Computational and In Vitro Investigation of (-)-Epicatechin and Proanthocyanidin B2 as Inhibitors of Human Matrix Metalloproteinase 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodology
2.1. Receptor and Ligands Collection
2.2. ADME and Quantum Chemical Calculations
2.3. Molecular Docking Simulation
2.4. Explicit Molecular Dynamics Simulations
2.5. Post Molecular Simulation Quantum Chemical Calculations
2.6. Molecular Mechanics Generalized Born Surface Area (MM/GBSA) Calculations
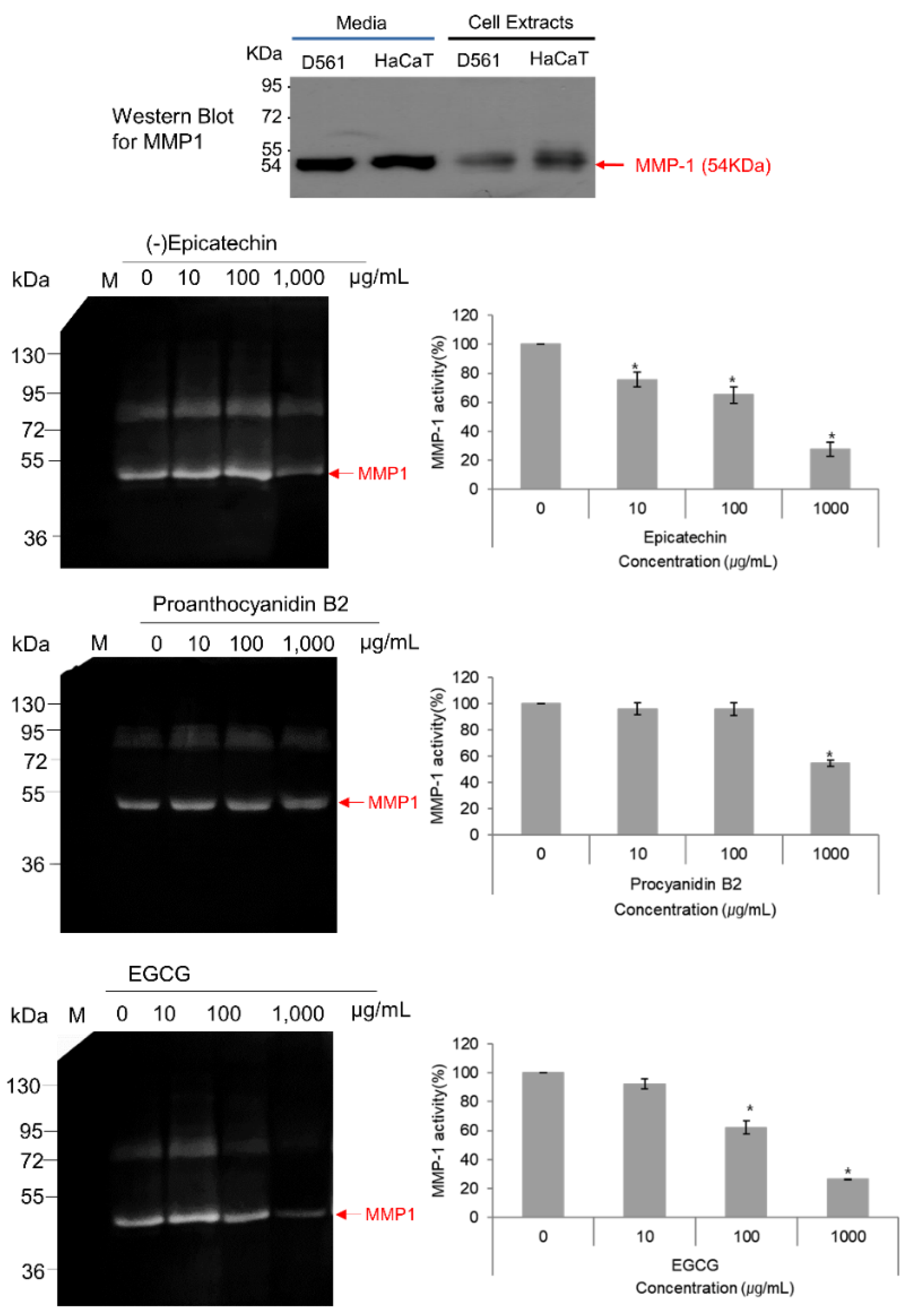
2.7. MMP-1 Zymography
2.8. Western Blot Analysis for MMP-1
3. Results and Discussion
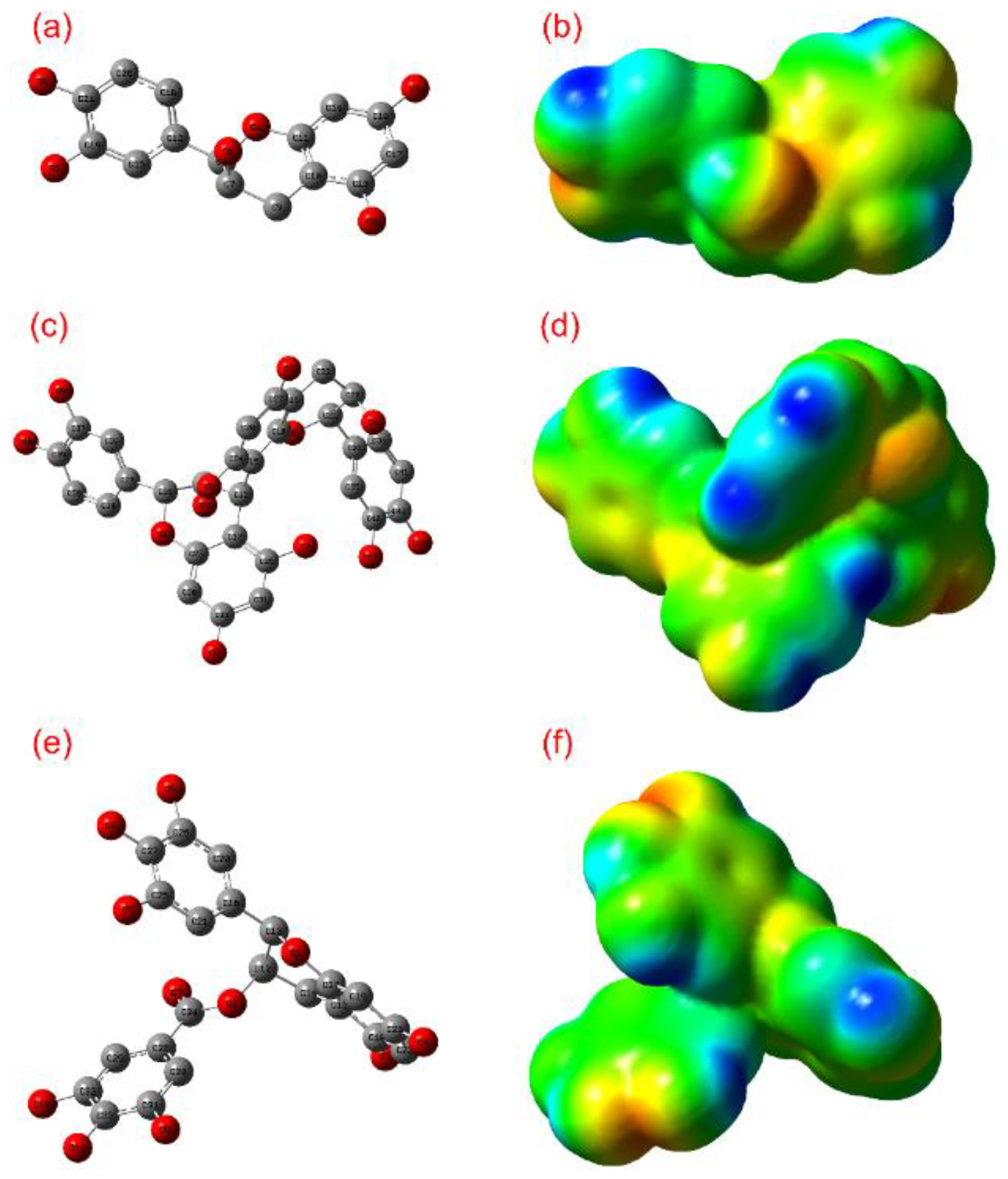
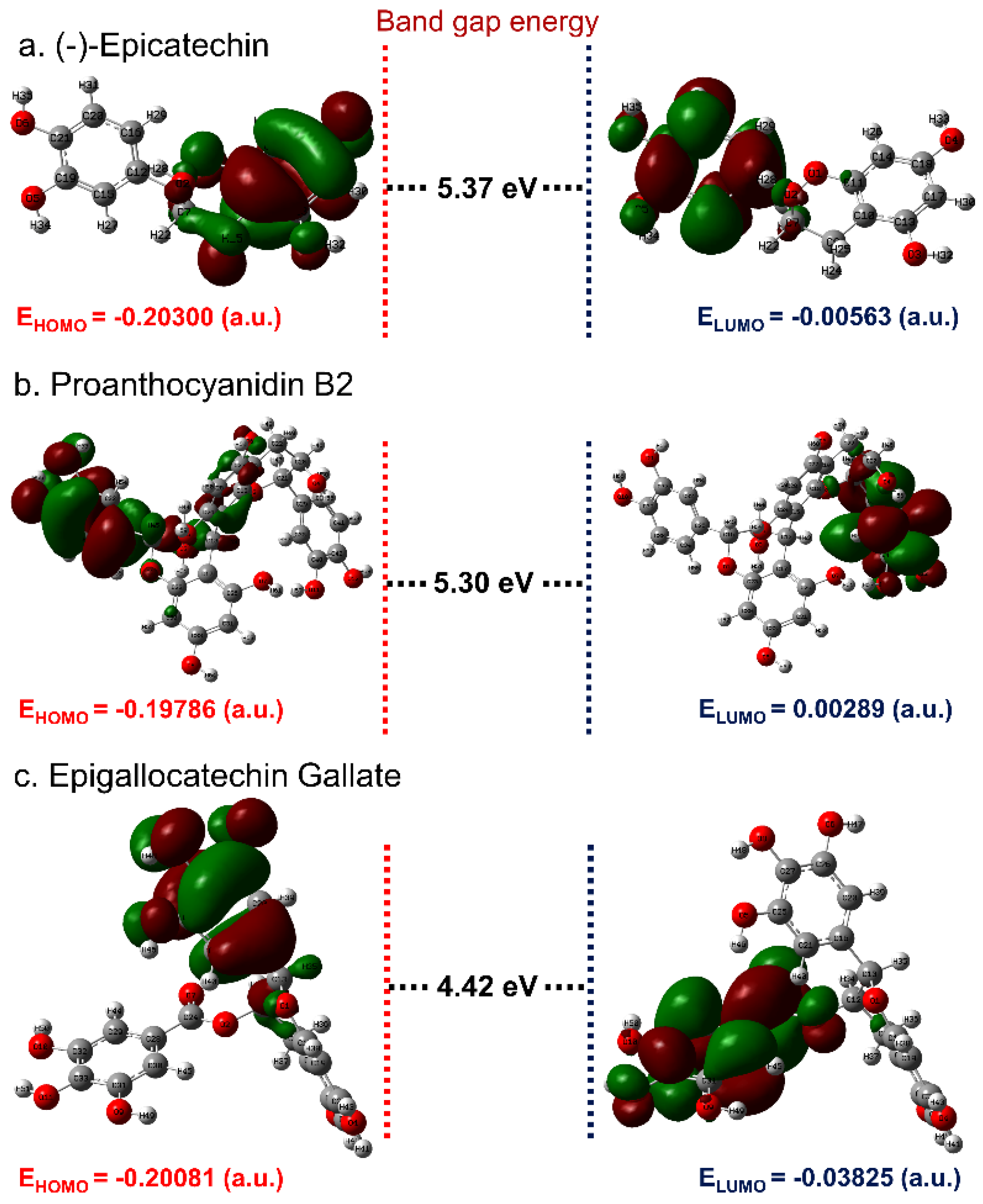
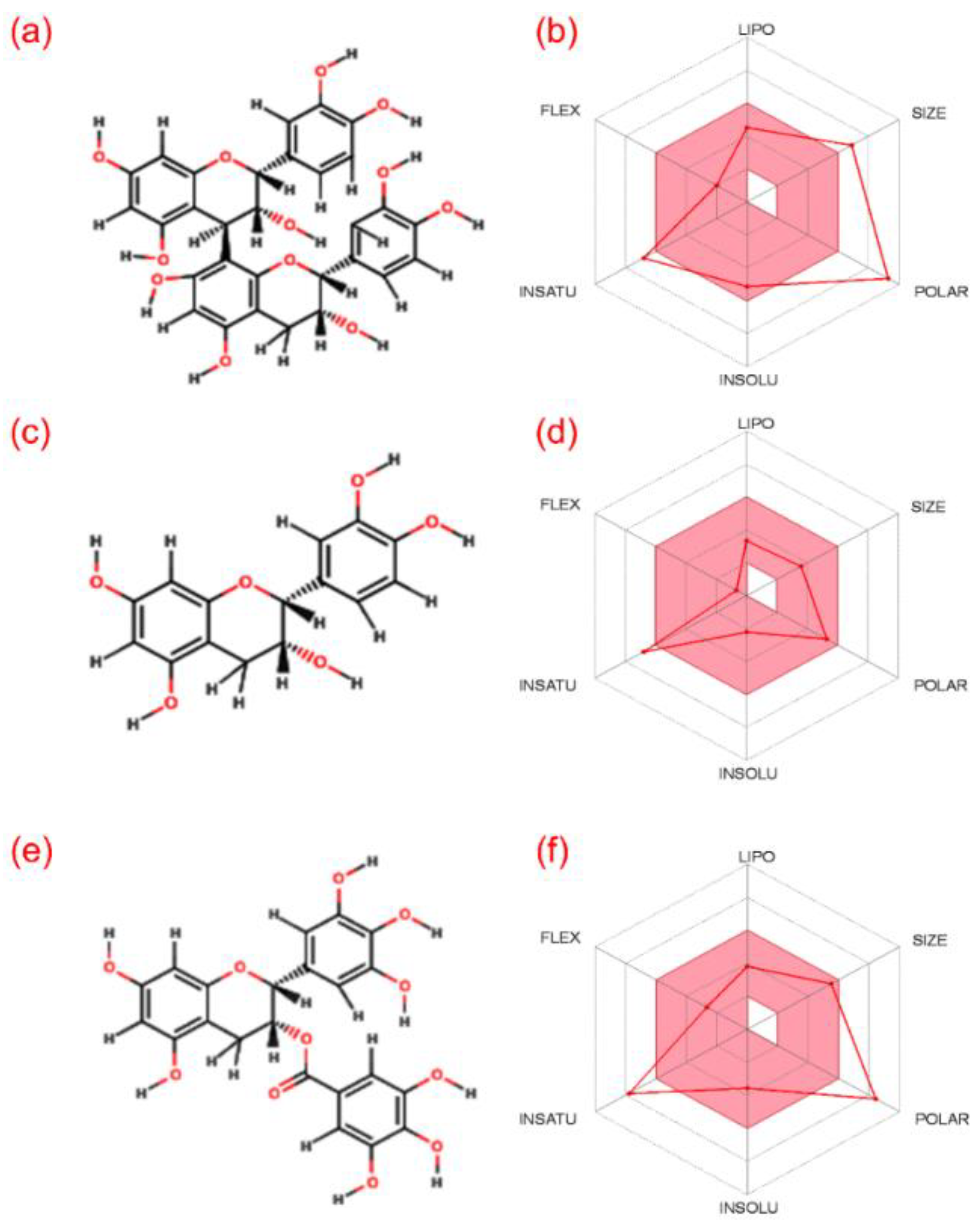
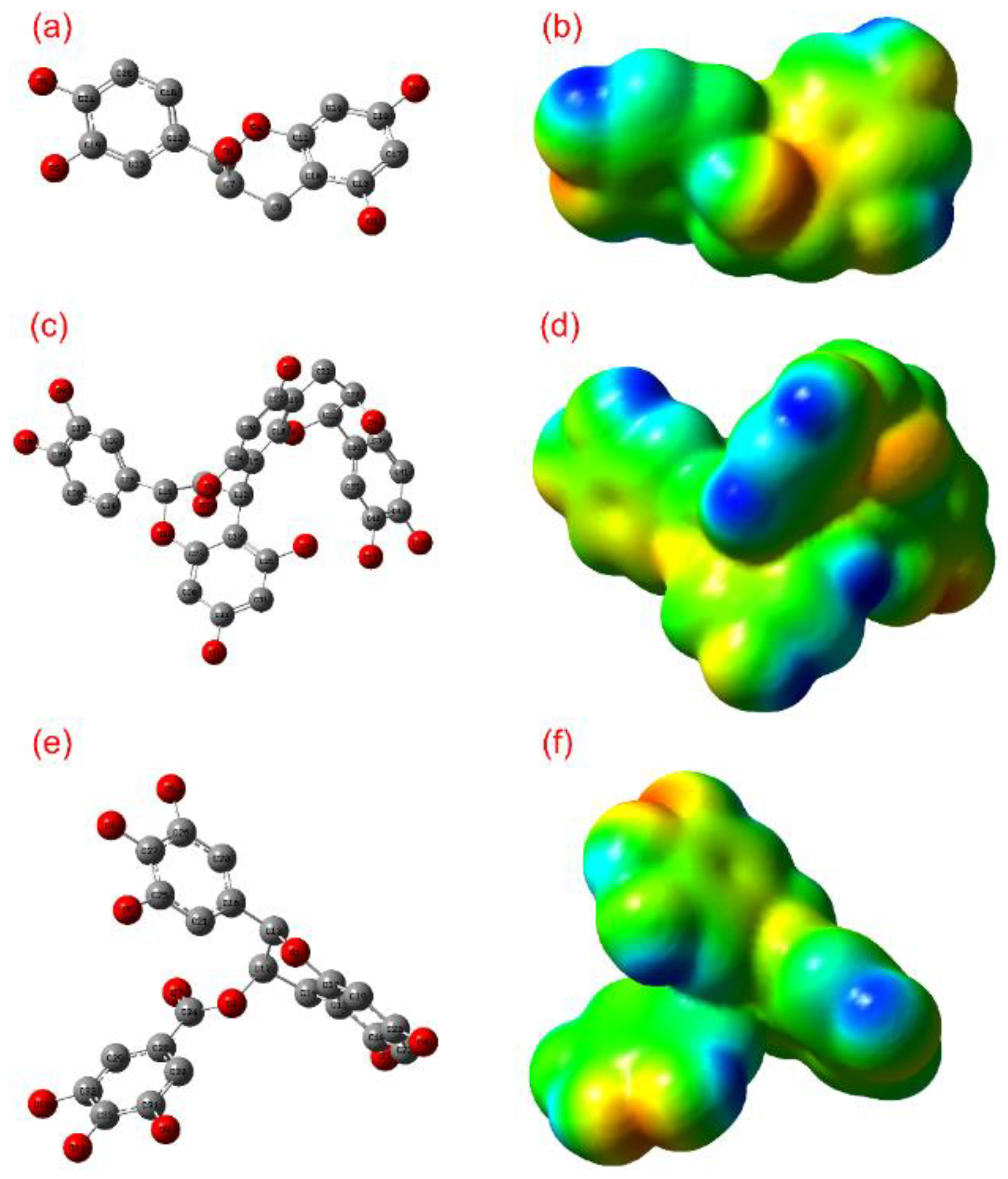
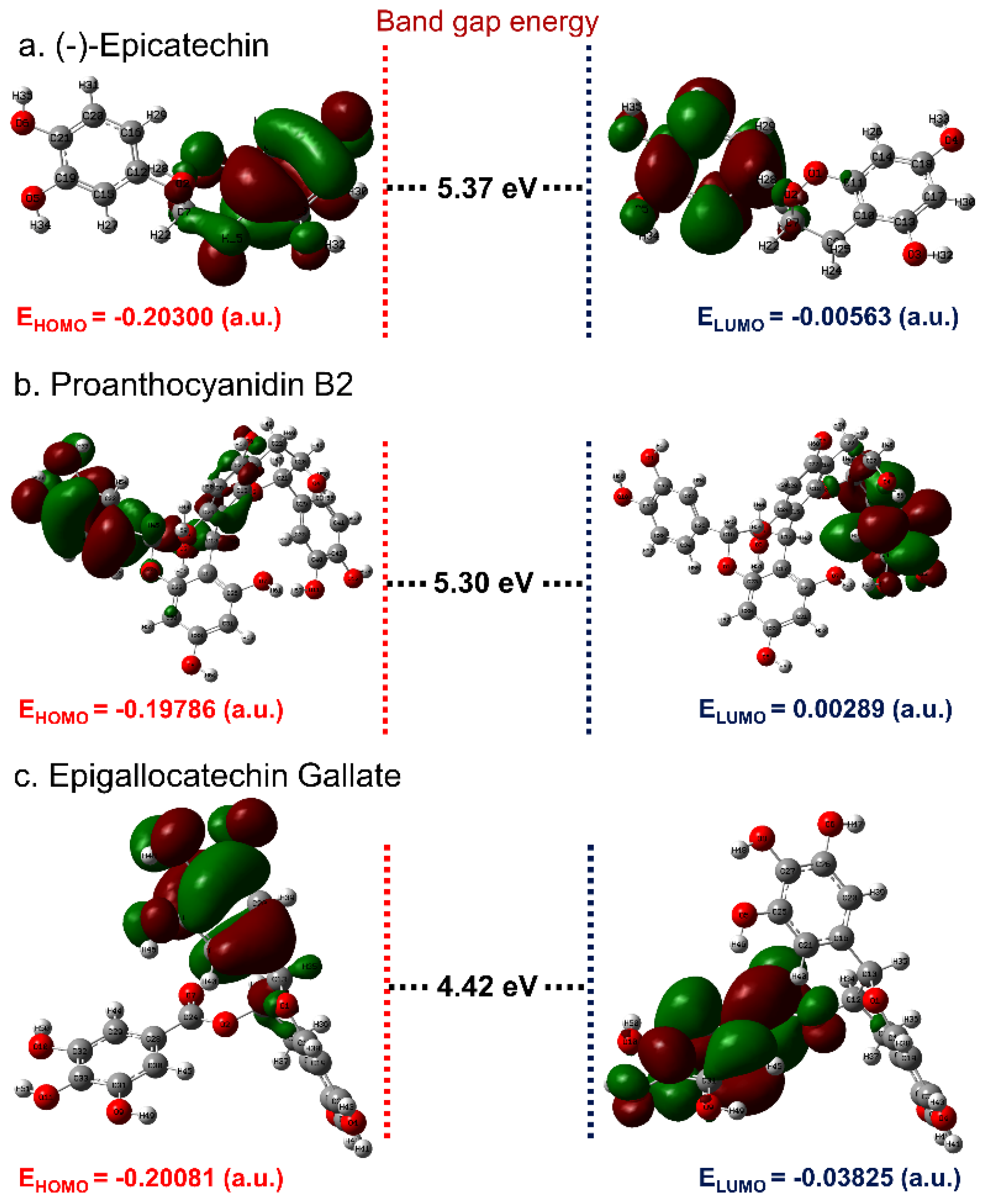
3.1. ADME and Quantum Chemical Calculation Analysis
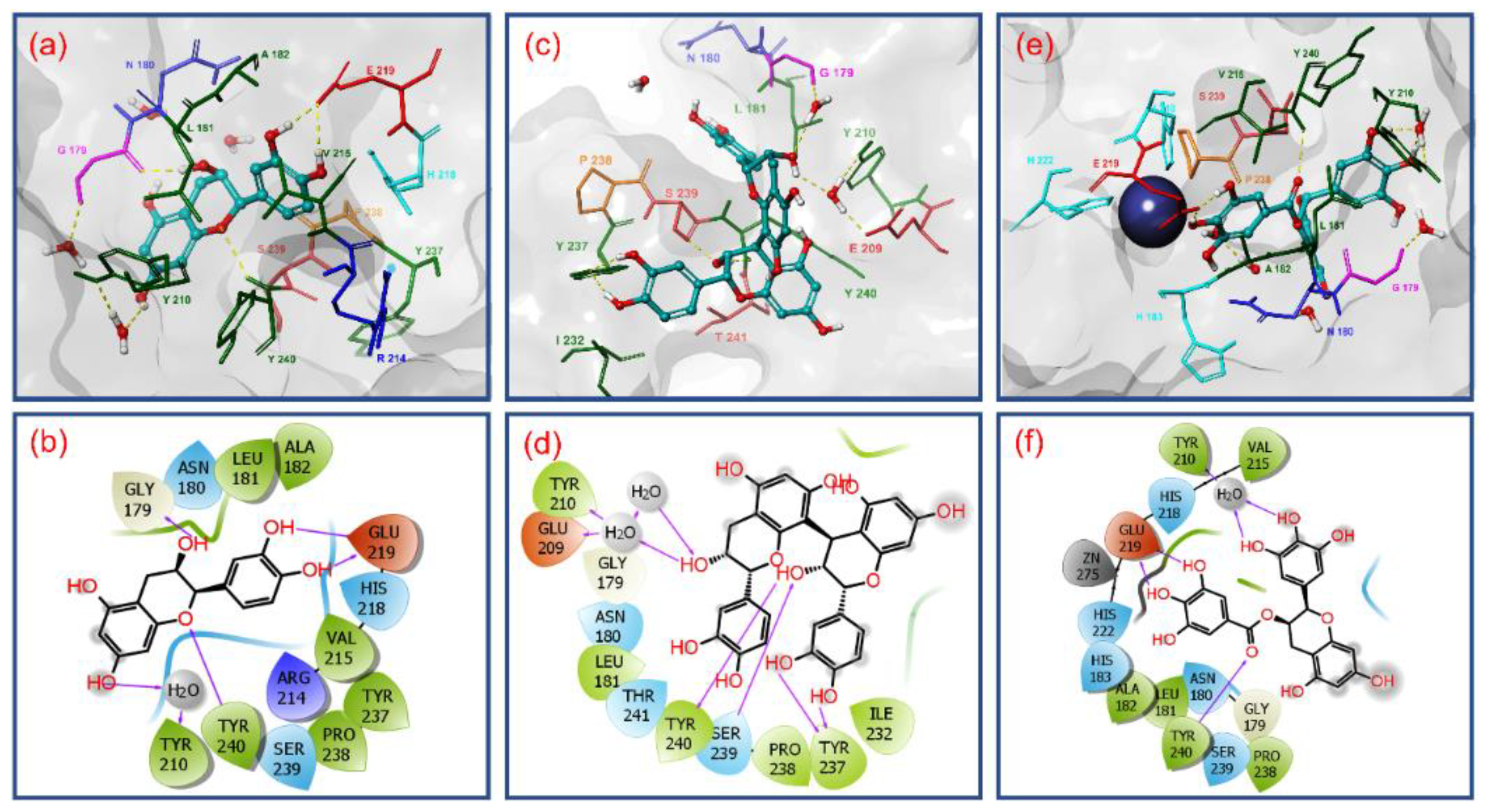
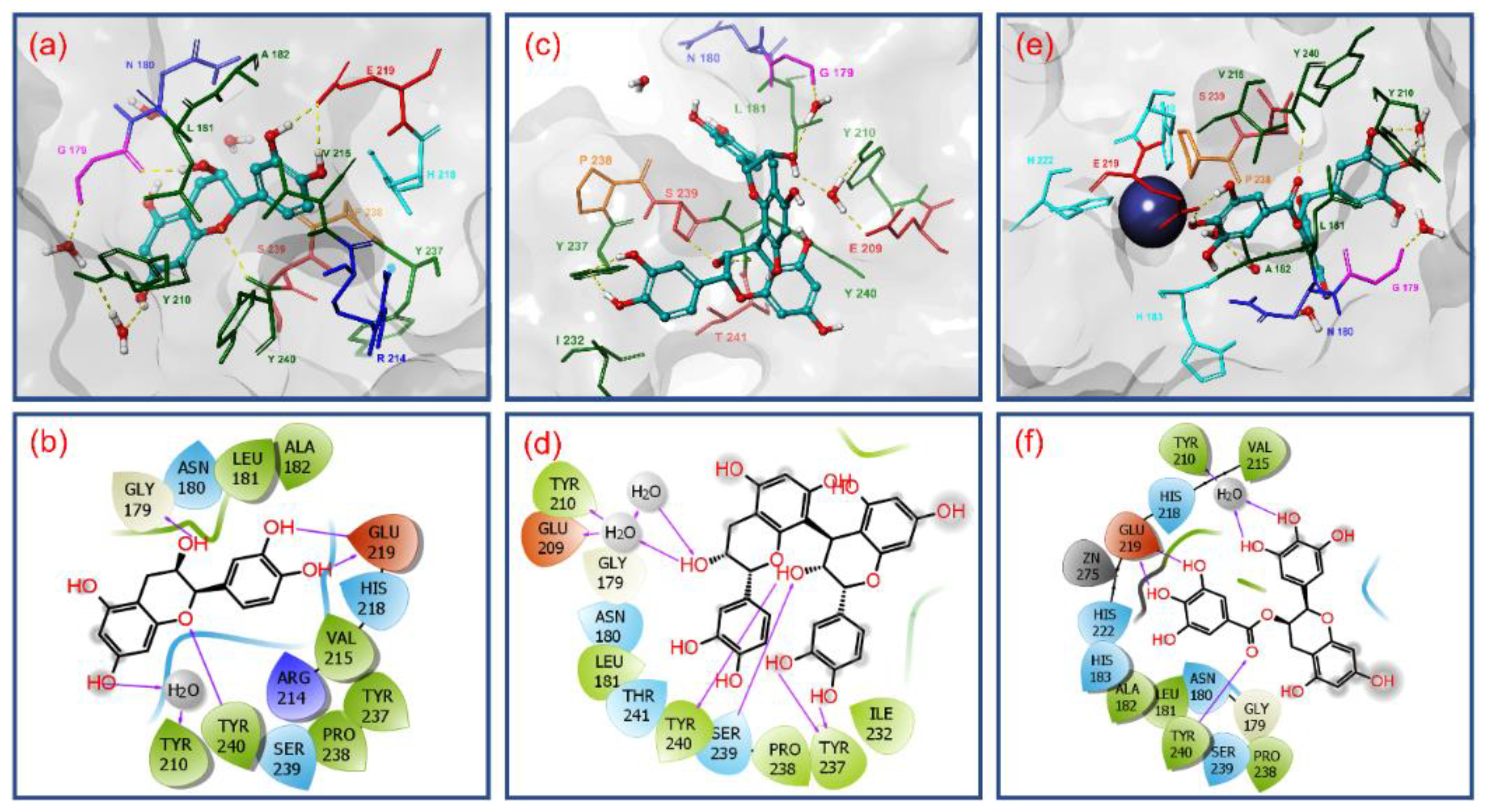
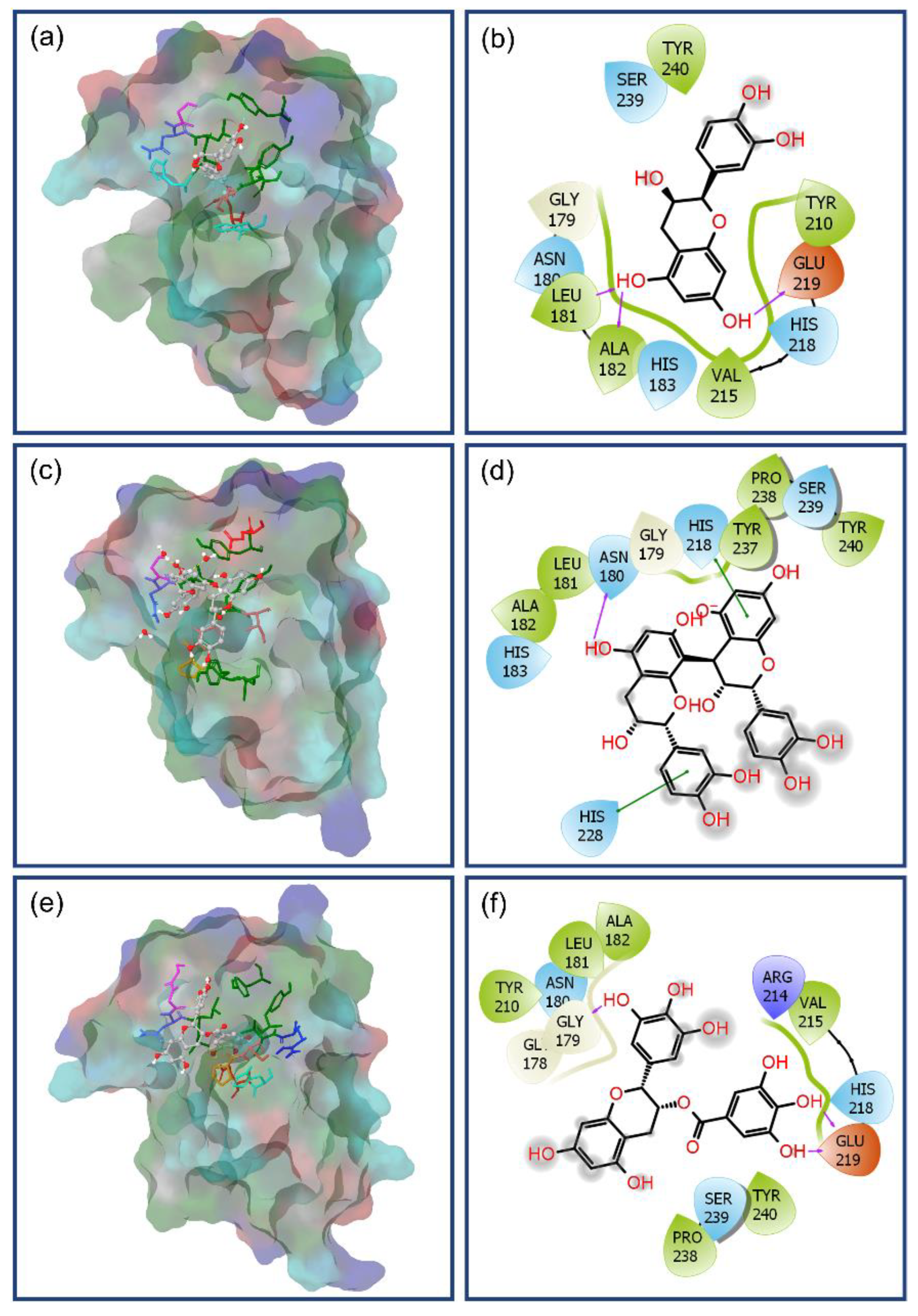
3.2. Molecular Docking Simulation and Intermolecular Interaction Analysis
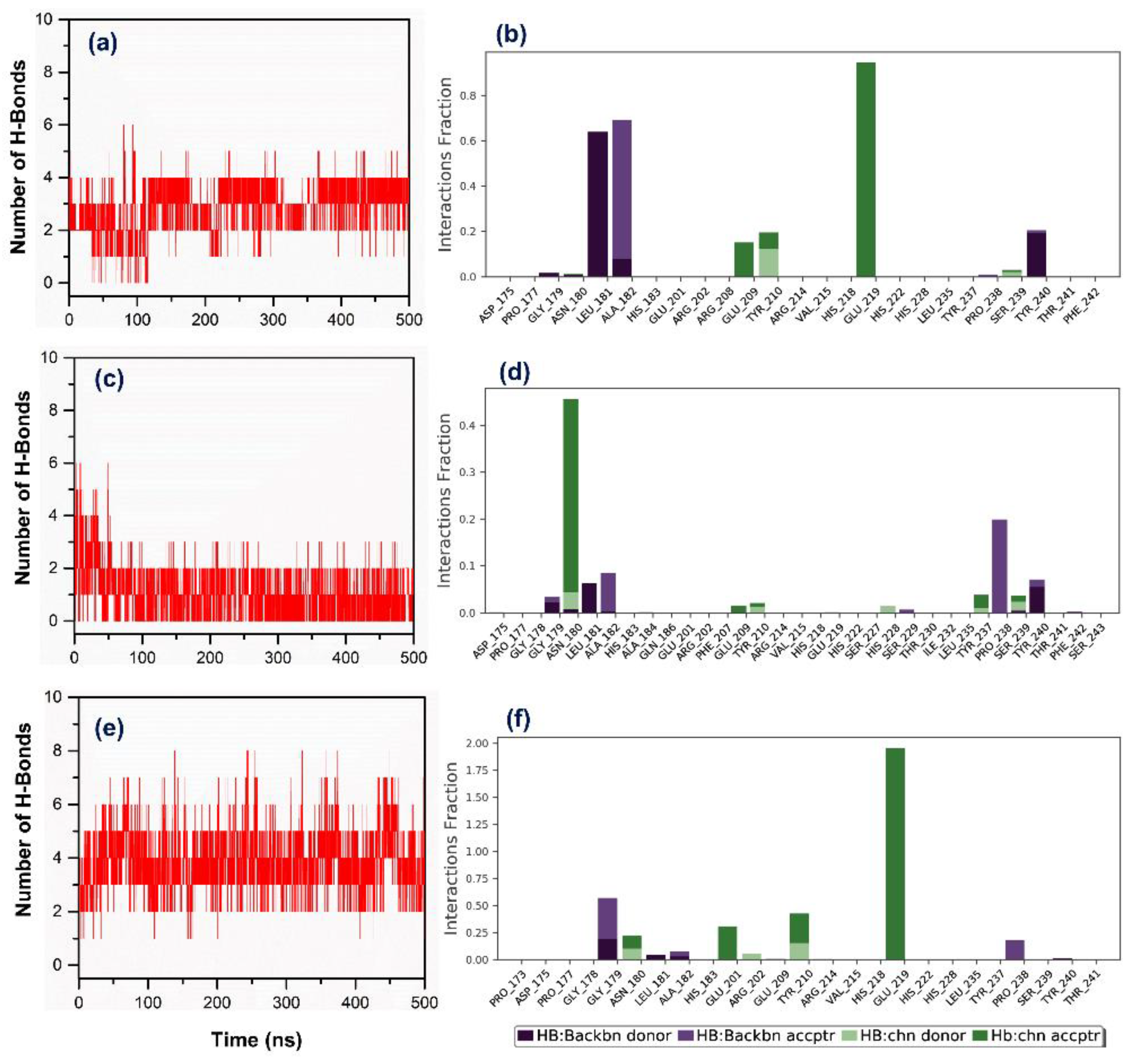
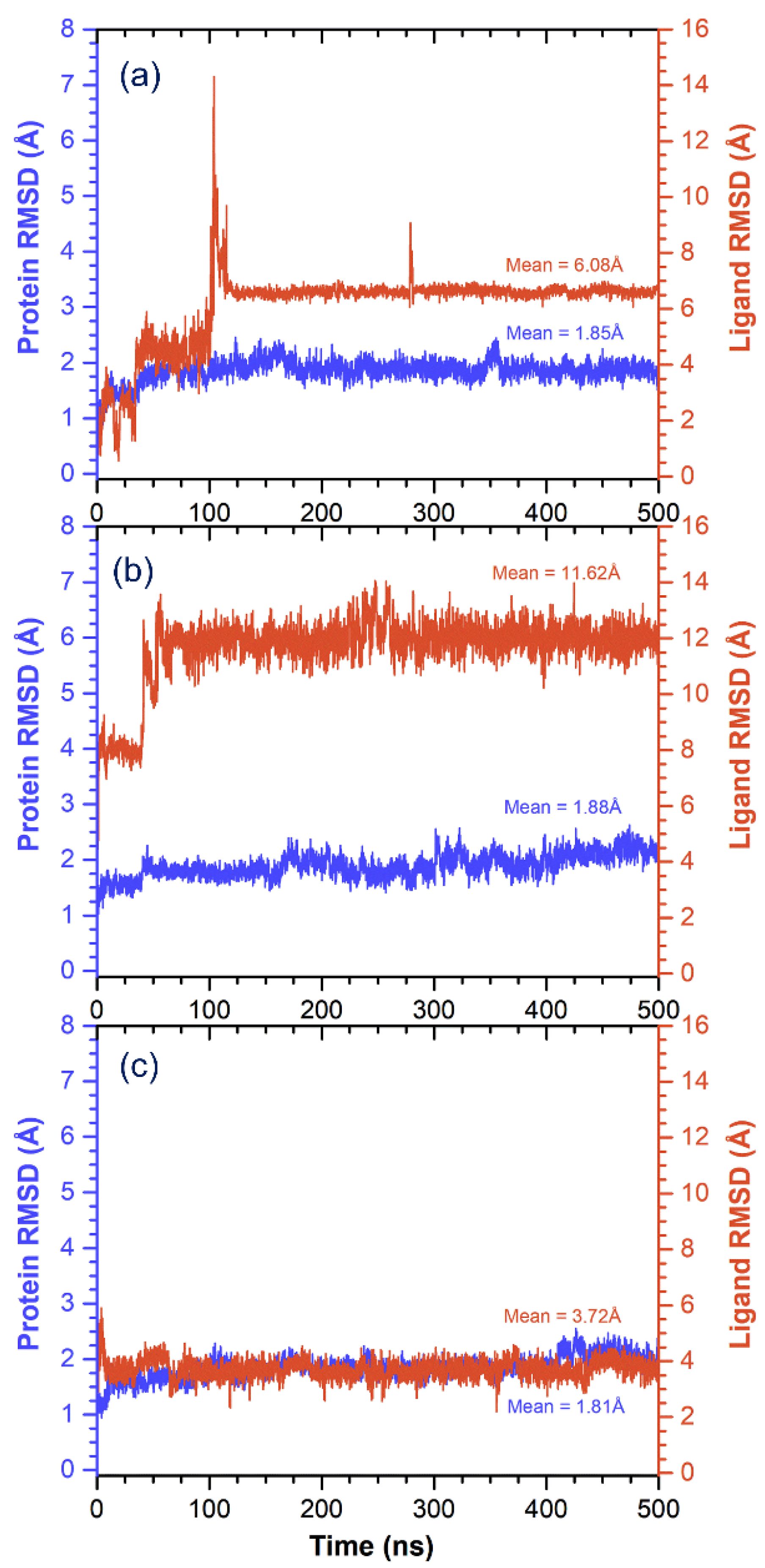
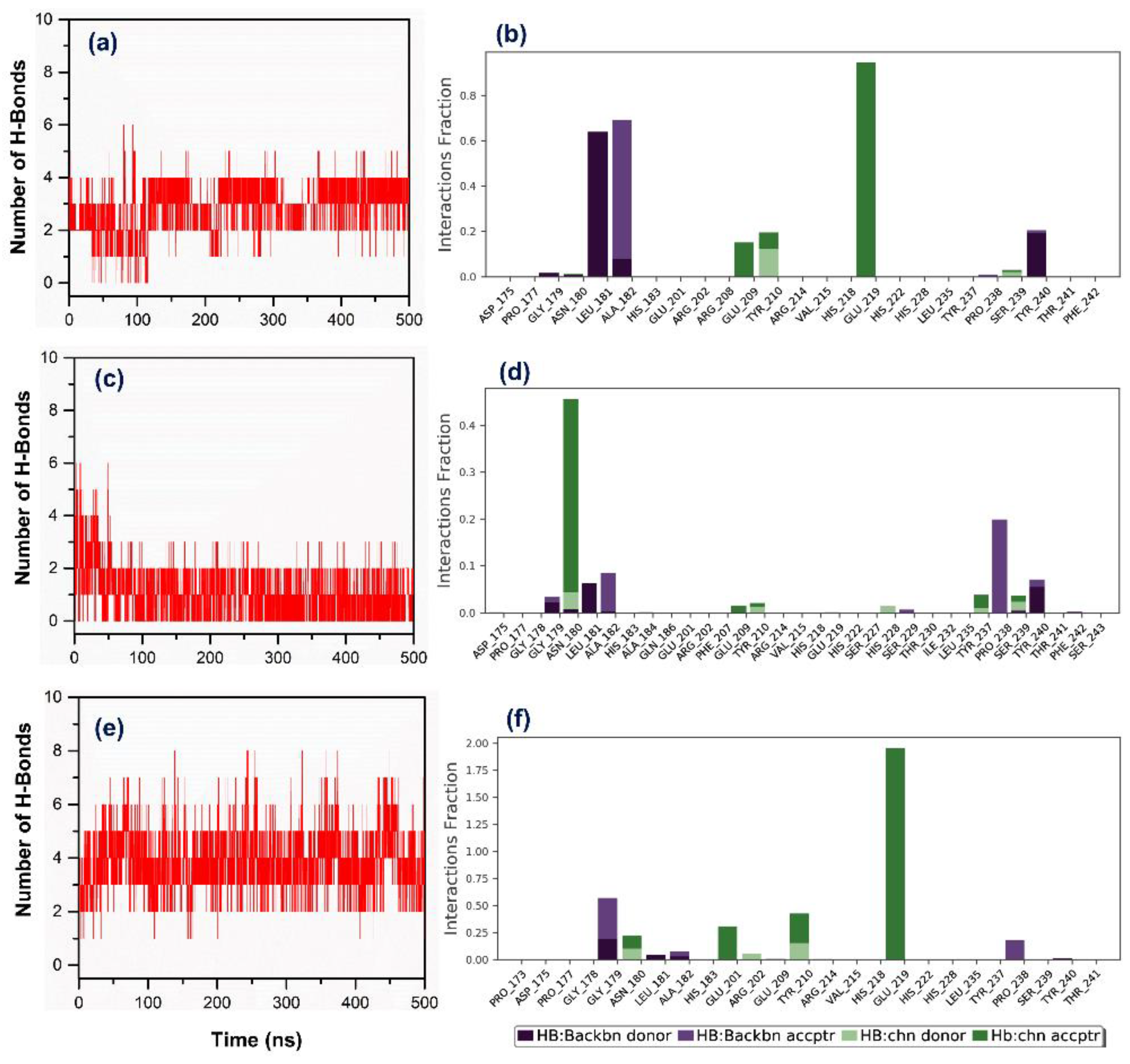
3.3. Molecular Dynamics Simulation Analysis
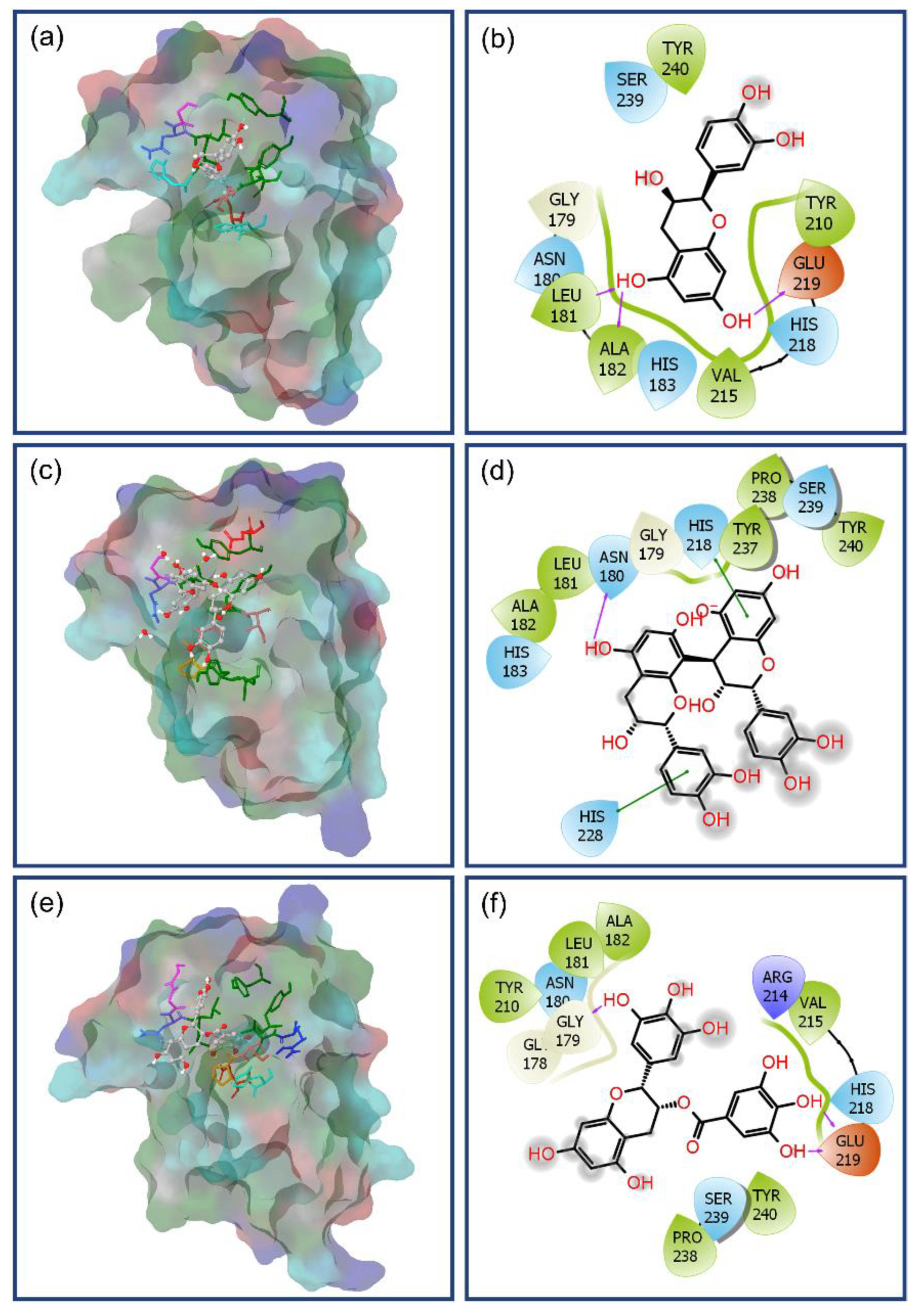
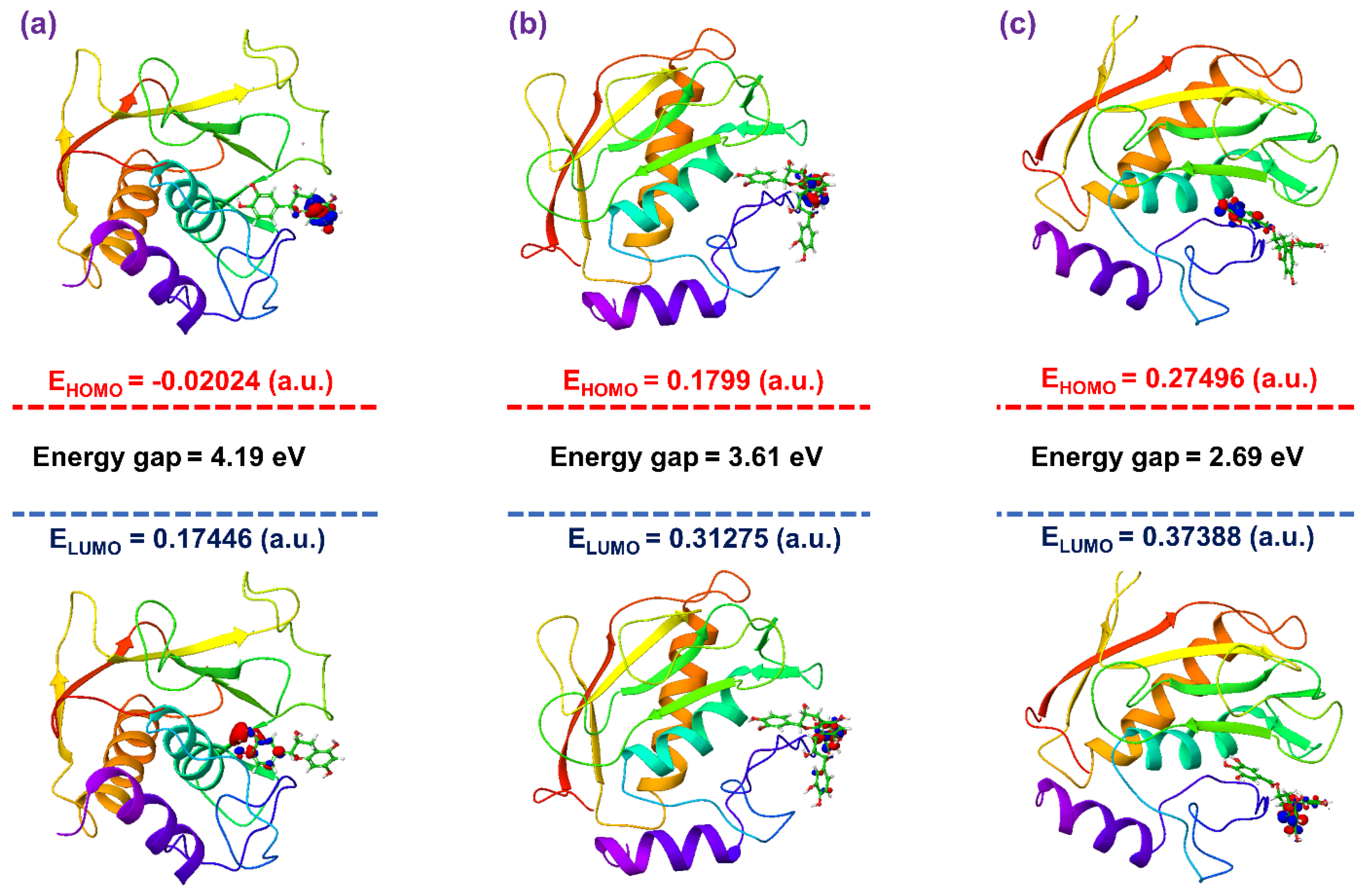
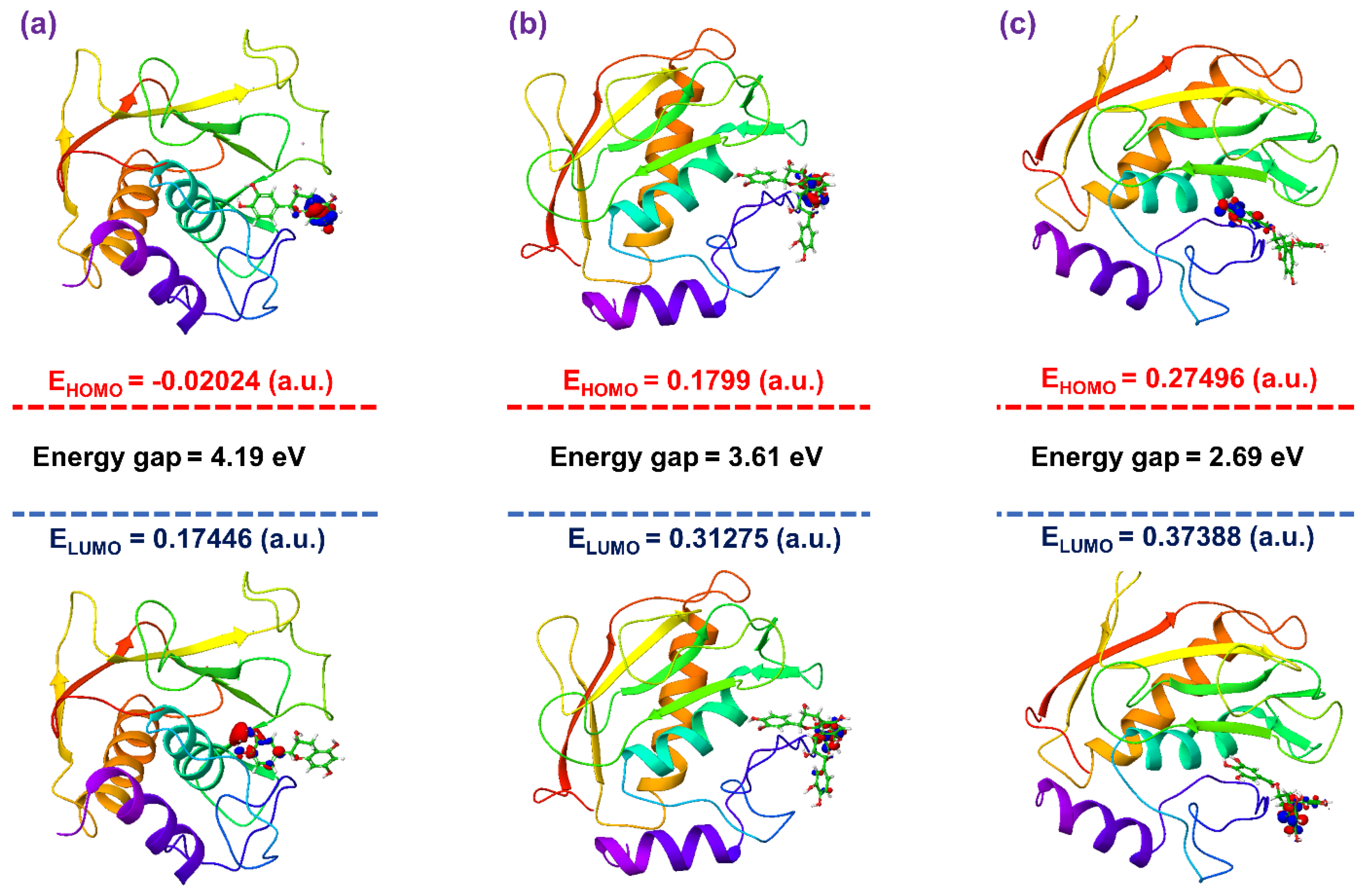
3.4. Post-Molecular Simulation Quantum Chemical Calculations
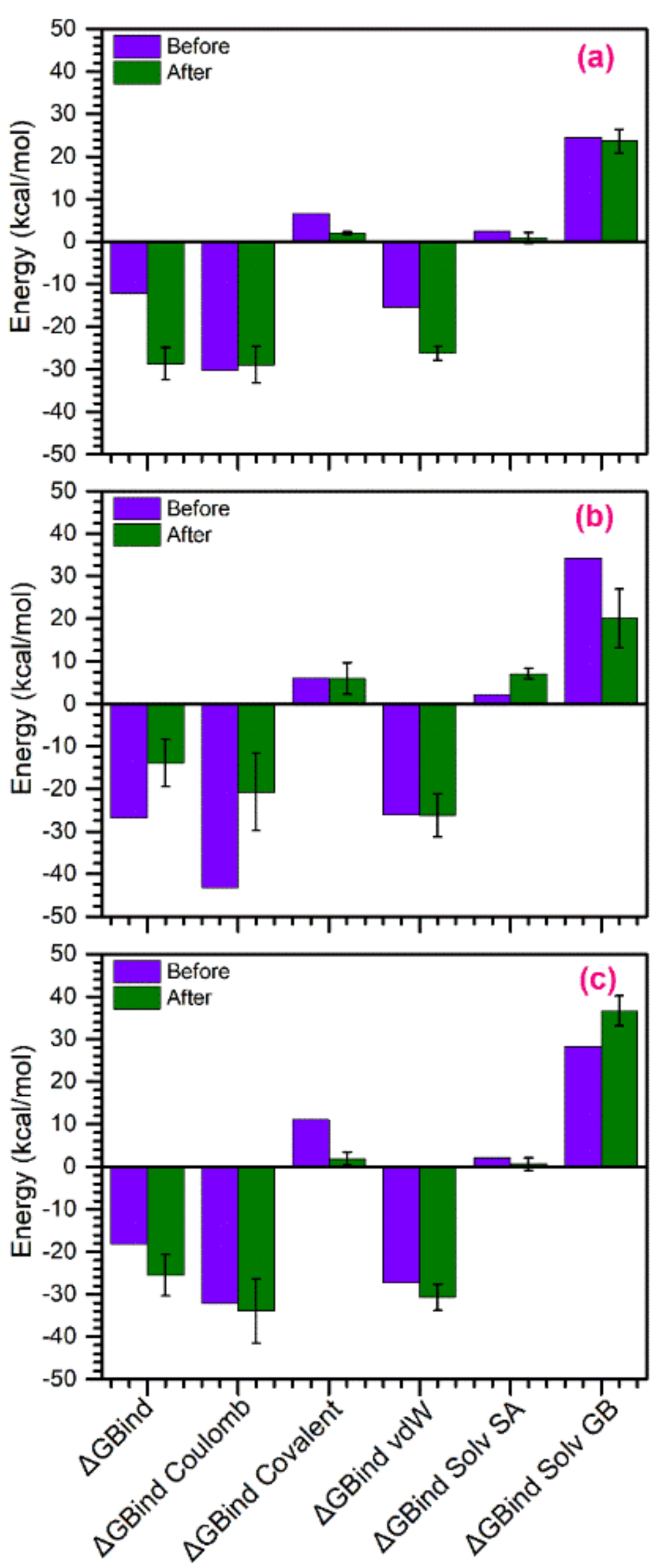
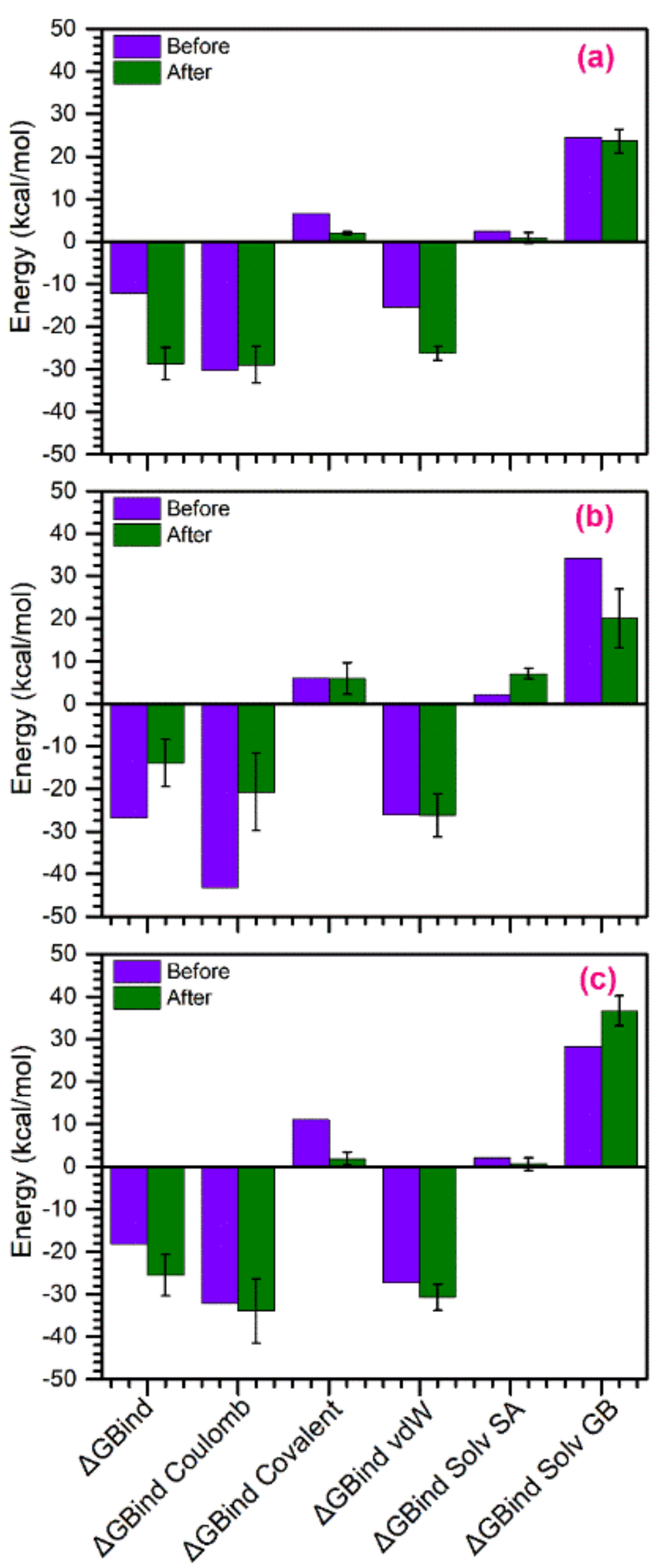
3.5. Binding Free Energy Calculations
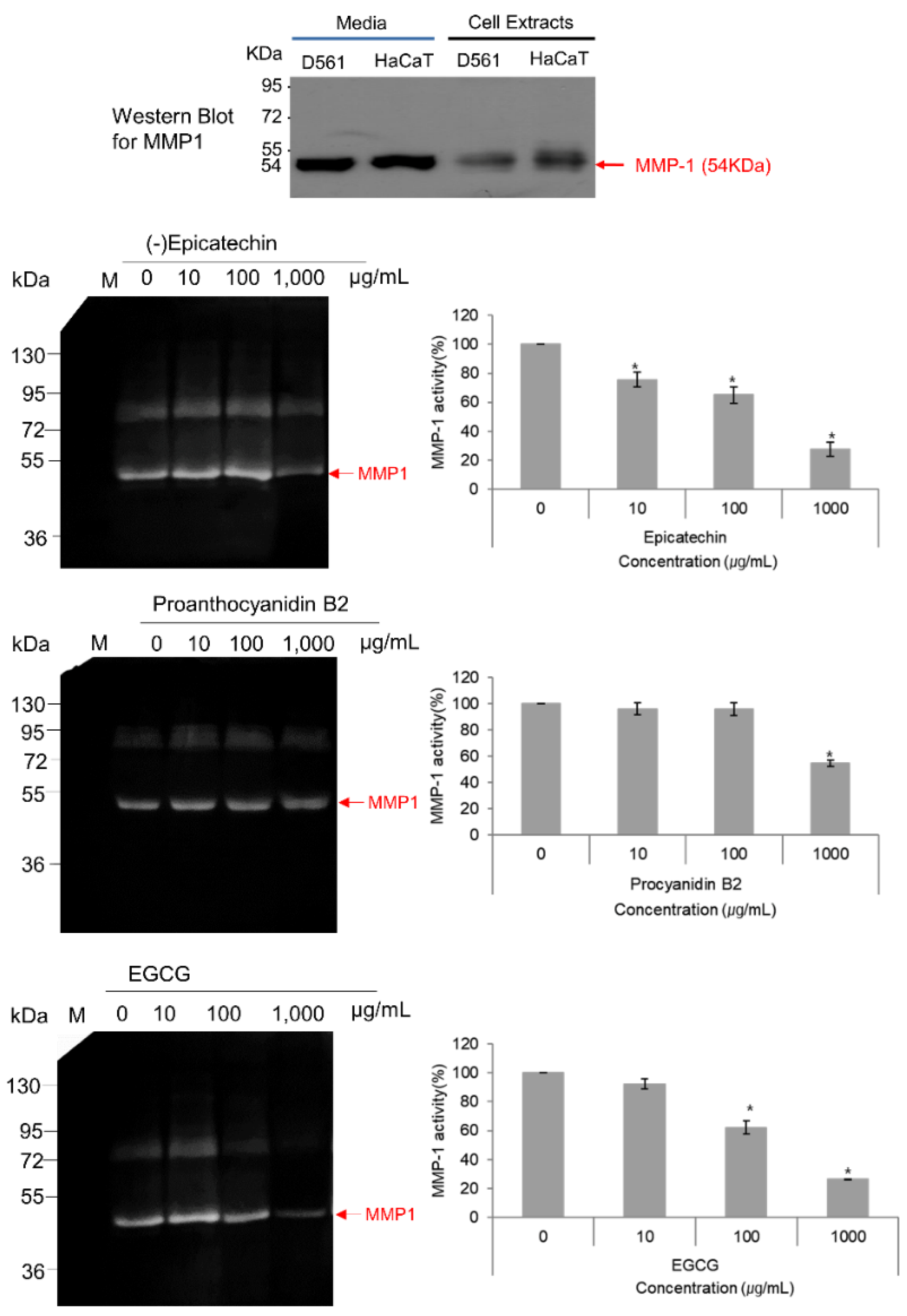
3.6. MMP-1–Zymography Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sriram, G.; Bigliardi, P.L.; Bigliardi-Qi, M. Fibroblast heterogeneity and its implications for engineering organotypic skin models in vitro. Eur. J. Cell Biol. 2015, 94, 483–512. [Google Scholar] [CrossRef] [Green Version]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef]
- McAnulty, R.J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J. Biochem. Cell B 2007, 39, 666–671. [Google Scholar] [CrossRef]
- Desmouliere, A.; Darby, I.A.; Gabbiani, G. Normal and pathologic soft tissue remodeling: Role of the myofibroblast, with special emphasis on liver and kidney fibrosis. Lab. Investig. 2003, 83, 1689–1707. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wlaschek, M.; Tantcheva-Poor, I.; Schneider, L.A.; Naderi, L.; Razi-Wolf, Z.; Schuller, J.; Scharffetter-Kochanek, K. Chronological ageing and photoageing of the fibroblasts and the dermal connective tissue. Clin. Exp. Dermatol. 2001, 26, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Rabe, J.H.; Mamelak, A.J.; McElgunn, P.J.; Morison, W.L.; Sauder, D.N. Photoaging: Mechanisms and repair. J. Am. Acad. Dermatol. 2006, 55, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.A.; Yoo, T.; Hwang, J.S.; Kang, E.S.; Paek, K.S.; Park, C.; Kim, J.H.; Do, J.T.; Seo, H.G. Peroxisome proliferator-activated receptor delta modulates MMP-2 secretion and elastin expression in human dermal fibroblasts exposed to ultraviolet B radiation. J. Dermatol. Sci. 2014, 76, 44–50. [Google Scholar] [CrossRef]
- Hwang, Y.P.; Choi, J.H.; Kim, H.G.; Choi, J.M.; Hwang, S.K.; Chung, Y.C.; Jeong, H.G. Cultivated ginseng suppresses ultraviolet B-induced collagenase activation via mitogen-activated protein kinases and nuclear factor kappaB/activator protein-1-dependent signaling in human dermal fibroblasts. Nutr. Res. 2012, 32, 428–438. [Google Scholar] [CrossRef]
- Quan, T.H.; Qin, Z.P.; Xia, W.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Matrix-Degrading Metalloproteinases in Photoaging. J. Investig. Derm. Symp. P 2009, 14, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.K.; Lee, K.W.; Kim, H.Y.; Oh, M.H.; Byun, S.; Lim, S.H.; Heo, Y.S.; Kang, N.J.; Bode, A.M.; Dong, Z.G.; et al. Myricetin suppresses UVB-induced wrinkle formation and MMP-9 expression by inhibiting Raf. Biochem. Pharmacol. 2010, 79, 1455–1461. [Google Scholar] [CrossRef] [Green Version]
- Sbardella, D.; Fasciglione, G.F.; Gioia, M.; Ciaccio, C.; Tundo, G.R.; Marini, S.; Coletta, M. Human matrix metalloproteinases: An ubiquitarian class of enzymes involved in several pathological processes. Mol. Aspects Med. 2012, 33, 119–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.; Werb, Z. The many faces of metalloproteases: Cell growth, invasion, angiogenesis and metastasis. Trends Cell. Biol. 2001, 11, S37–S43. [Google Scholar] [CrossRef]
- Piao, M.J.; Kumara, M.H.S.R.; Kim, K.C.; Kang, K.A.; Kang, H.K.; Lee, N.H.; Hyun, J.W. Diphlorethohydroxycarmalol Suppresses Ultraviolet B-Induced Matrix Metalloproteinases via Inhibition of JNK and ERK Signaling in Human Keratinocytes. Biomol. Ther. 2015, 23, 557–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiros, G.J.; Kusinsky, A.G.; Balana, M.E.; Hagelin, K. Triolein reduces MMP-1 upregulation in dermal fibroblasts generated by ROS production in UVB-irradiated keratinocytes. J. Derm. Sci. 2017, 85, 124–130. [Google Scholar] [CrossRef]
- Clark, I.M.; Cawston, T.E. Fragments of human fibroblast collagenase. Purification and characterization. Biochem. J. 1989, 263, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Guo, J.H.; Tu, X.L.; Zhang, C.; Zhao, M.; Zhang, Q.W.; Gao, F.H. Tiron Inhibits UVB-Induced AP-1 Binding Sites Transcriptional Activation on MMP-1 and MMP-3 Promoters by MAPK Signaling Pathway in Human Dermal Fibroblasts. PLoS ONE 2016, 11, e0159998. [Google Scholar] [CrossRef]
- Chiocchio, I.; Mandrone, M.; Sanna, C.; Maxia, A.; Tacchini, M.; Poli, F. Screening of a hundred plant extracts as tyrosinase and elastase inhibitors, two enzymatic targets of cosmetic interest. Ind. Crop. Prod. 2018, 122, 498–505. [Google Scholar] [CrossRef]
- Garbisa, S.; Sartor, L.; Biggin, S.; Salvato, B.; Benelli, R.; Albini, A. Tumor gelatinases and invasion inhibited by the green tea flavanol epigallocatechin-3-gallate. Cancer 2001, 91, 822–832. [Google Scholar] [CrossRef]
- Huang, B.; Chen, H.Q. (-)-Epigallocatechin-3-gallate inhibits matrix metalloproteinases in oral ulcers. RSC Adv. 2015, 5, 23758–23766. [Google Scholar] [CrossRef]
- La, V.D.; Bergeron, C.; Gafner, S.; Grenier, D. Grape seed extract suppresses lipopolysaccharide-induced matrix metalloproteinase (MMP) secretion by macrophages and inhibits human MMP-1 and -9 activities. J. Periodontol. 2009, 80, 1875–1882. [Google Scholar] [CrossRef]
- Yamakoshi, J.; Saito, M.; Kataoka, S.; Kikuchi, M. Safety evaluation of proanthocyanidin-rich extract from grape seeds. Food Chem. Toxicol. 2002, 40, 599–607. [Google Scholar] [CrossRef]
- Yu, F.; Li, B.Y.; Li, X.L.; Cai, Q.; Zhang, Z.; Cheng, M.; Yin, M.; Wang, J.F.; Zhang, J.H.; Lu, W.D.; et al. Proteomic analysis of aorta and protective effects of grape seed procyanidin B2 in db/db mice reveal a critical role of milk fat globule epidermal growth factor-8 in diabetic arterial damage. PLoS ONE 2012, 7, e52541. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.M.; Cai, X.; Kwik-Uribe, C.L.; Zeng, R.; Zhu, X.Z. Inhibitory effects of procyanidin B(2) dimer on lipid-laden macrophage formation. J. Cardiovasc. Pharmacol. 2006, 48, 54–70. [Google Scholar] [CrossRef] [PubMed]
- Li, B.Y.; Li, X.L.; Cai, Q.; Gao, H.Q.; Cheng, M.; Zhang, J.H.; Wang, J.F.; Yu, F.; Zhou, R.H. Induction of lactadherin mediates the apoptosis of endothelial cells in response to advanced glycation end products and protective effects of grape seed procyanidin B2 and resveratrol. Apoptosis 2011, 16, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, G.G.; Adamo, A.M.; Decker, N.P.; Oteiza, P.I. Dimeric procyanidin B2 inhibits constitutively active NF-kappaB in Hodgkin’s lymphoma cells independently of the presence of IkappaB mutations. Biochem. Pharmacol. 2008, 75, 1461–1471. [Google Scholar] [CrossRef]
- Spurlino, J.C.; Smallwood, A.M.; Carlton, D.D.; Banks, T.M.; Vavra, K.J.; Johnson, J.S.; Cook, E.R.; Falvo, J.; Wahl, R.C.; Pulvino, T.A.; et al. 1.56 A structure of mature truncated human fibroblast collagenase. Proteins 1994, 19, 98–109. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision B. 04; Gaussian Inc.: Wallingford, CT, USA, 2003. [Google Scholar]
- Bharadwaj, S.; Lee, K.E.; Dwivedi, V.D.; Yadava, U.; Nees, M.; Kang, S.G. Density functional theory and molecular dynamics simulation support Ganoderma lucidum triterpenoids as broad range antagonist of matrix metalloproteinases. J. Mol. Liq. 2020, 311, 113322. [Google Scholar] [CrossRef]
- Yadava, U.; Gupta, H.; Roychoudhury, M. A comparison of crystallographic and DFT optimized geometries on two taxane diterpenoids and docking studies with phospholipase A2. Med. Chem. Res. 2012, 21, 2162–2168. [Google Scholar] [CrossRef]
- Yadava, U.; Gupta, H.; Roychoudhury, M. Stabilization of microtubules by taxane diterpenoids: Insight from docking and MD simulations. J. Biol. Phys. 2015, 41, 117–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release, Version 2015-2; Schrödinger, LLC: New York, NY, USA, 2015.
- Iyer, S.; Visse, R.; Nagase, H.; Acharya, K.R. Crystal structure of an active form of human MMP-1. J. Mol. Biol. 2006, 362, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, A.C.; Kumar, A.; Rehman, A.U.; Junaid, M.; Khan, A.; Bharadwaj, S.; Sahi, S.; Wei, D.Q. Deciphering G-Protein-Coupled Receptor 119 Agonists as Promising Strategy against Type 2 Diabetes Using Systems Biology Approach. ACS Omega 2018, 3, 18214–18226. [Google Scholar] [CrossRef]
- Li, L.; Li, C.; Zhang, Z.; Alexov, E. On the Dielectric “Constant” of Proteins: Smooth Dielectric Function for Macromolecular Modeling and Its Implementation in DelPhi. J. Chem. Theory Comput. 2013, 9, 2126–2136. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 84. [Google Scholar]
- Guo, Z.; Mohanty, U.; Noehre, J.; Sawyer, T.K.; Sherman, W.; Krilov, G. Probing the alpha-helical structural stability of stapled p53 peptides: Molecular dynamics simulations and analysis. Chem. Biol. Drug Des. 2010, 75, 348–359. [Google Scholar] [CrossRef]
- Dapprich, S.; Komáromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives1Dedicated to Professor Keiji Morokuma in celebration of his 65th birthday.1. J. Mol. Struct. 1999, 461–462, 1–21. [Google Scholar] [CrossRef]
- Genheden, S.; Kuhn, O.; Mikulskis, P.; Hoffmann, D.; Ryde, U. The Normal-Mode Entropy in the MM/GBSA Method: Effect of System Truncation, Buffer Region, and Dielectric Constant. J. Chem. Inf. Model. 2012, 52, 2079–2088. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shalom, I.Y.; Pfeiffer-Marek, S.; Baringhaus, K.H.; Gohlke, H. Efficient Approximation of Ligand Rotational and Translational Entropy Changes upon Binding for Use in MM-PBSA Calculations. J. Chem. Inf. Model. 2017, 57, 170–189. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.L.; Liu, X.; Zhang, J.Z.H. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein-Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Akke, M.; Ryde, U. Conformational Entropies and Order Parameters: Convergence, Reproducibility, and Transferability. J. Chem. Theory Comput. 2014, 10, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Hikiri, S.; Yoshidome, T.; Ikeguchi, M. Computational Methods for Configurational Entropy Using Internal and Cartesian Coordinates. J. Chem. Theory Comput. 2016, 12, 5990–6000. [Google Scholar] [CrossRef] [Green Version]
- Sharp, K. Calculation of Molecular Entropies Using Temperature Integration. J. Chem. Theory Comput. 2013, 9, 1164–1172. [Google Scholar] [CrossRef]
- Choi, H.; Kang, H.; Park, H. Computational prediction of molecular hydration entropy with hybrid scaled particle theory and free-energy perturbation method. J. Chem. Theory Comput. 2015, 11, 4933–4942. [Google Scholar] [CrossRef]
- Gyimesi, G.; Zavodszky, P.; Szilagyi, A. Calculation of Configurational Entropy Differences from Conformational Ensembles Using Gaussian Mixtures. J. Chem. Theory Comput. 2017, 13, 29–41. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Rao, A.K.; Dwivedi, V.D.; Mishra, S.K.; Yadava, U. Structure-based screening and validation of bioactive compounds as Zika virus methyltransferase (MTase) inhibitors through first-principle density functional theory, classical molecular simulation and QM/MM affinity estimation. J. Biomol. Struct. Dynam. 2020, 1–14. [Google Scholar] [CrossRef]
- Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Study of the Differential Activity of Thrombin Inhibitors Using Docking, QSAR, Molecular Dynamics, and MM-GBSA. PLoS ONE 2015, 10, e0142774. [Google Scholar] [CrossRef] [Green Version]
- Di, L.; Kerns, E.H. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Kramer, C.; Ting, A.; Zheng, H.; Hert, J.; Schindler, T.; Stahl, M.; Robb, G.; Crawford, J.J.; Blaney, J.; Montague, S.; et al. Learning Medicinal Chemistry Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Rules from Cross-Company Matched Molecular Pairs Analysis (MMPA). J. Med. Chem. 2018, 61, 3277–3292. [Google Scholar] [CrossRef] [PubMed]
- Macarron, R. Critical review of the role of HTS in drug discovery. Drug Discov. Today 2006, 11, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Darcel, L.; Djibo, M.; Gaillard, M.; Raviglione, D.; Bonnard, I.; Banaigs, B.; Inguimbert, N. Trichormamide C Structural Confirmation through Total Synthesis and Extension to Analogs. Org. Lett. 2020, 22, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Prabavathi, N.; Nilufer, A.; Krishnakumar, V. Vibrational spectroscopic (FT-IR and FT-Raman) studies, natural bond orbital analysis and molecular electrostatic potential surface of Isoxanthopterin. Spectrochim. Acta A 2013, 114, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Yearley, E.J.; Zhurova, E.A.; Zhurov, V.V.; Pinkerton, A.A. Experimental electron density studies of non-steroidal synthetic estrogens: Diethylstilbestrol and dienestrol. J. Mol. Struct. 2008, 890, 240–248. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Relationships between dissociation energies and electrostatic potentials of CNO2 bonds: Applications to impact sensitivities. J. Mol. Struct. 1996, 376, 419–424. [Google Scholar] [CrossRef]
- Fukui, K. Role of frontier orbitals in chemical reactions. Science 1982, 218, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Streitwieser, A., Jr. The 1981 nobel prize in chemistry. Science 1981, 214, 627–629. [Google Scholar] [CrossRef]
- Pearson, R.G. Electronic spectra and chemical reactivity. J. Am. Chem. Soc. 1988, 110, 2092–2097. [Google Scholar] [CrossRef]
- Thanthiriwatte, K.S.; de Silva, K.M.N. Non-linear optical properties of novel fluorenyl derivatives - ab initio quantum chemical calculations. J. Mol. Struc. 2002, 617, 169–175. [Google Scholar] [CrossRef]
- Aihara, J. Reduced HOMO-LUMO gap as an index of kinetic stability for polycyclic aromatic hydrocarbons. J. Phys. Chem. A 1999, 103, 7487–7495. [Google Scholar] [CrossRef]
- Shoichet, B.K.; McGovern, S.L.; Wei, B.Q.; Irwin, J.J. Lead discovery using molecular docking. Curr. Opin. Chem. Biol. 2002, 6, 439–446. [Google Scholar] [CrossRef]
- Gohlke, H.; Klebe, G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew. Chem. Int. Ed. Engl. 2002, 41, 2644–2676. [Google Scholar] [CrossRef]
- Mohankumar, T.; Chandramohan, V.; Lalithamba, H.S.; Jayaraj, R.L.; Kumaradhas, P.; Sivanandam, M.; Hunday, G.; Vijayakumar, R.; Balakrishnan, R.; Manimaran, D.; et al. Design and Molecular dynamic Investigations of 7,8-Dihydroxyflavone Derivatives as Potential Neuroprotective Agents Against Alpha-synuclein. Sci. Rep. 2020, 10, 599. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Nakashima, Y.; Tsukamoto, S.; Kurohara, T.; Suzuki, M.; Sakae, Y.; Oda, M.; Okamoto, Y.; Suzuki, T. N(+)-C-H...O Hydrogen bonds in protein-ligand complexes. Sci. Rep. 2019, 9, 767. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer Science & Business Media: Heidelberg, Germany, 2012. [Google Scholar]
- Desiraju, G.R. Hydrogen bridges in crystal engineering: Interactions without borders. Acc. Chem. Res. 2002, 35, 565–573. [Google Scholar] [CrossRef]
- Sanphui, P.; Rajput, L.; Gopi, S.P.; Desiraju, G.R. New multi-component solid forms of anti-cancer drug Erlotinib: Role of auxiliary interactions in determining a preferred conformation. Acta Crystallogr. B 2016, 72, 291–300. [Google Scholar] [CrossRef]
- Connelly, P.R.; Snyder, P.W.; Zhang, Y.; McClain, B.; Quinn, B.P.; Johnston, S.; Medek, A.; Tanoury, J.; Griffith, J.; Patrick Walters, W.; et al. The potency-insolubility conundrum in pharmaceuticals: Mechanism and solution for hepatitis C protease inhibitors. Biophys. Chem. 2015, 196, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Panigrahi, S.K. Strong and weak hydrogen bonds in protein-ligand complexes of kinases: A comparative study. Amino Acids 2008, 34, 617–633. [Google Scholar] [CrossRef]
- Derewenda, Z.S.; Lee, L.; Derewenda, U. The occurrence of C-H...O hydrogen bonds in proteins. J. Mol. Biol. 1995, 252, 248–262. [Google Scholar] [CrossRef] [PubMed]
- McDonald, I.K.; Thornton, J.M. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994, 238, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Lovejoy, B.; Cleasby, A.; Hassell, A.M.; Longley, K.; Luther, M.A.; Weigl, D.; McGeehan, G.; McElroy, A.B.; Drewry, D.; Lambert, M.H.; et al. Structure of the catalytic domain of fibroblast collagenase complexed with an inhibitor. Science 1994, 263, 375–377. [Google Scholar] [CrossRef]
- Thi, T.H.N.; Moon, Y.H.; Ryu, Y.B.; Kim, Y.M.; Nam, S.H.; Kim, M.S.; Kimura, A.; Kim, D. The influence of flavonoid compounds on the in vitro inhibition study of a human fibroblast collagenase catalytic domain expressed in E. coli. Enzyme Microb. Tech. 2013, 52, 26–31. [Google Scholar] [CrossRef]
- Madhan, B.; Krishnamoorthy, G.; Rao, J.R.; Nair, B.U. Role of green tea polyphenols in the inhibition of collagenolytic activity by collagenase. Int. J. Biol. Macromol. 2007, 41, 16–22. [Google Scholar] [CrossRef]
- Singh, A.N.; Baruah, M.M.; Sharma, N. Structure Based docking studies towards exploring potential anti-androgen activity of selected phytochemicals against Prostate Cancer. Sci. Rep. 2017, 7, 1955. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, V.D.; Tripathi, I.P.; Bharadwaj, S.; Kaushik, A.C.; Mishra, S.K. Identification of new potent inhibitors of dengue virus NS3 protease from traditional Chinese medicine database. Virusdisease 2016, 27, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, S.; Lee, K.E.; Dwivedi, V.D.; Yadava, U.; Kang, S.G. Computational aided mechanistic understanding of Camellia sinensis bioactive compounds against co-chaperone p23 as potential anticancer agent. J. Cell Biochem. 2019, 120, 19064–19075. [Google Scholar] [CrossRef]
- Chaires, J.B. Calorimetry and thermodynamics in drug design. Annu. Rev. Biophys. 2008, 37, 135–151. [Google Scholar] [CrossRef] [Green Version]
- Ruben, A.J.; Kiso, Y.; Freire, E. Overcoming roadblocks in lead optimization: A thermodynamic perspective. Chem. Biol. Drug Design 2006, 67, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Freire, E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov. Today 2008, 13, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Campoy, A.; Kiso, Y.; Freire, E. The binding energetics of first- and second-generation HIV-1 protease inhibitors: Implications for drug design. Arch. Biochem. Biophys. 2001, 390, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Freire, E. Isothermal titration calorimetry: Controlling binding forces in lead optimization. Drug Discov. Today Technol. 2004, 1, 295–299. [Google Scholar] [CrossRef]
- Lonsdale, R.; Harvey, J.N.; Mulholland, A.J. Effects of Dispersion in Density Functional Based Quantum Mechanical/Molecular Mechanical Calculations on Cytochrome P450 Catalyzed Reactions. J. Chem. Theory Comput. 2012, 8, 4637–4645. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Lee, K.E.; Dwivedi, V.D.; Yadava, U.; Panwar, A.; Lucas, S.J.; Pandey, A.; Kang, S.G. Discovery of Ganoderma lucidum triterpenoids as potential inhibitors against Dengue virus NS2B-NS3 protease. Sci. Rep. 2019, 9, 19059. [Google Scholar] [CrossRef] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.E.; Bharadwaj, S.; Yadava, U.; Kang, S.G. Computational and In Vitro Investigation of (-)-Epicatechin and Proanthocyanidin B2 as Inhibitors of Human Matrix Metalloproteinase 1. Biomolecules 2020, 10, 1379. https://doi.org/10.3390/biom10101379
Lee KE, Bharadwaj S, Yadava U, Kang SG. Computational and In Vitro Investigation of (-)-Epicatechin and Proanthocyanidin B2 as Inhibitors of Human Matrix Metalloproteinase 1. Biomolecules. 2020; 10(10):1379. https://doi.org/10.3390/biom10101379
Chicago/Turabian StyleLee, Kyung Eun, Shiv Bharadwaj, Umesh Yadava, and Sang Gu Kang. 2020. "Computational and In Vitro Investigation of (-)-Epicatechin and Proanthocyanidin B2 as Inhibitors of Human Matrix Metalloproteinase 1" Biomolecules 10, no. 10: 1379. https://doi.org/10.3390/biom10101379
APA StyleLee, K. E., Bharadwaj, S., Yadava, U., & Kang, S. G. (2020). Computational and In Vitro Investigation of (-)-Epicatechin and Proanthocyanidin B2 as Inhibitors of Human Matrix Metalloproteinase 1. Biomolecules, 10(10), 1379. https://doi.org/10.3390/biom10101379