Nonhistone Proteins HMGB1 and HMGB2 Differentially Modulate the Response of Human Embryonic Stem Cells and the Progenitor Cells to the Anticancer Drug Etoposide


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning of Inducible HMGB-shRNA Constructs
2.2. Generation of Stably Transfected hESCs with Inducible HMGB-shRNA Constructs
2.3. Human Neuroectodermal Lineage Commitment from hESCs
2.4. Crystal Violet Staining
2.5. Western Blotting
2.6. Cell Cycle Assay by Flow Cytometry
2.7. Immunocytochemistry
2.8. Confocal Microscopy
2.9. Annexin V-FITC/Propidium Iodide Double-Staining Assay
2.10. Cell Cycle and Apoptosis of Etoposide-Treated Cells
2.11. Image Acquisition and Analysis
2.12. Determination of Telomerase Activity by TRAP Assay
2.13. Statistical Analysis
3. Results
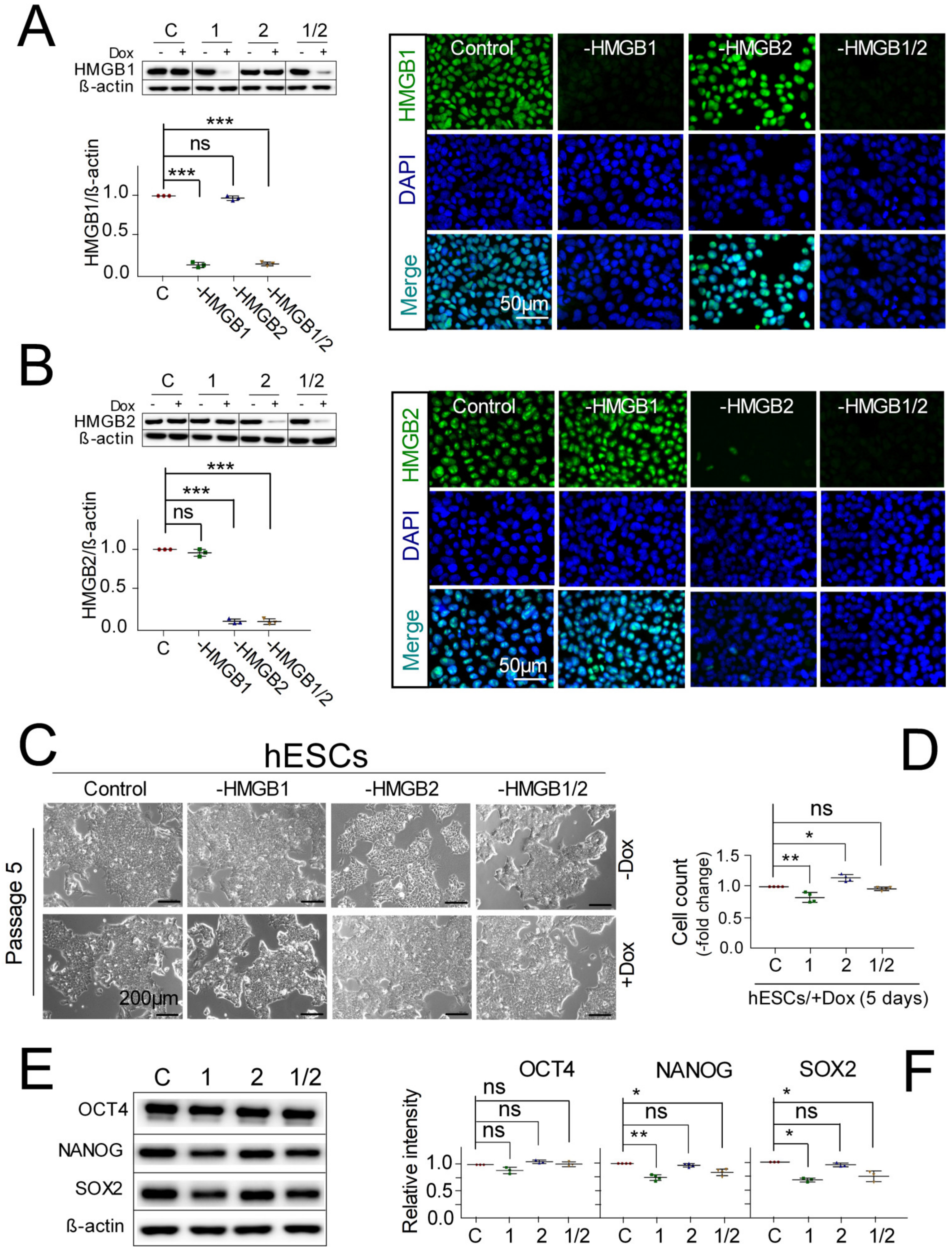
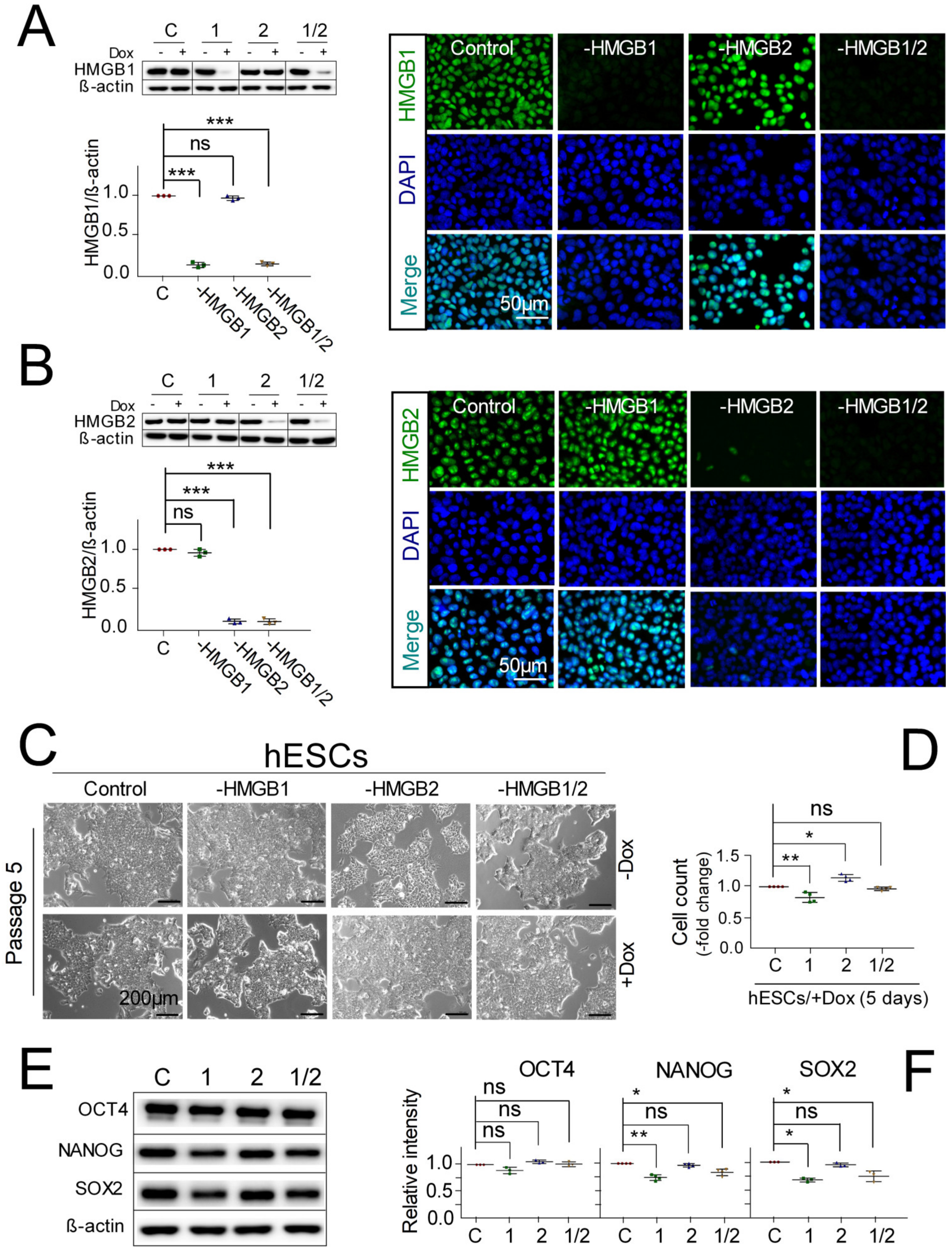
3.1. Pluripotent hESCs with Downregulated HMGB1/2 Expression Retain Stemness and Pluripotent State
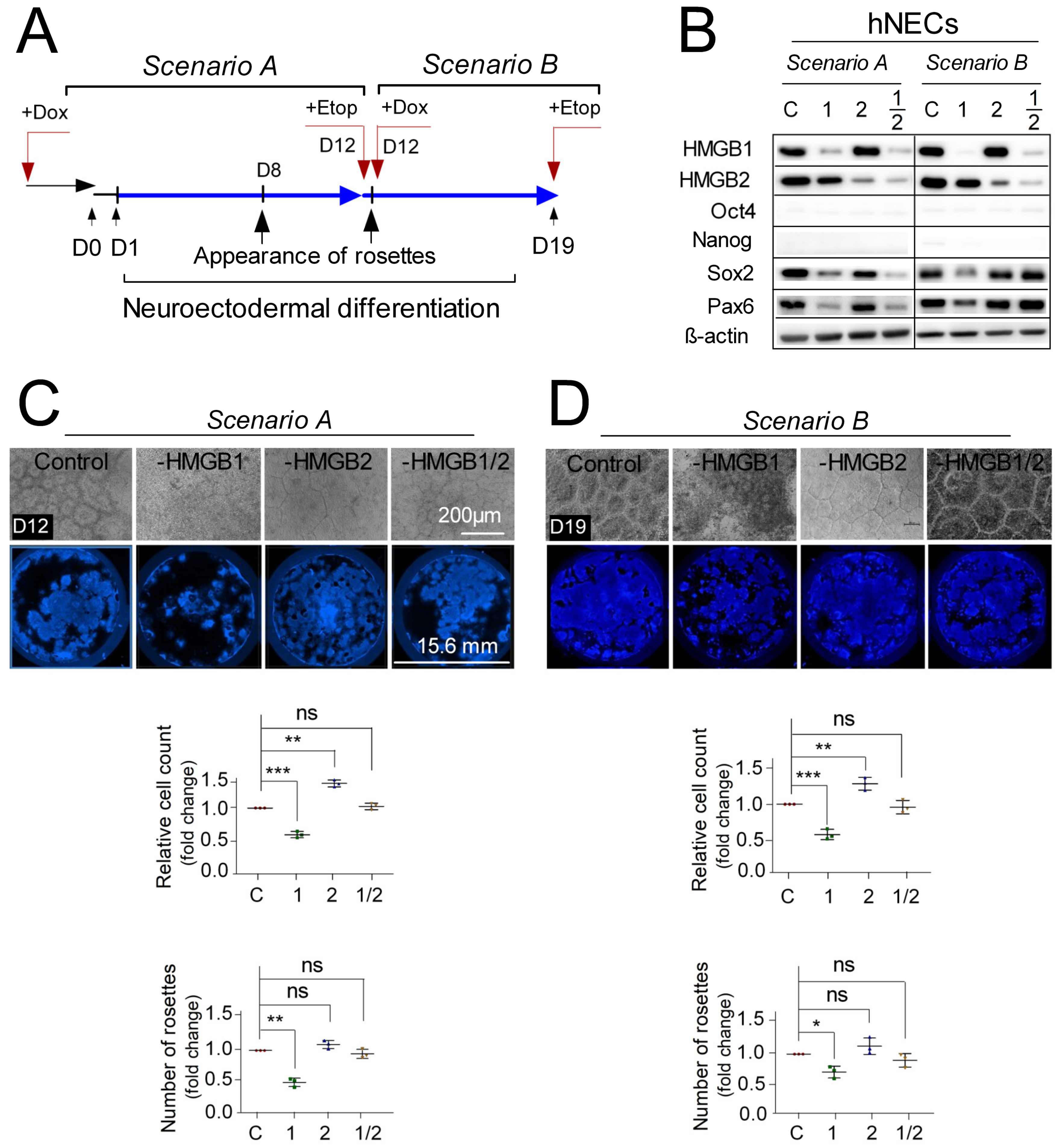
3.2. Distinct Impact of HMGB1/2 KD on Survival and Neuroectodermal Lineage Commitment
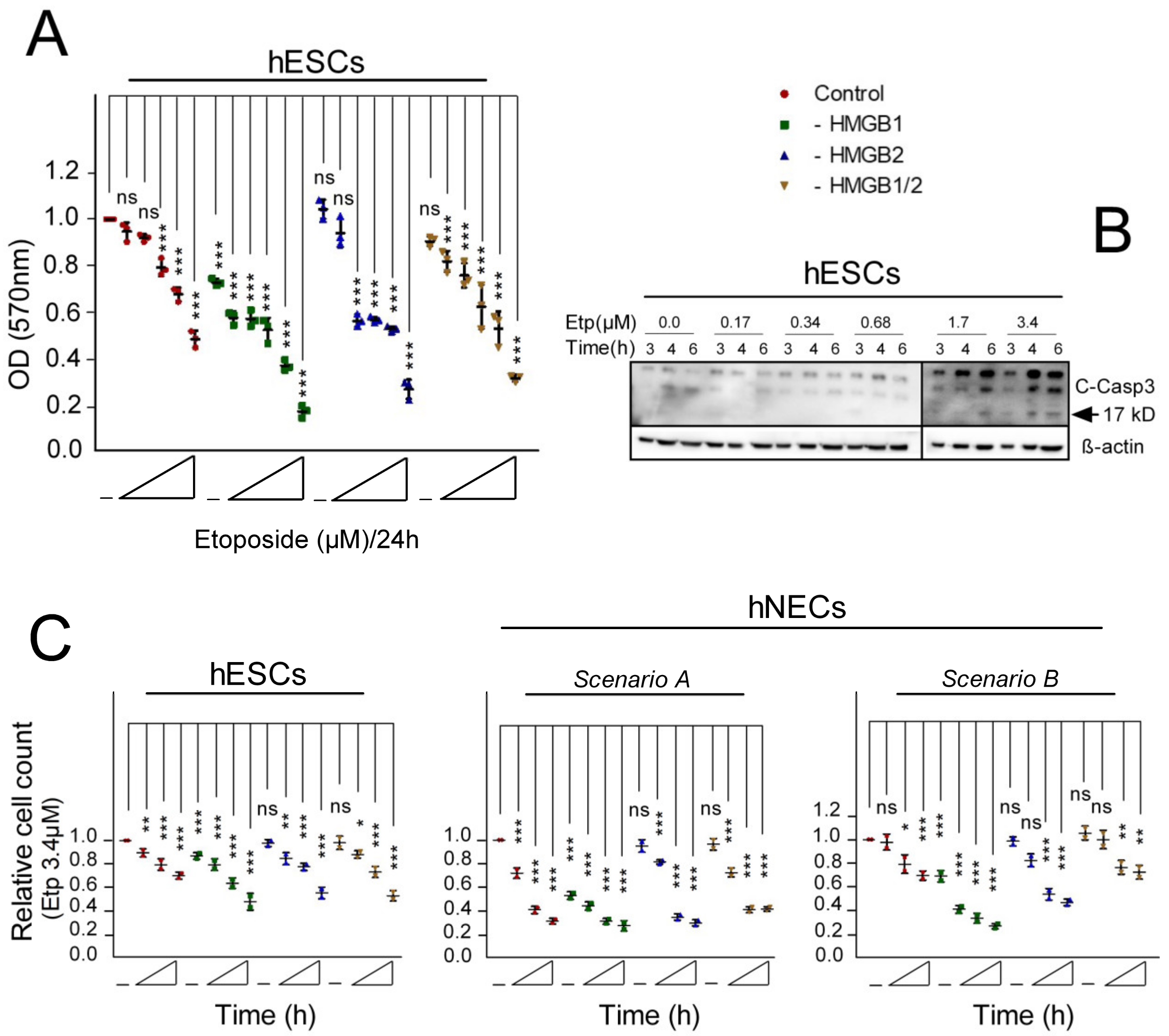
3.3. Pluripotent hESCs Exposed to Etoposide are more Susceptible to Cell Death than the hESC-Derived Progenitor Cells
3.4. Cell Cycle Progression in HMGB KD hESCs and hNECs Treated with Etoposide
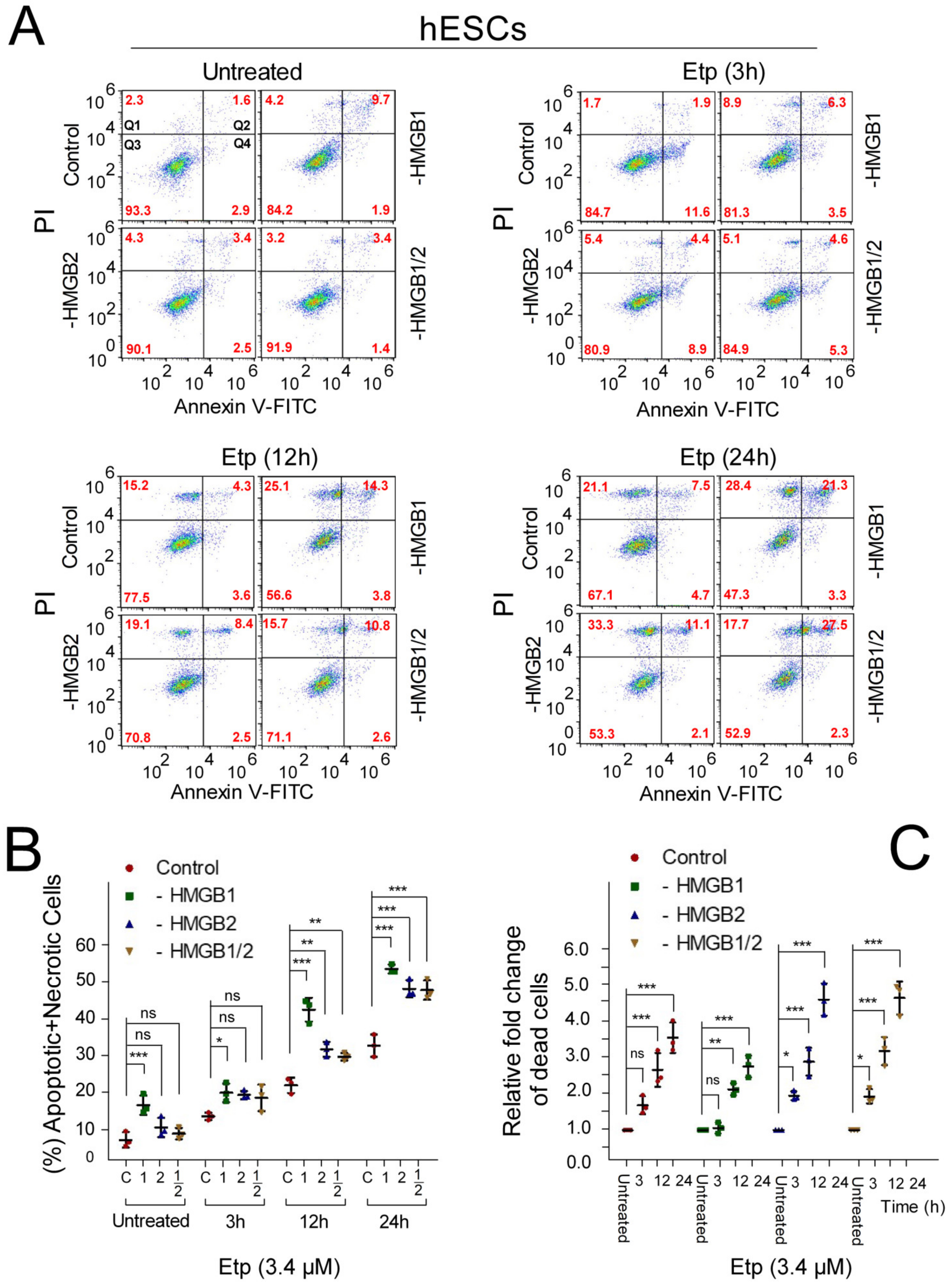
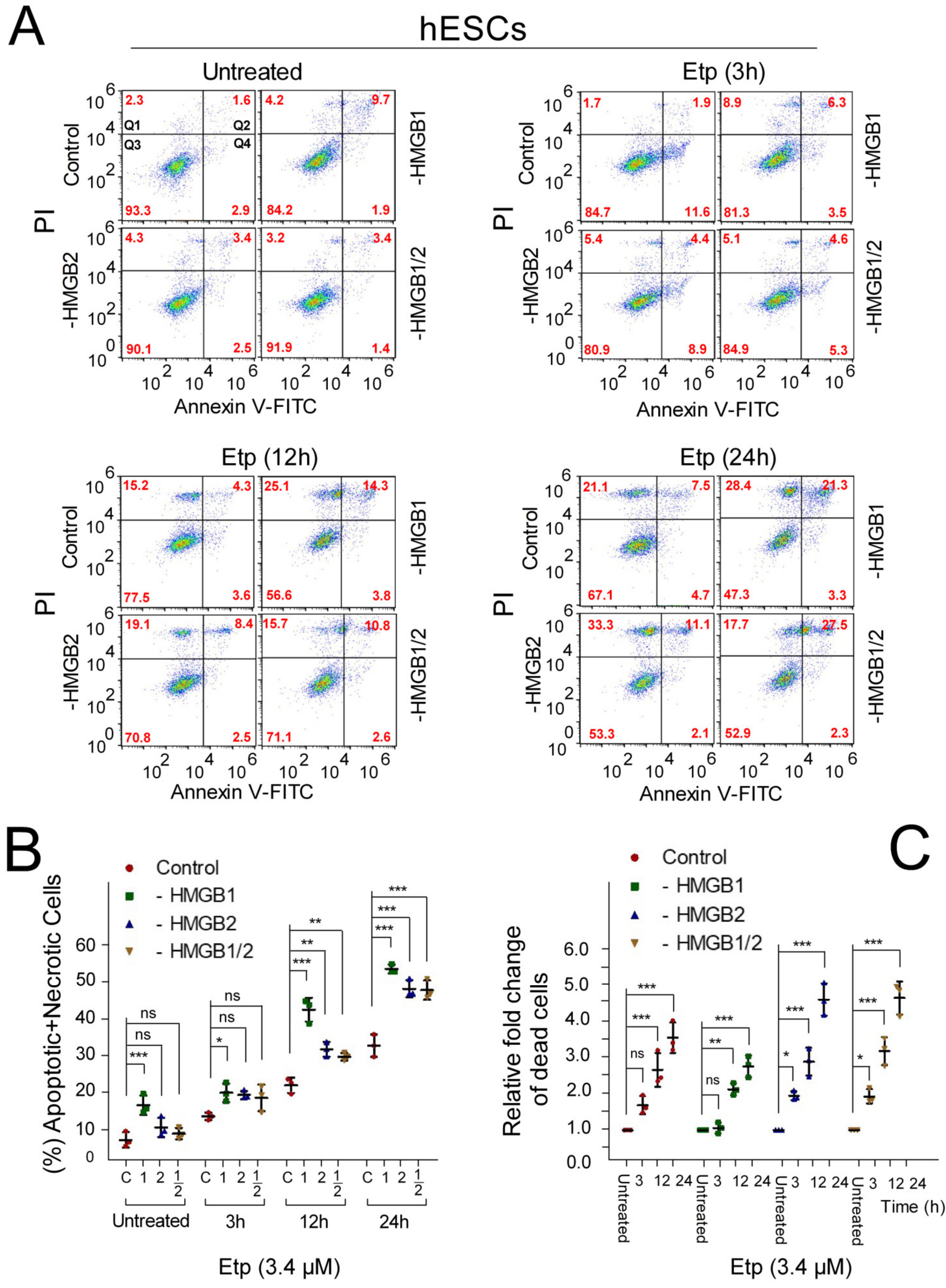
3.5. Apoptosis of Etoposide-Treated hESCs is Enhanced upon HMGB1 KD
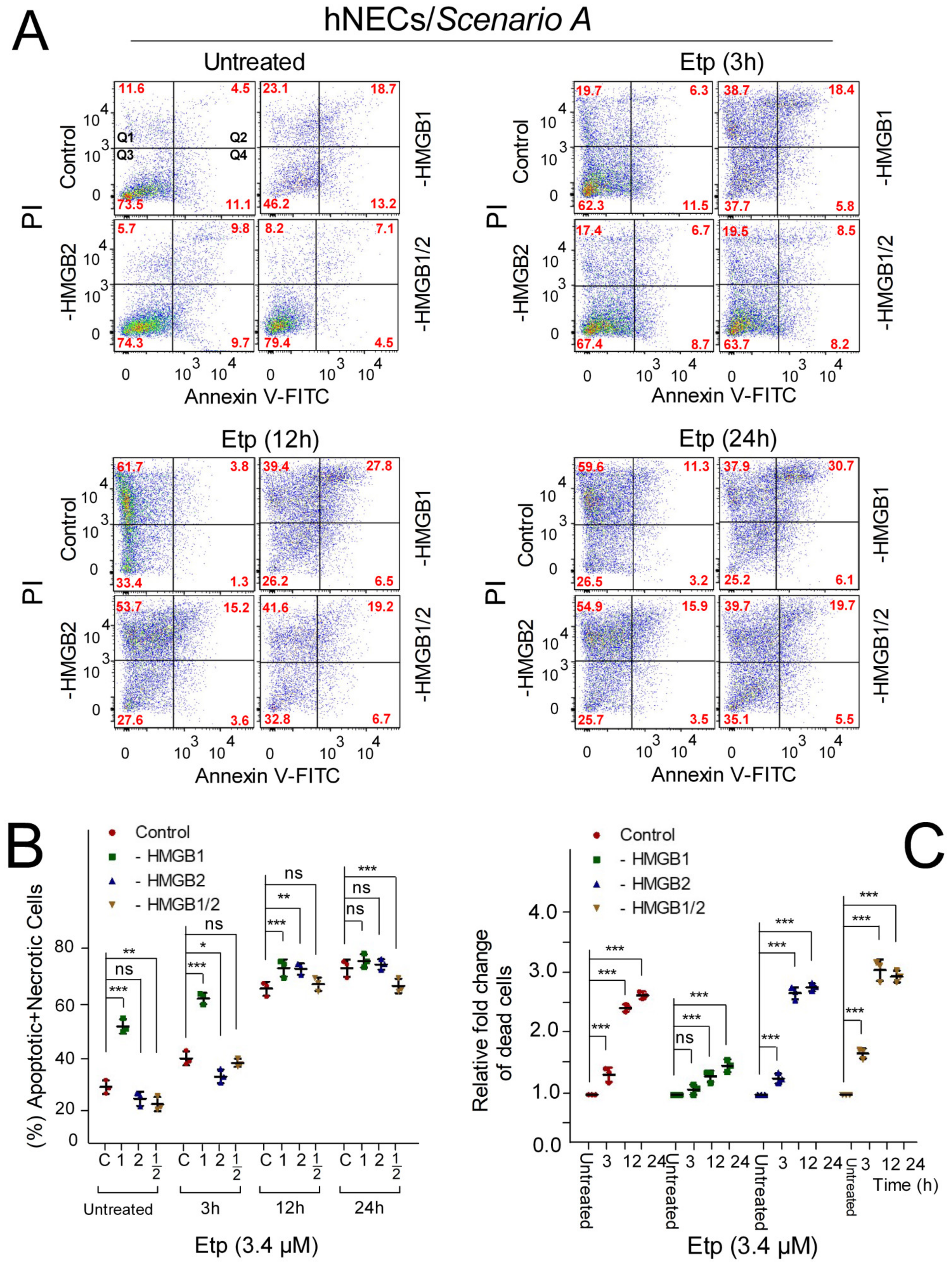
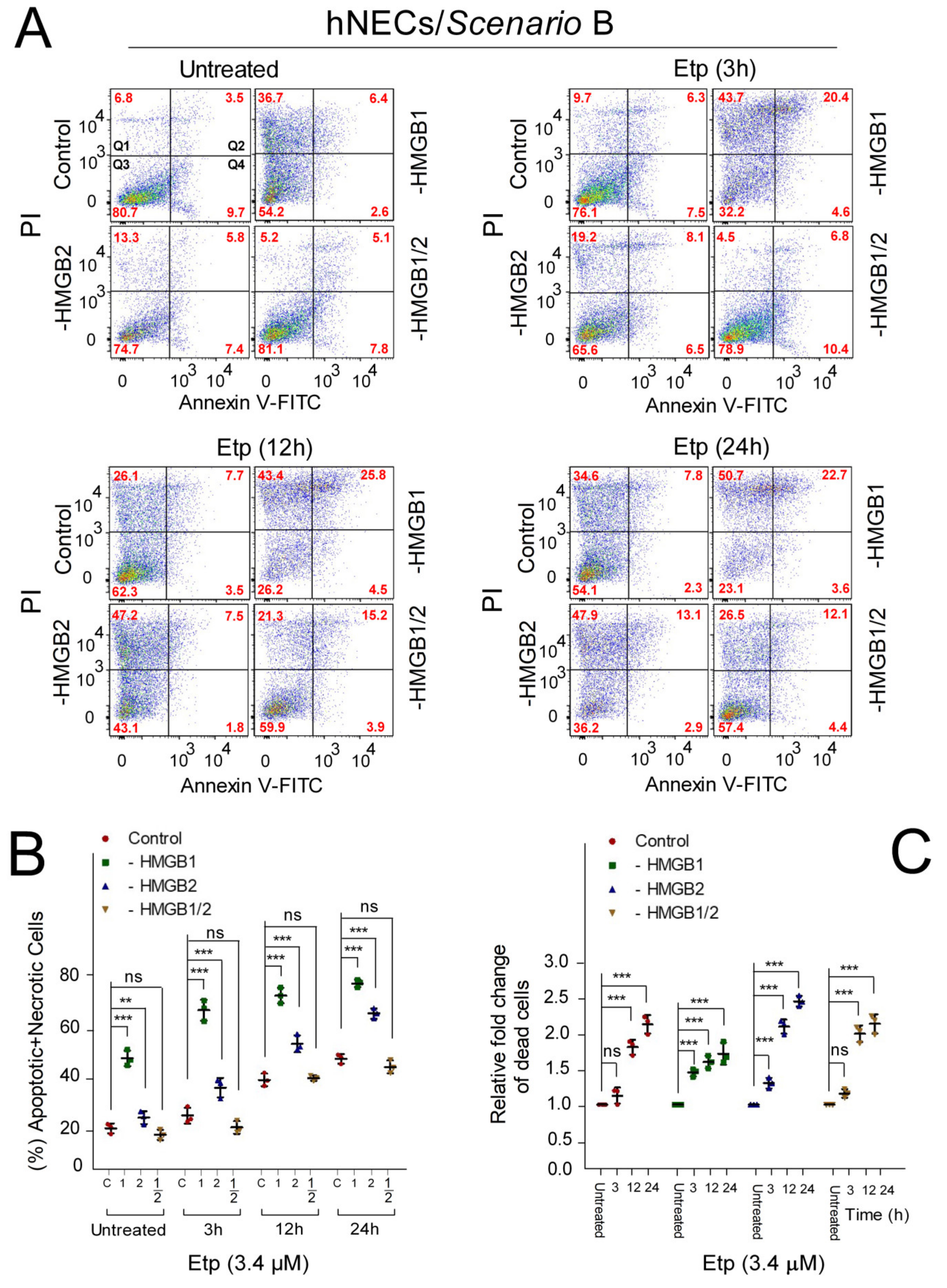
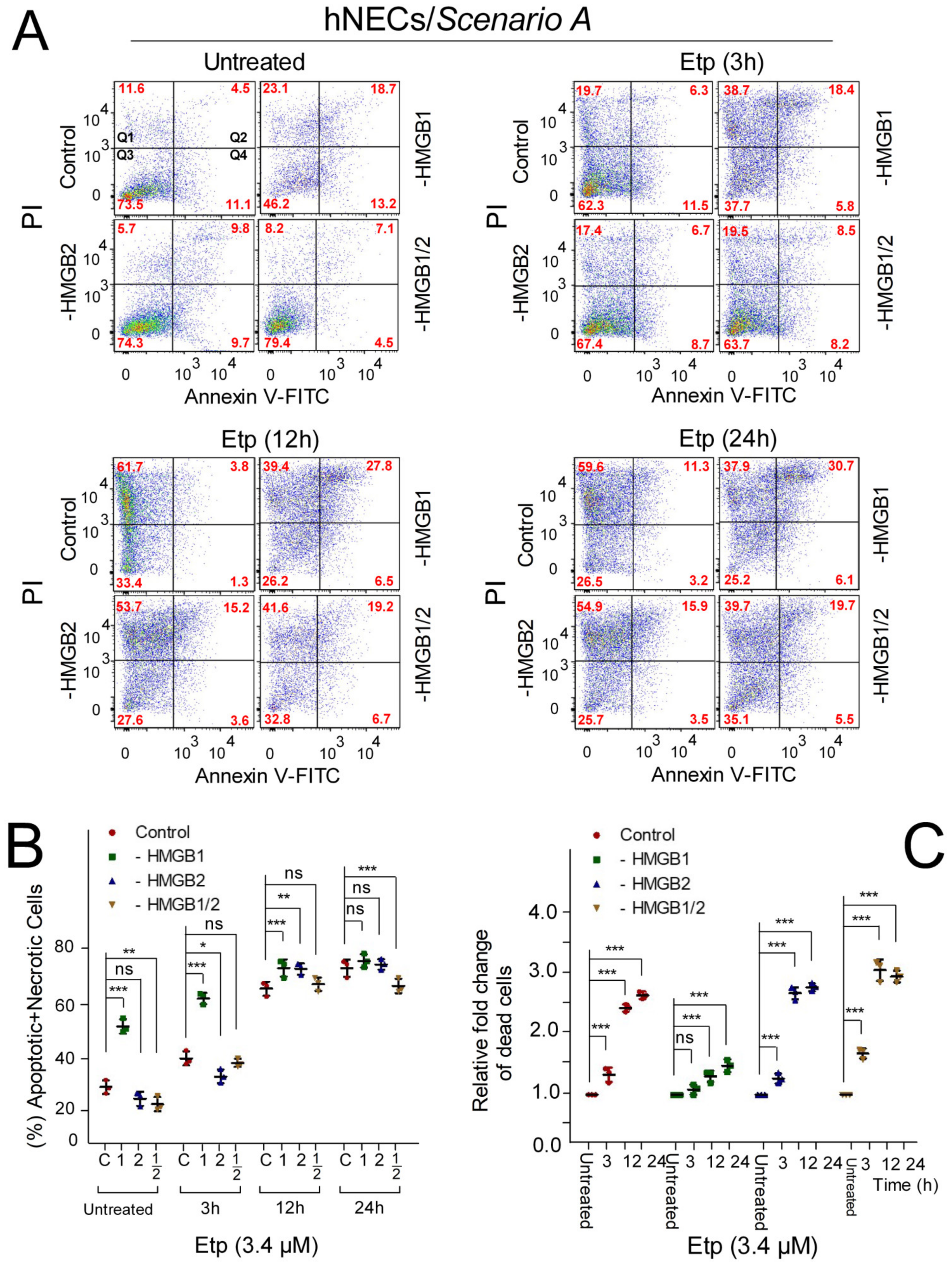
3.6. The Impact of HMGB1/2 Knockdown on Apoptosis of Etoposide-Treated Progenitor Cells Depends on the Differentiation Protocol of hESCs
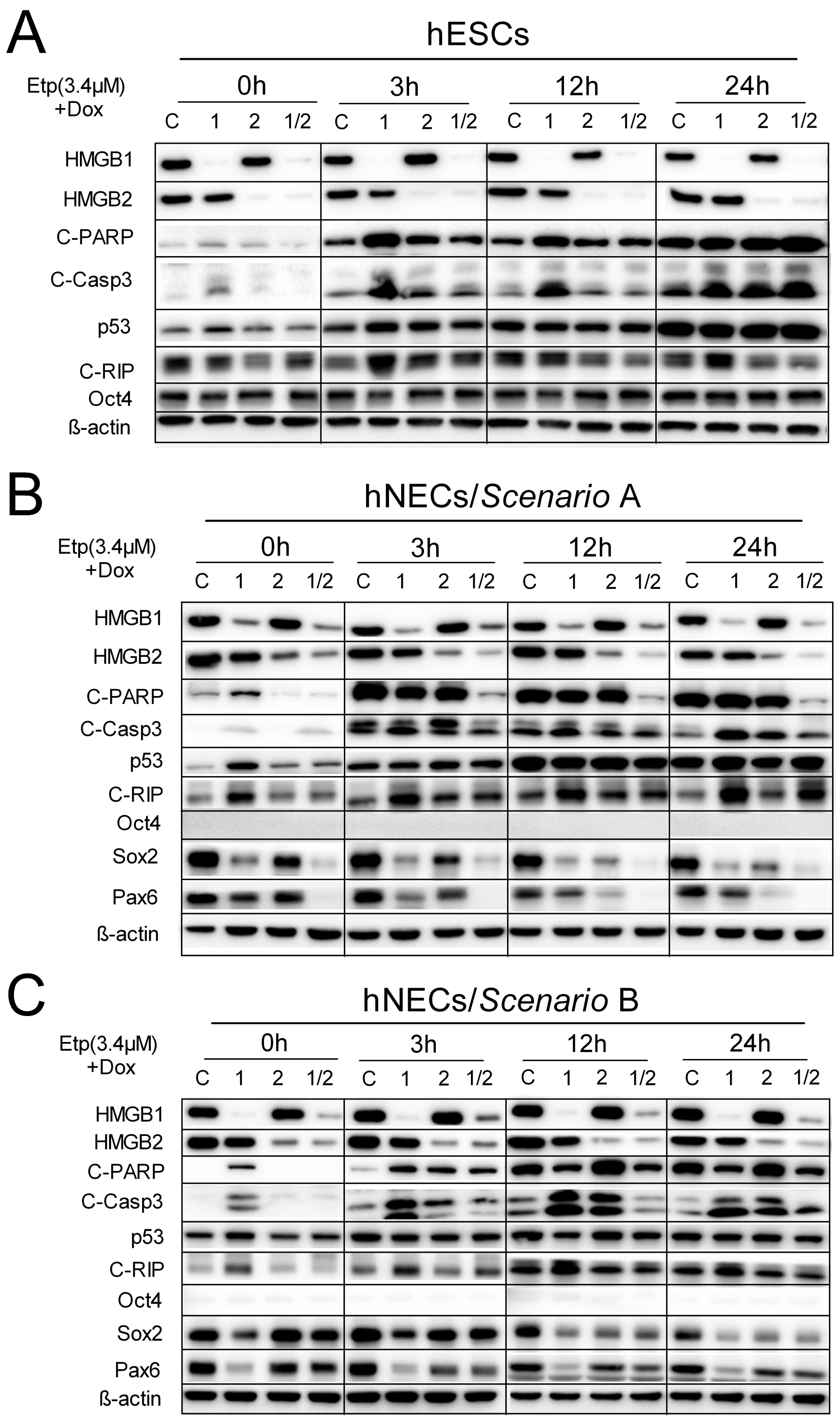
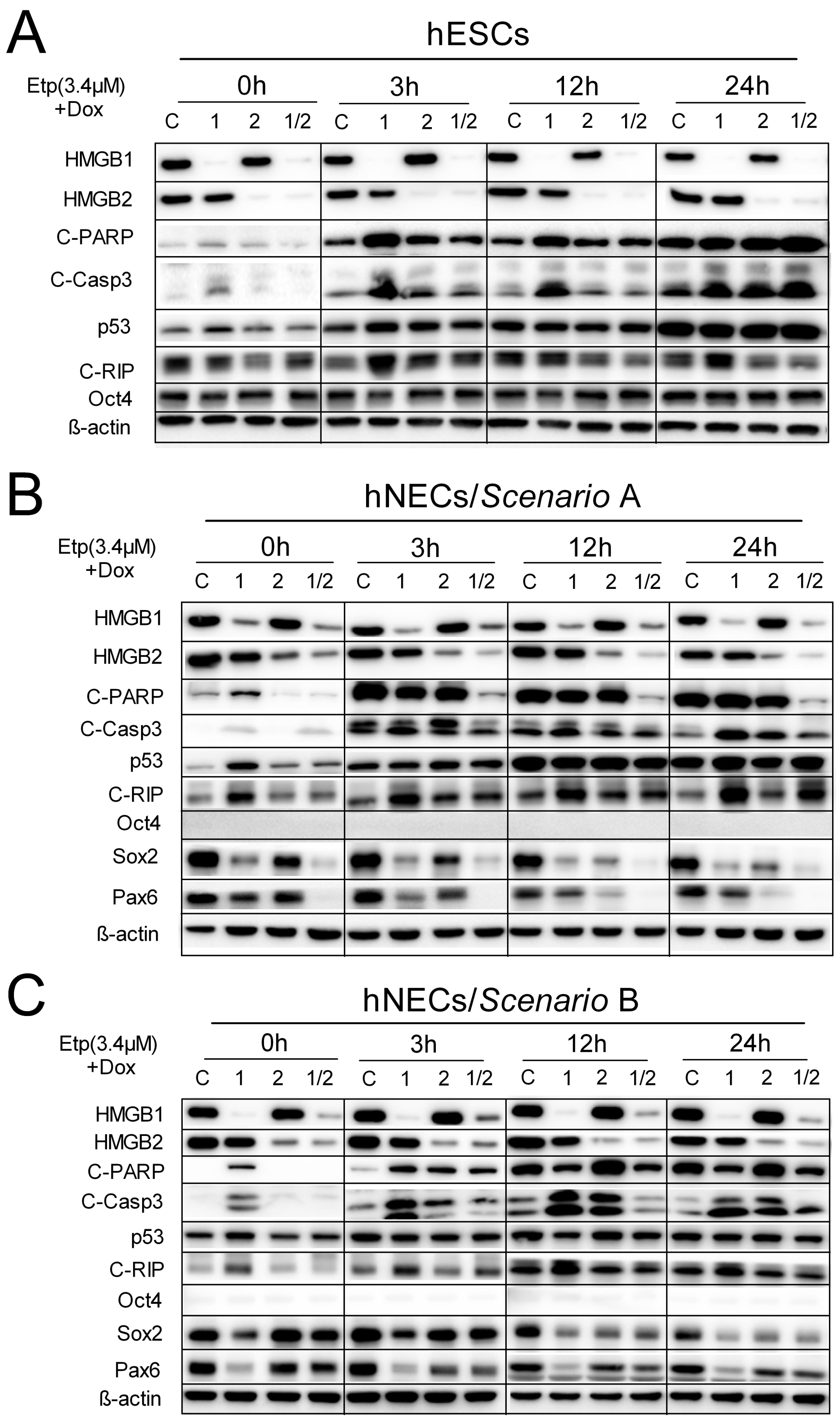
3.7. HMGB1 KD Promotes Cell Death Induction in Etoposide-Treated hESCs and hNECs via Activation of Caspase-3 Pathway
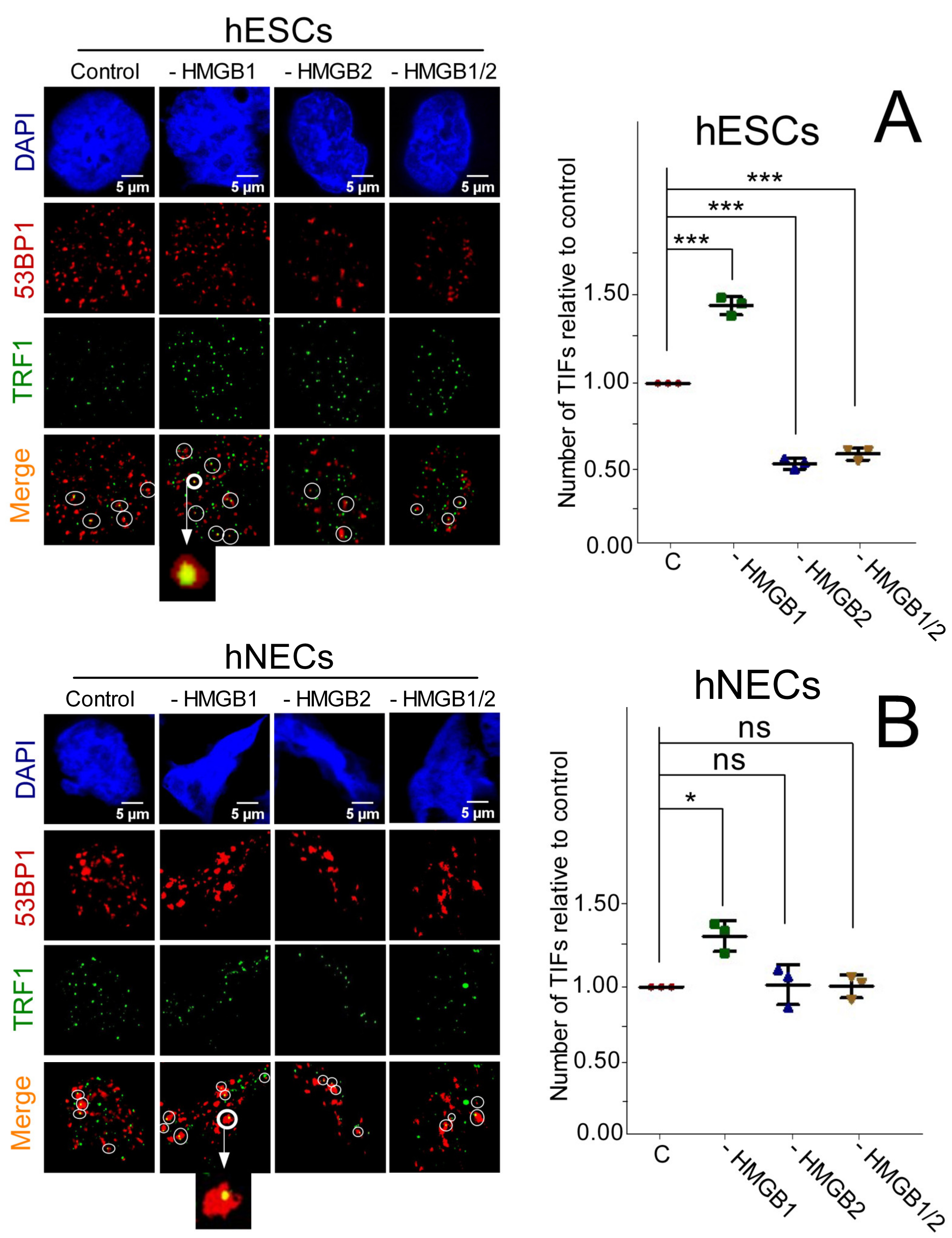
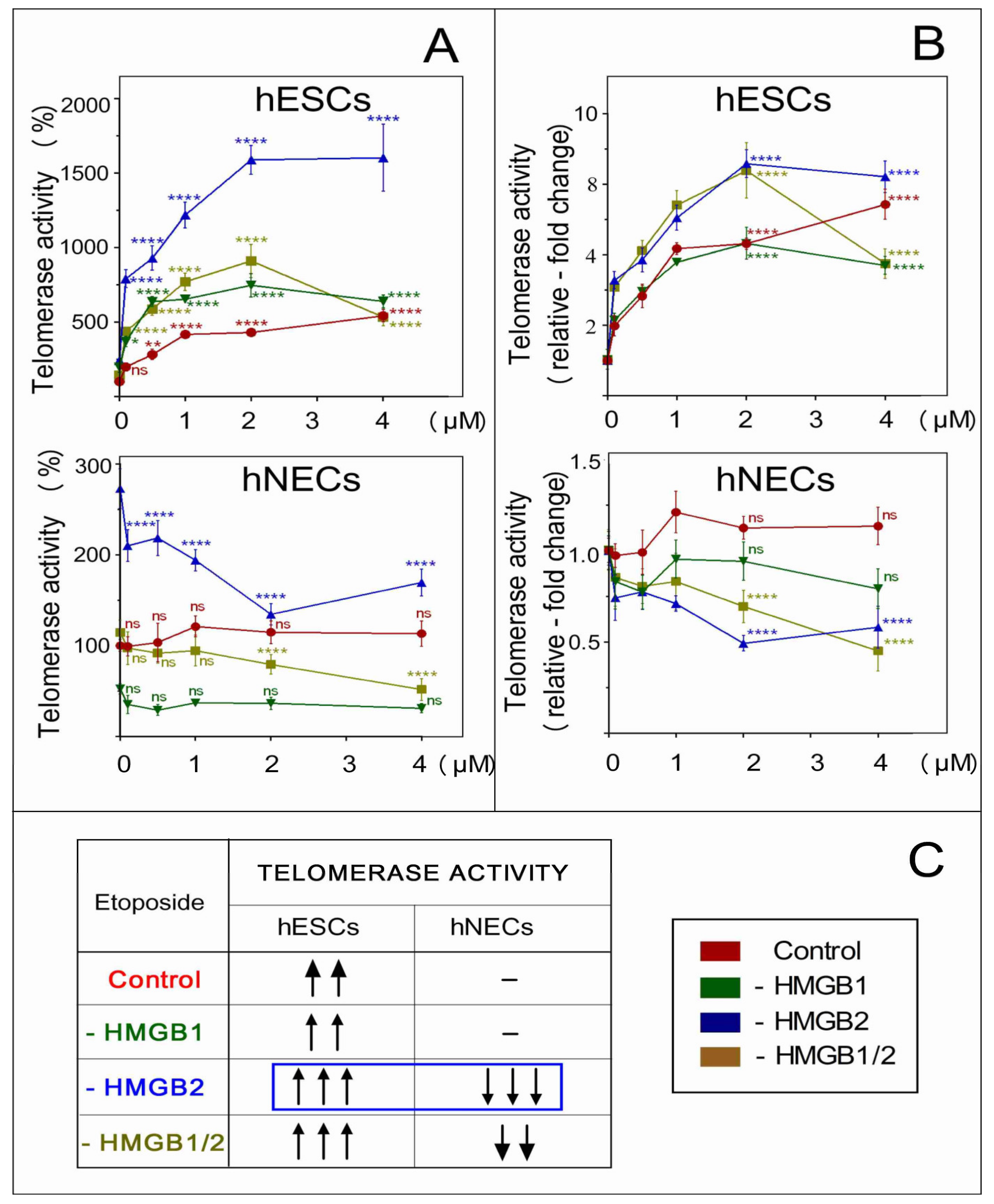
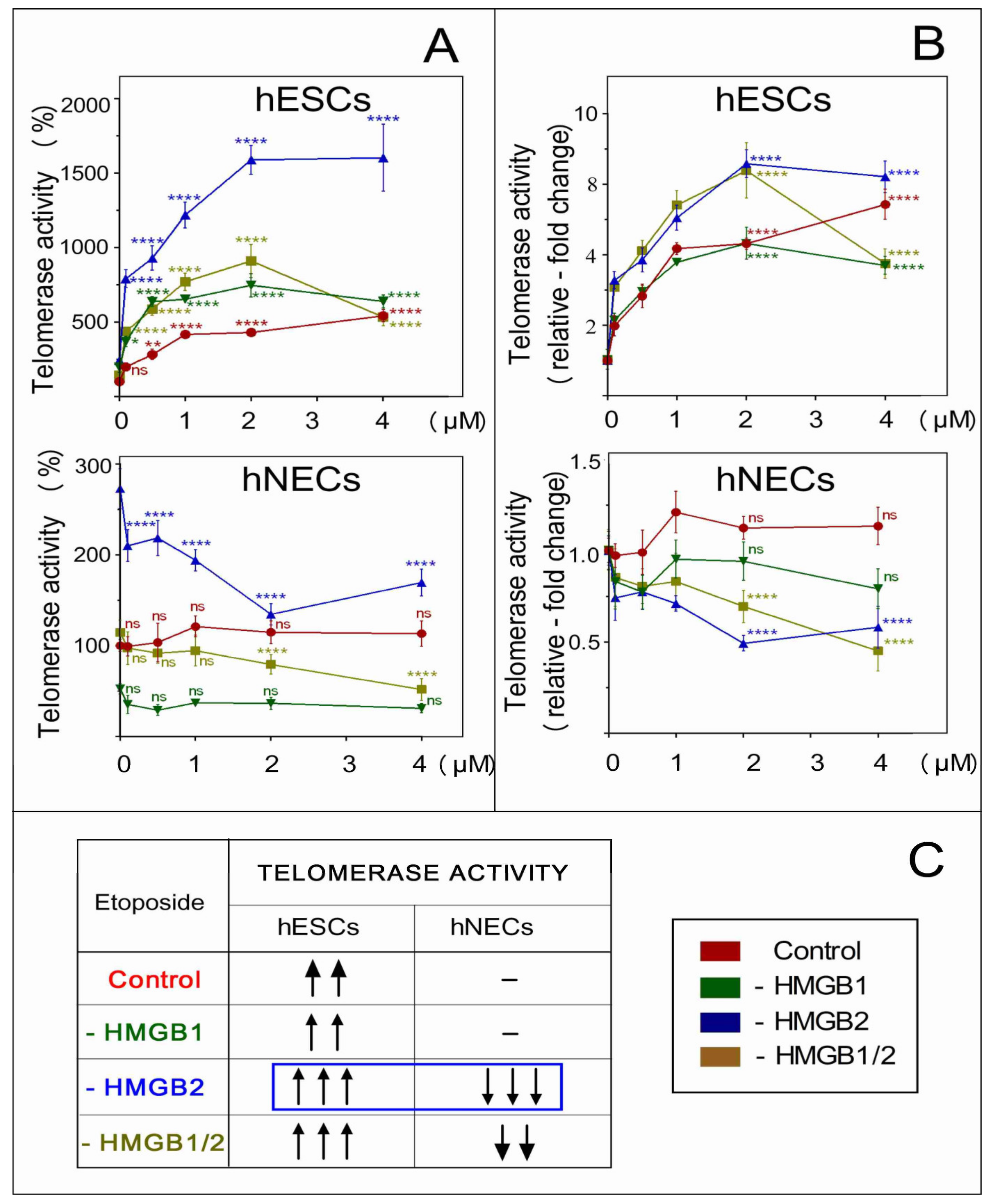
3.8. Impact of HMGB1/2 KD on Telomere Integrity is Distinct in Etoposide-Treated hESCs and hNECs
3.9. The Distinct Impact of HMGB2 KD on Telomerase Activity in Etoposide-Treated hESCs and hNECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shroff, G. Therapeutic potential of human embryonic stem cells in type 2 diabetes mellitus. World J. Stem Cells 2016, 8, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filion, T.M.; Qiao, M.; Ghule, P.N.; Mandeville, M.; van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Altieri, D.C.; Stein, G.S. Survival responses of human embryonic stem cells to DNA damage. J. Cell. Physiol. 2009, 220, 586–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Štros, M. HMGB proteins: Interactions with DNA and chromatin. Biochim Biophys Acta 2010, 1799, 101–113. [Google Scholar] [CrossRef]
- Tripathi, A.; Shrinet, K.; Kumar, A. HMGB1 protein as a novel target for cancer. Toxicol. Rep. 2019, 6, 253–261. [Google Scholar] [CrossRef]
- Mukherjee, A.; Vasquez, K.M. Targeting Chromosomal Architectural HMGB Proteins Could Be the Next Frontier in Cancer Therapy. Cancer Res. 2020, 80, 2075–2082. [Google Scholar] [CrossRef] [Green Version]
- Muller, S.; Ronfani, L.; Bianchi, M.E. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J. Intern. Med. 2004, 255, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.G.; Lee, J.J.; Jung, D.Y.; Jeon, J.; Heo, H.S.; Kang, H.C.; Shin, J.H.; Cho, Y.S.; Cha, K.J.; Kim, C.G.; et al. Profiling of differentially expressed genes in human stem cells by cDNA microarray. Mol. Cells 2006, 21, 343–355. [Google Scholar]
- Adjaye, J.; Huntriss, J.; Herwig, R.; BenKahla, A.; Brink, T.C.; Wierling, C.; Hultschig, C.; Groth, D.; Yaspo, M.L.; Picton, H.M.; et al. Primary differentiation in the human blastocyst: Comparative molecular portraits of inner cell mass and trophectoderm cells. Stem Cells 2005, 23, 1514–1525. [Google Scholar] [CrossRef]
- Bagherpoor, A.J.; Dolezalova, D.; Barta, T.; Kučírek, M.; Sani, S.A.; Esner, M.; Bosakova, M.K.; Vinarsky, V.; Peskova, L.; Hampl, A.; et al. Properties of Human Embryonic Stem Cells and Their Differentiated Derivatives Depend on Nonhistone DNA-Binding HMGB1 and HMGB2 Proteins. Stem Cells Dev. 2017, 26, 328–340. [Google Scholar] [CrossRef]
- Yanai, H.; Ban, T.; Wang, Z.; Choi, M.K.; Kawamura, T.; Negishi, H.; Nakasato, M.; Lu, Y.; Hangai, S.; Koshiba, R.; et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nat. Cell Biol. 2009, 462, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Malarkey, C.S.; Churchill, M.E. The high mobility group box: The ultimate utility player of a cell. Trends Biochem. Sci. 2012, 37, 553–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, A.B.; Bronstein, R.; Reddy, A.S.; Maletic-Savatic, M.; Aguirre, A.; Tsirka, S.E. Aberrant neural stem cell proliferation and increased adult neurogenesis in mice lacking chromatin protein HMGB2. PLoS ONE 2013, 8, e84838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, N.; Carames, B.; Hsu, E.; Cherqui, S.; Kawakami, Y.; Lotz, M. Expression patterns and function of chromatin protein HMGB2 during mesenchymal stem cell differentiation. J. Biol. Chem. 2011, 286, 41489–41498. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yu, L.; Zhang, T.; Wang, L.; Leng, Z.; Guan, Y.; Wang, X. HMGB1 enhances embryonic neural stem cell proliferation by activating the MAPK signaling pathway. Biotechnol. Lett. 2014, 36, 1631–1639. [Google Scholar] [CrossRef]
- Guazzi, S.; Strangio, A.; Franzi, A.T.; Bianchi, M.E. HMGB1, an architectural chromatin protein and extracellular signalling factor, has a spatially and temporally restricted expression pattern in mouse brain. Gene Expr. Patterns GEP 2003, 3, 29–33. [Google Scholar] [CrossRef]
- Kučírek, M.; Bagherpoor, A.J.; Jaroš, J.; Hampl, A.; Štros, M. HMGB2 is a negative regulator of telomerase activity in human embryonic stem and progenitor cells. FASEB J. 2019, 33, 14307–14324. [Google Scholar] [CrossRef] [Green Version]
- Agresti, A.; Bianchi, M.E. HMGB proteins and gene expression. Curr. Opin. Genet. Dev. 2003, 13, 170–178. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef] [Green Version]
- Hiyama, E.; Hiyama, K. Telomere and telomerase in stem cells. Br. J. Cancer 2007, 96, 1020–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.A.; Upton, H.E.; Vogan, J.M.; Collins, K. Telomerase Mechanism of Telomere Synthesis. Ann. Rev. Biochem. 2017, 86, 439–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, N.; Mizumoto, K.; Kusumoto, M.; Nishio, S.; Maehara, N.; Urashima, T.; Ogawa, T.; Tanaka, M. Up-regulation of telomerase activity in human pancreatic cancer cells after exposure to etoposide. Br. J. Cancer 2000, 82, 1819–1826. [Google Scholar] [CrossRef]
- Moriarty, T.J.; Dupuis, S.; Autexier, C. Rapid upregulation of telomerase activity in human leukemia HL-60 cells treated with clinical doses of the DNA-damaging drug etoposide. Leukemia 2002, 16, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Grandela, C.; Pera, M.F.; Grimmond, S.M.; Kolle, G.; Wolvetang, E.J. p53 is required for etoposide-induced apoptosis of human embryonic stem cells. Stem Cell Res. 2007, 1, 116–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, S.; Kondo, Y.; Li, G.; Silverman, R.H.; Cowell, J.K. Targeted therapy of human malignant glioma in a mouse model by 2-5A antisense directed against telomerase RNA. Oncogene 1998, 16, 3323–3330. [Google Scholar] [CrossRef] [Green Version]
- Bashash, D.; Zareii, M.; Safaroghli-Azar, A.; Omrani, M.D.; Ghaffari, S.H. Inhibition of telomerase using BIBR1532 enhances doxorubicin-induced apoptosis in pre-B acute lymphoblastic leukemia cells. Hematology 2017, 22, 330–340. [Google Scholar] [CrossRef] [Green Version]
- Polanská, E.; Dobsakova, Z.; Dvorackova, M.; Fajkus, J.; Štros, M. HMGB1 gene knockout in mouse embryonic fibroblasts results in reduced telomerase activity and telomere dysfunction. Chromosoma 2012, 121, 419–431. [Google Scholar] [CrossRef]
- Štros, M.; Polanská, E.; Štruncová, S.; Pospíšilová, S. HMGB1 and HMGB2 proteins up-regulate cellular expression of human topoisomerase IIalpha. Nucleic Acids Res. 2009, 37, 2070–2086. [Google Scholar] [CrossRef] [Green Version]
- Adewumi, O.; Aflatoonian, B.; Ahrlund-Richter, L.; Amit, M.; Andrews, P.W.; Beighton, G.; Bello, P.A.; Benvenisty, N.; Berry, L.S.; Bevan, S.; et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 2007, 25, 803–816. [Google Scholar]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.A.; Logan, D.J.; Madden, K.L.; Ljosa, V.; Rueden, C.; Eliceiri, K.W.; Carpenter, A.E. Improved structure, function and compatibility for CellProfiler: Modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.A.; Ghule, P.N.; Therrien, J.A.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J. Cell. Physiol. 2006, 209, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.B.; Bronstein, R.; Chen, E.I.; Koller, A.; Ronfani, L.; Maletic-Savatic, M.; Tsirka, S.E. Members of the high mobility group B protein family are dynamically expressed in embryonic neural stem cells. Proteome Sci. 2013, 11, 18. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A.; Matsuda, T.; Sakai, A.; Murao, N.; Nakashima, K. HMGB2 expression is associated with transition from a quiescent to an activated state of adult neural stem cells. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2018, 247, 229–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pibiri, V.; Ravarino, A.; Gerosa, C.; Pintus, M.C.; Fanos, V.; Faa, G. Stem/progenitor cells in the developing human cerebellum: An immunohistochemical study. Eur. J. Histochem. EJH 2016, 60, 2686. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Yu, L.; Ren, J.; Wang, M.; Liu, Z.; Hu, X.; Hu, D.; Chen, Y.; Chen, L.; Zhang, Y.; et al. Efficient and Fast Differentiation of Human Neural Stem Cells from Human Embryonic Stem Cells for Cell Therapy. Stem Cells Int. 2017, 2017, 9405204. [Google Scholar] [CrossRef] [Green Version]
- Pauklin, S.; Vallier, L. The Cell-Cycle State of Stem Cells Determines Cell Fate Propensity. Cell 2014, 156, 1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzales, K.A.; Liang, H.; Lim, Y.S.; Chan, Y.S.; Yeo, J.C.; Tan, C.P.; Gao, B.; Le, B.; Tan, Z.Y.; Low, K.Y.; et al. Deterministic Restriction on Pluripotent State Dissolution by Cell-Cycle Pathways. Cell 2015, 162, 564–579. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.H.; Yoon, Y.G.; Lee, J.S.; Song, Y.S.; Oh, J.S.; Park, B.S.; Kwon, T.K.; Park, C.; Choi, Y.H.; Yoo, Y.H. Etoposide induces a mixed type of programmed cell death and overcomes the resistance conferred by Bcl-2 in Hep3B hepatoma cells. Int. J. Oncol. 2012, 41, 1443–1454. [Google Scholar] [CrossRef] [Green Version]
- Meyn, R.E.; Meistrich, M.L.; White, R.A. Cycle-dependent anticancer drug cytotoxicity in mammalian cells synchronized by centrifugal elutriation. J. Natl. Cancer Inst. 1980, 64, 1215–1219. [Google Scholar]
- Liu, J.C.; Guan, X.; Ryan, J.A.; Rivera, A.G.; Mock, C.; Agrawal, V.; Letai, A.; Lerou, P.H.; Lahav, G. High mitochondrial priming sensitizes hESCs to DNA-damage-induced apoptosis. Cell Stem Cell 2013, 13, 483–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Zhang, Q.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. HMGB1 in cancer: Good, bad, or both? Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Antoine, D.J.; Andersson, U.; Tracey, K.J. The many faces of HMGB1: Molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 2013, 93, 865–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantzer, F.; de la Rubia, G.; Ménissier-de Murcia, J.; Hostomsky, Z.; de Murcia, G.; Schreiber, V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53, Nature reviews. Cancer 2002, 2, 594–604. [Google Scholar]
- de Lange, T. Shelterin-Mediated Telomere Protection. Ann. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- Tutton, S.; Azzam, G.A.; Stong, N.; Vladimirova, O.; Wiedmer, A.; Monteith, J.A.; Beishline, K.; Wang, Z.; Deng, Z.; Riethman, H.; et al. Subtelomeric p53 binding prevents accumulation of DNA damage at human telomeres. Embo J. 2016, 35, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef] [Green Version]
- Reeves, R. High mobility group (HMG) proteins: Modulators of chromatin structure and DNA repair in mammalian cells. DNA Repair 2015, 36, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Barreiro-Alonso, A.; Lamas-Maceiras, M.; Rodriguez-Belmonte, E.; Vizoso-Vazquez, A.; Quindos, M.; Cerdan, M.E. High Mobility Group B Proteins, Their Partners, and Other Redox Sensors in Ovarian and Prostate Cancer. Oxidative Med. Cell. Longev. 2016, 2016, 5845061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calogero, S.; Grassi, F.; Aguzzi, A.; Voigtlander, T.; Ferrier, P.; Ferrari, S.; Bianchi, M.E. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 1999, 22, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Ronfani, L.; Ferraguti, M.; Croci, L.; E Ovitt, C.; Schöler, H.R.; Consalez, G.G.; E Bianchi, M. Reduced fertility and spermatogenesis defects in mice lacking chromosomal protein Hmgb2. Development 2001, 128, 1265–1273. [Google Scholar] [PubMed]
- Laurent, B.; Randrianarison-Huetz, V.; Marechal, V.; Mayeux, P.; Dusanter-Fourt, I.; Dumenil, D. High-mobility group protein HMGB2 regulates human erythroid differentiation through trans-activation of GFI1B transcription. Blood 2010, 115, 687–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, G.; Cai, F.; Ding, Z.; Gao, L. HMGB2 promotes the malignancy of human gastric Cancer and indicates poor survival outcome. Hum. Pathol. 2019, 84, 133–141. [Google Scholar] [CrossRef]
- Fu, D.; Li, J.; Wei, J.; Zhang, Z.; Luo, Y.; Tan, H.; Ren, C. HMGB2 is associated with malignancy and regulates Warburg effect by targeting LDHB and FBP1 in breast cancer. Cell Commun. Signal. CCS 2018, 16, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.J.; Cheng, J.; Feng, X.; Yu, Y.; Tian, L.; Huang, Q. The dual role and therapeutic potential of high-mobility group box 1 in cancer. Oncotarget 2017, 8, 64534–64550. [Google Scholar] [CrossRef] [Green Version]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Biniossek, M.L.; Lechel, A.; Rudolph, K.L.; Martens, U.M.; Zimmermann, S. Quantitative proteomic profiling of tumor cell response to telomere dysfunction using isotope-coded protein labeling (ICPL) reveals interaction network of candidate senescence markers. J. Proteom. 2013, 91, 515–535. [Google Scholar] [CrossRef]
- Cai, X.; Ding, H.; Liu, Y.; Pan, G.; Li, Q.; Yang, Z.; Liu, W. Expression of HMGB2 indicates worse survival of patients and is required for the maintenance of Warburg effect in pancreatic cancer. Acta Biochim. Biophys. Sin. 2017, 49, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Yang, Z.; Wu, J.; Wu, R.; Keshipeddy, S.K.; Wright, D.; Wang, L. High-mobility-group protein 2 regulated by microRNA-127 and small heterodimer partner modulates pluripotency of mouse embryonic stem cells and liver tumor initiating cells. Hepatol. Commun. 2017, 1, 816–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seita, J.; Rossi, D.J.; Weissman, I.L. Differential DNA damage response in stem and progenitor cells. Cell Stem Cell 2010, 7, 145–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Ge, Y.; Liu, D.; Songyang, Z. The role of telomere-binding modulators in pluripotent stem cells. Protein Cell 2020, 11, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Giuffrida, D.; Rogers, I.M.; Nagy, A.; Calogero, A.E.; Brown, T.J.; Casper, R.F. Human embryonic stem cells secrete soluble factors that inhibit cancer cell growth. Cell Prolif. 2009, 42, 788–798. [Google Scholar] [CrossRef]
- Lotem, J.; Sachs, L. Epigenetics and the plasticity of differentiation in normal and cancer stem cells. Oncogene 2006, 25, 7663–7672. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Abdouh, M.; Arena, V.; Arena, M.; Arena, G.O. Reprogramming Malignant Cancer Cells toward a Benign Phenotype following Exposure to Human Embryonic Stem Cell Microenvironment. PLoS ONE 2017, 12, e0169899. [Google Scholar] [CrossRef] [Green Version]
- Camara-Quilez, M.; Barreiro-Alonso, A.; Vizoso-Vazquez, A.; Rodriguez-Belmonte, E.; Quindos-Varela, M.; Lamas-Maceiras, M.; Cerdan, M.E. The HMGB1-2 Ovarian Cancer Interactome. The Role of HMGB Proteins and Their Interacting Partners MIEN1 and NOP53 in Ovary Cancer and Drug-Response. Cancers 2020, 12, 2435. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagherpoor, A.J.; Kučírek, M.; Fedr, R.; Sani, S.A.; Štros, M. Nonhistone Proteins HMGB1 and HMGB2 Differentially Modulate the Response of Human Embryonic Stem Cells and the Progenitor Cells to the Anticancer Drug Etoposide. Biomolecules 2020, 10, 1450. https://doi.org/10.3390/biom10101450
Bagherpoor AJ, Kučírek M, Fedr R, Sani SA, Štros M. Nonhistone Proteins HMGB1 and HMGB2 Differentially Modulate the Response of Human Embryonic Stem Cells and the Progenitor Cells to the Anticancer Drug Etoposide. Biomolecules. 2020; 10(10):1450. https://doi.org/10.3390/biom10101450
Chicago/Turabian StyleBagherpoor, Alireza Jian, Martin Kučírek, Radek Fedr, Soodabeh Abbasi Sani, and Michal Štros. 2020. "Nonhistone Proteins HMGB1 and HMGB2 Differentially Modulate the Response of Human Embryonic Stem Cells and the Progenitor Cells to the Anticancer Drug Etoposide" Biomolecules 10, no. 10: 1450. https://doi.org/10.3390/biom10101450
APA StyleBagherpoor, A. J., Kučírek, M., Fedr, R., Sani, S. A., & Štros, M. (2020). Nonhistone Proteins HMGB1 and HMGB2 Differentially Modulate the Response of Human Embryonic Stem Cells and the Progenitor Cells to the Anticancer Drug Etoposide. Biomolecules, 10(10), 1450. https://doi.org/10.3390/biom10101450