Chalancon, G.; de Groot, N.S.; Gemayel, R.; Latysheva, N.S.; Ing-Simmons, E.; Verstrepen, K.J.; Balaji, S.; Babu, M.M. Constraints and consequences of the emergence of amino acid repeats in eukaryotic proteins. Nat. Struct. Mol. Biol. 2017, 24, 765–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavali, S.; Singh, A.K.; Santhanam, B.; Babu, M.M. Amino acid homorepeats in proteins. Nat. Rev. Chem. 2020. [Google Scholar] [CrossRef]
- Spirin, A.S. Cell-Free Translation Systems; Spirin, A.S., Ed.; Springer: Berlin/Heidelberg, Germany, 2002; ISBN 978-3-642-63956-2. [Google Scholar]
- Bravo-Arredondo, J.M.; Kegulian, N.C.; Schmidt, T.; Pandey, N.K.; Situ, A.J.; Ulmer, T.S.; Langen, R. The folding equilibrium of huntingtin exon 1 monomer depends on its polyglutamine tract. J. Biol. Chem. 2018, 293, 19613–19623. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, Y.; Kanamori, T.; Ueda, T. Protein synthesis by pure translation systems. Methods 2005, 36, 299–304. [Google Scholar] [CrossRef]
- Lavickova, B.; Maerkl, S.J. A Simple, Robust, and Low-Cost Method to Produce the PURE Cell-Free System. ACS Synth. Biol. 2019, 8, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 1992, 31, 1647–1651. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D. The 13C chemical-shift index: A simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 1994, 4, 171–180. [Google Scholar] [CrossRef]
- Zhang, H.; Neal, S.; Wishart, D.S. RefDB: A database of uniformly referenced protein chemical shifts. J. Biomol. NMR 2003, 25, 173–195. [Google Scholar] [CrossRef] [PubMed]
- Tamiola, K.; Acar, B.; Mulder, F.A.A. Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J. Am. Chem. Soc. 2010, 132, 18000–18003. [Google Scholar] [CrossRef]
- Tamiola, K.; Mulder, F.A.A. Using NMR chemical shifts to calculate the propensity for structural order and disorder in proteins. Biochem. Soc. Trans. 2012, 40, 1014–1020. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morató, A.; Elena-Real, C.A.; Popovic, M.; Fournet, A.; Zhang, K.; Allemand, F.; Sibille, N.; Urbanek, A.; Bernadó, P. Robust Cell-Free Expression of Sub-Pathological and Pathological Huntingtin Exon-1 for NMR Studies. General Approaches for the Isotopic Labeling of Low-Complexity Proteins. Biomolecules 2020, 10, 1458. https://doi.org/10.3390/biom10101458
Morató A, Elena-Real CA, Popovic M, Fournet A, Zhang K, Allemand F, Sibille N, Urbanek A, Bernadó P. Robust Cell-Free Expression of Sub-Pathological and Pathological Huntingtin Exon-1 for NMR Studies. General Approaches for the Isotopic Labeling of Low-Complexity Proteins. Biomolecules. 2020; 10(10):1458. https://doi.org/10.3390/biom10101458
Chicago/Turabian StyleMorató, Anna, Carlos A. Elena-Real, Matija Popovic, Aurélie Fournet, Karen Zhang, Frédéric Allemand, Nathalie Sibille, Annika Urbanek, and Pau Bernadó. 2020. "Robust Cell-Free Expression of Sub-Pathological and Pathological Huntingtin Exon-1 for NMR Studies. General Approaches for the Isotopic Labeling of Low-Complexity Proteins" Biomolecules 10, no. 10: 1458. https://doi.org/10.3390/biom10101458
APA StyleMorató, A., Elena-Real, C. A., Popovic, M., Fournet, A., Zhang, K., Allemand, F., Sibille, N., Urbanek, A., & Bernadó, P. (2020). Robust Cell-Free Expression of Sub-Pathological and Pathological Huntingtin Exon-1 for NMR Studies. General Approaches for the Isotopic Labeling of Low-Complexity Proteins. Biomolecules, 10(10), 1458. https://doi.org/10.3390/biom10101458