Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate



Abstract
1. Introduction

2. Sphingomyelinases (SMases)
2.1. Glioblastoma (GBM)
2.2. Metastatic Cancers
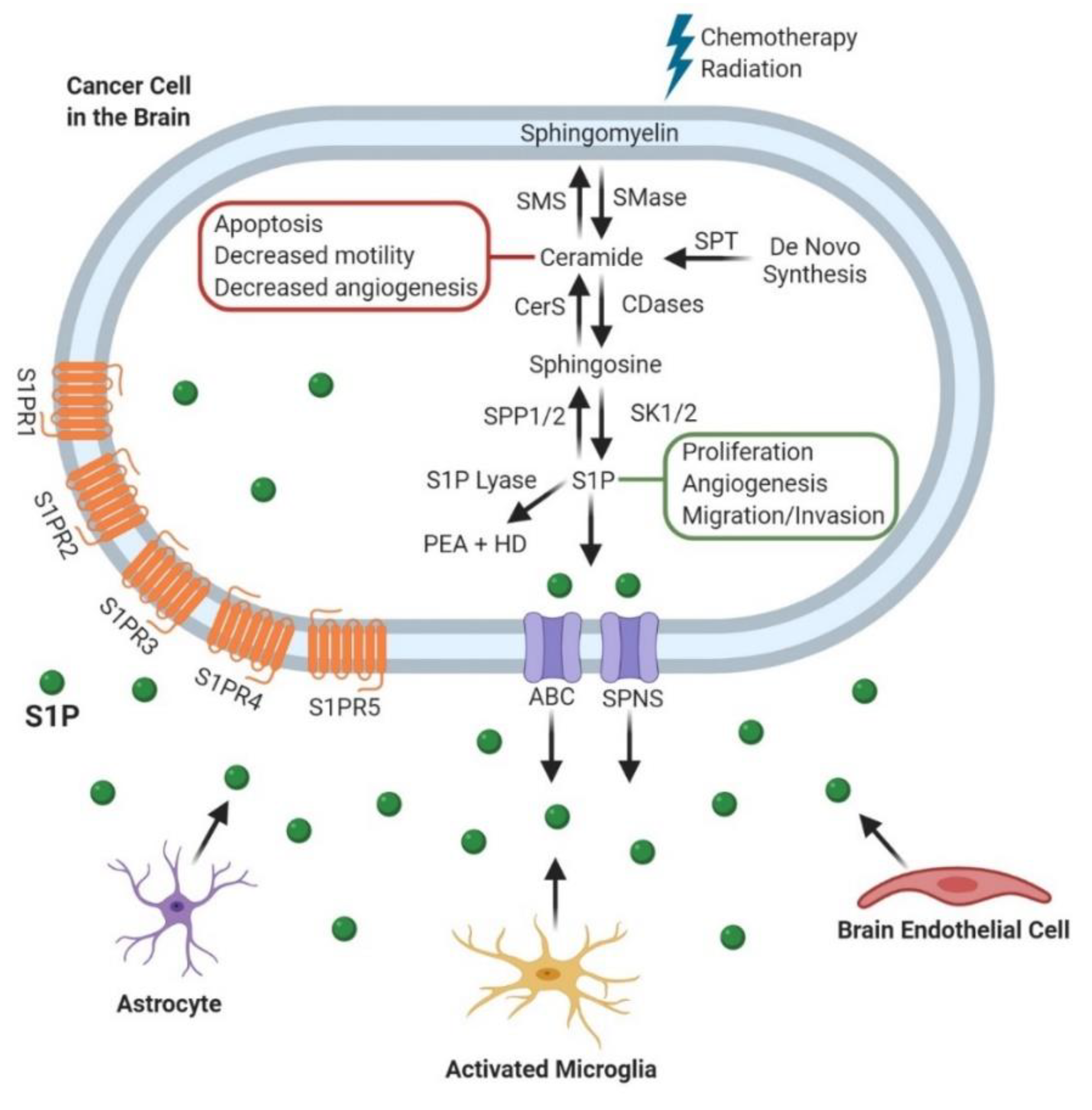
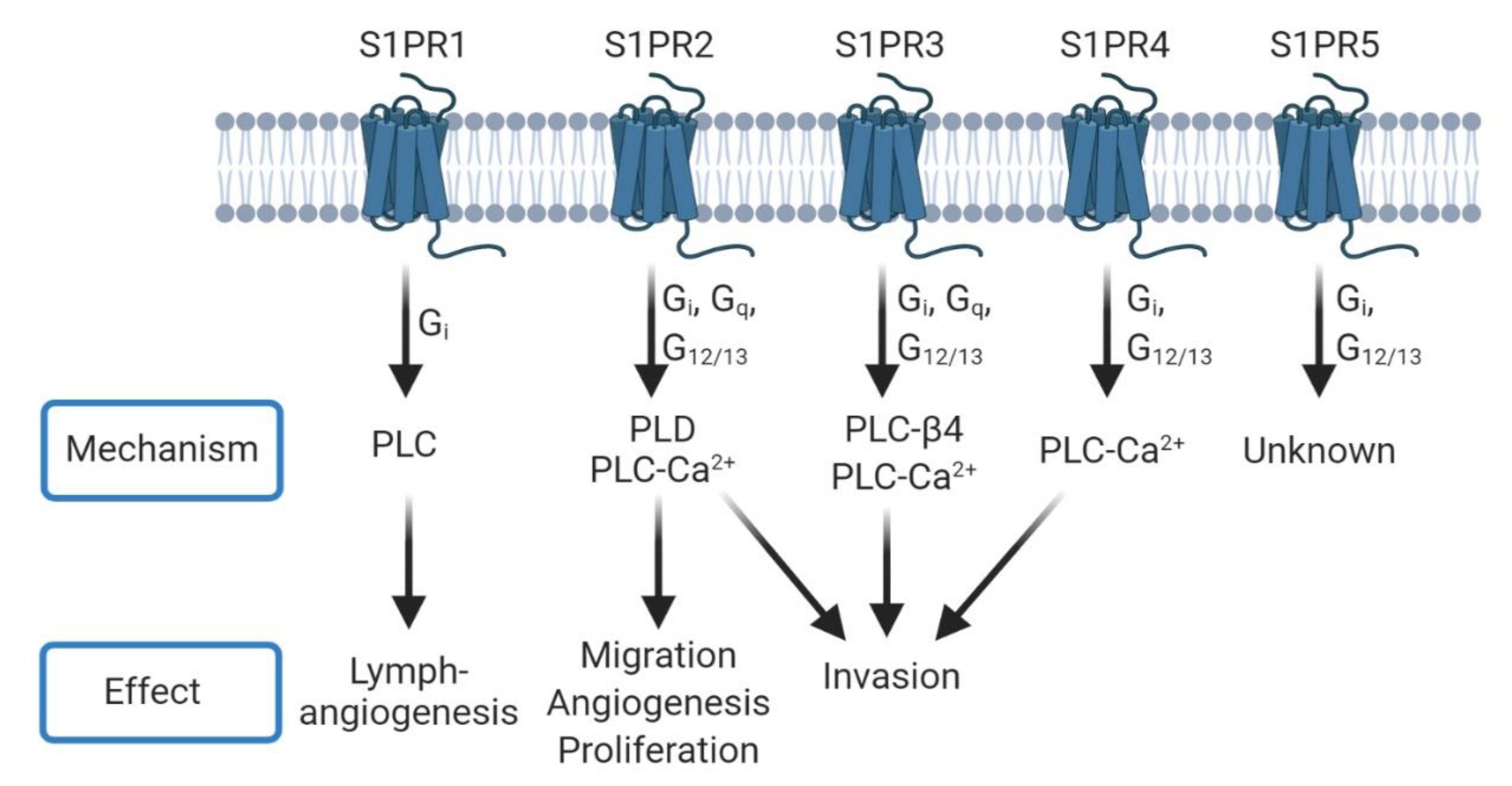
3. Sphingosine-1-Phosphate (S1P)
3.1. Glioblastoma (GBM)
3.2. Metastatic Cancers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S1PR | Cancer Type | Alteration | Phenotype | Study |
|---|---|---|---|---|
| S1PR1 | GBM | Absence | Increased proliferation | Yoshida et al., 2010 [81] |
| GBM | Presence | Increased proliferation | Young et al., 2007 [82] | |
| Breast cancer | Decrease | Decreased metastasis | Nagahashi et al., 2018 [97] | |
| Solid tumors | Decrease | Decreased migration | Liu et al., 2015 [102] | |
| S1PR2 | GBM | Absence | Increased migration | Lepley et al., 2005 [83]; Malchinkkuu et al., 2008 [84] |
| Melanoma | Decrease | Increased migration/invasion | Arikawa et al., 2003 [103] | |
| S1PR3 | GBM | Presence | Increased proliferation | Young et al., 2007 [82] |
| TNBC | Presence | Increased metastasis | Wang et al., 2018 [96] | |
| Breast cancer | Decrease | Decreased migration | Filipenko et al., 2016 [100] | |
| NSCLC | Decrease | Decreased metastasis | Zhao et al., 2016 [99] | |
| S1PR4 | Breast cancer | Presence | Decreased survival | Ohotski et al., 2012 [104] |
| S1PR5 | GBM | Presence | Increased proliferation | Young et al., 2007 [82] |
4. Phospholipase-Mediated Signaling
4.1. Glioblastoma (GBM)
4.2. Metastatic Cancers
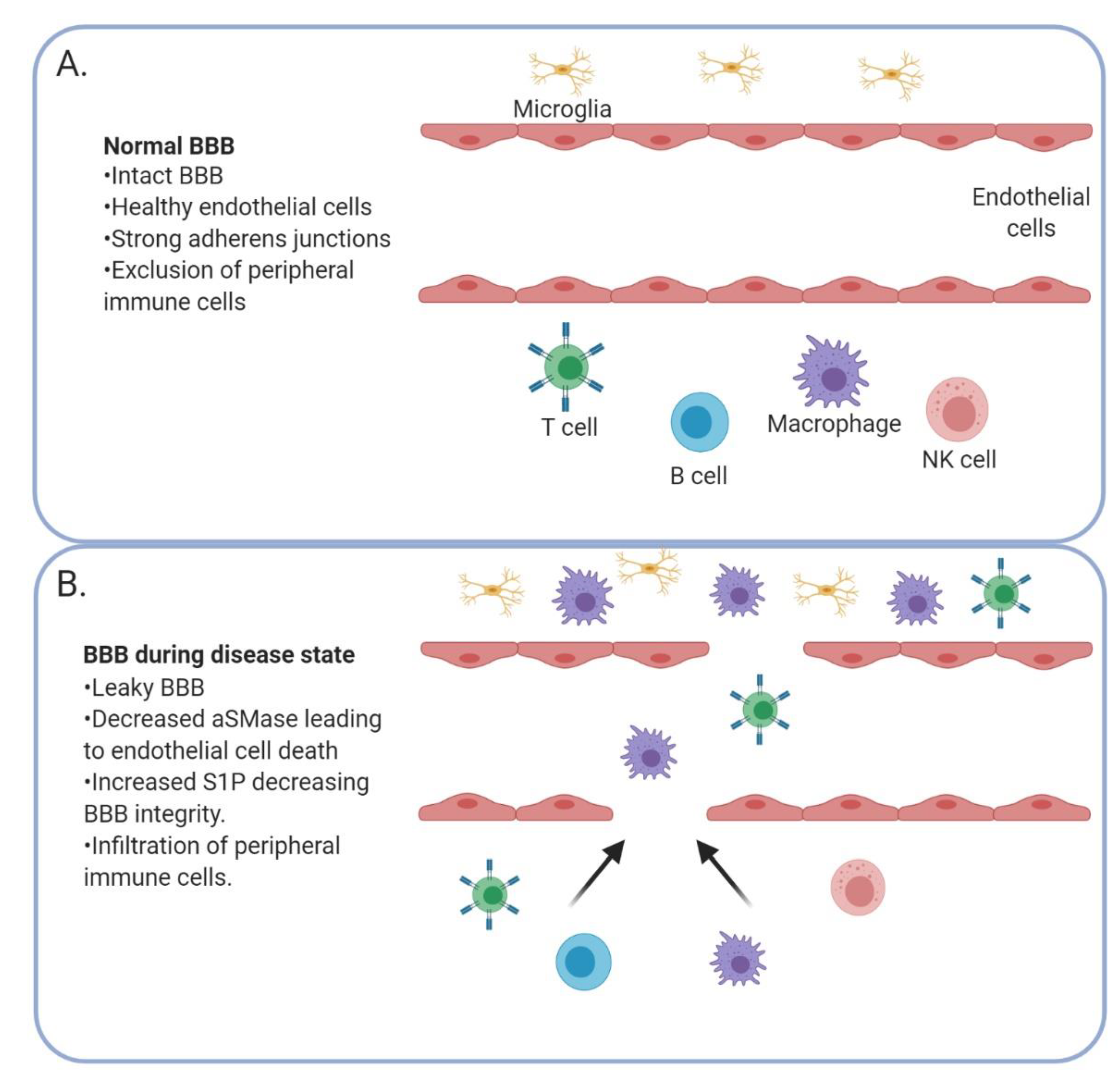
5. Blood–Brain Barrier Integrity
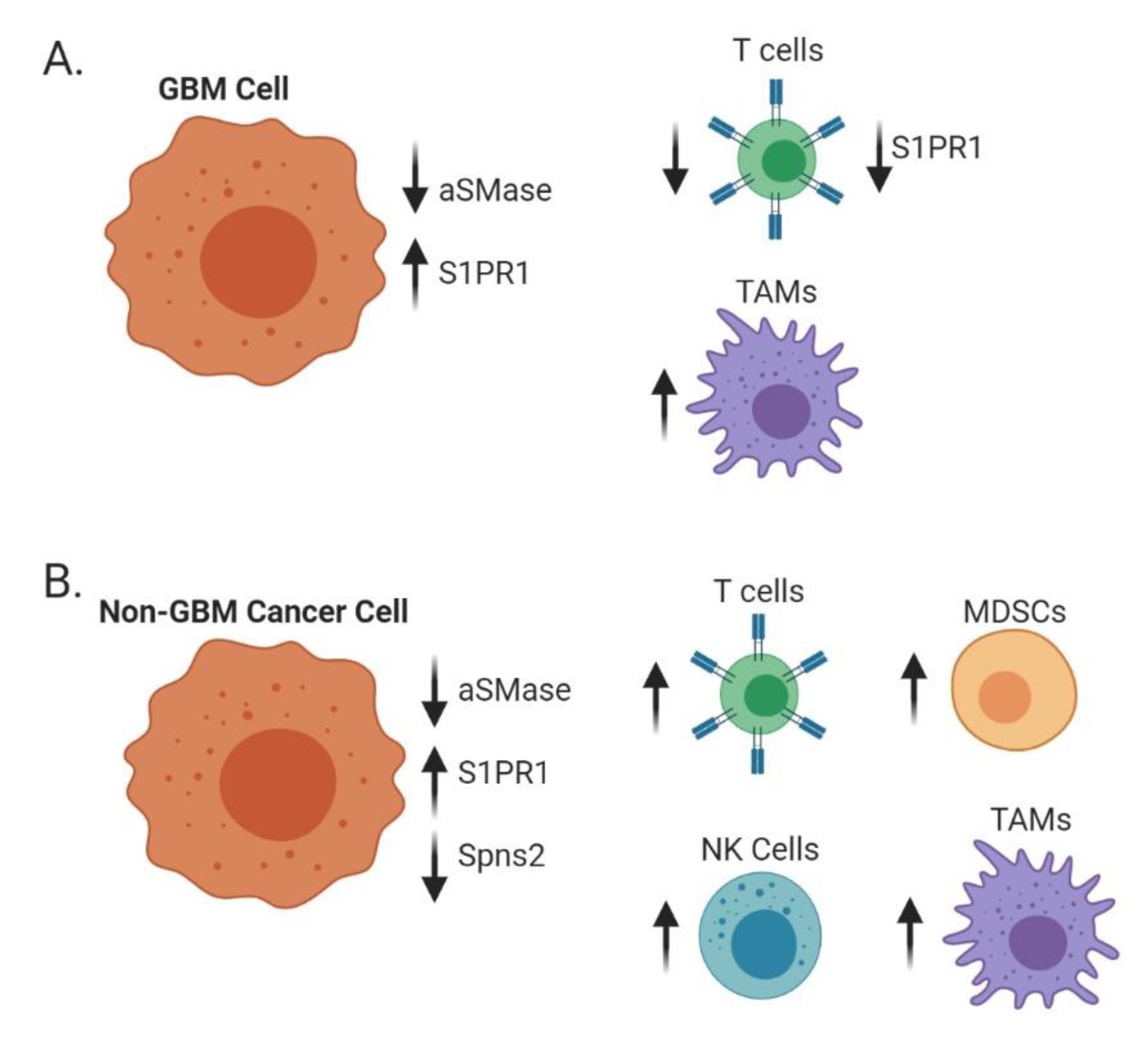
6. Immune Trafficking
6.1. Adaptive Immunity
6.2. Innate Immunity
7. Altering Sphingolipid Metabolism for Therapeutic Intervention
7.1. Glioblastoma
7.2. Metastatic Cancers
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aureli, M.; Murdica, V.; Loberto, N.; Samarani, M.; Prinetti, A.; Bassi, R.; Sonnino, S. Exploring the link between ceramide and ionizing radiation. Glycoconj. J. 2014, 31, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O.; Kanfer, J.N.; Mock, M.B.; Fredrickson, D.S. The metabolism of sphingomyelin. II. Evidence of an enzymatic deficiency in Niemann-Pick diseae. Proc. Natl. Acad. Sci. USA 1966, 55, 366. [Google Scholar] [CrossRef]
- Haimovitz-Friedman, A.; Kan, C.C.; Ehleiter, D.; Persaud, R.S.; McLoughlin, M.; Fuks, Z.; Kolesnick, R.N. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J. Exp. Med. 1994, 180, 525–535. [Google Scholar] [CrossRef]
- Senchenkov, A.; Litvak, D.A.; Cabot, M.C. Targeting Ceramide Metabolism—A Strategy for Overcoming Drug Resistance. J. Natl. Cancer Inst. 2001, 93, 347–357. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Luberto, C.; Argraves, K.M. Enzymes of sphingolipid metabolism: From modular to integrative signaling. Biochemistry 2001, 40, 4893–4903. [Google Scholar] [CrossRef]
- Wegner, M.S.; Schiffmann, S.; Parnham, M.J.; Geisslinger, G.; Grösch, S. The enigma of ceramide synthase regulation in mammalian cells. Prog. Lipid Res. 2016, 63, 93–119. [Google Scholar] [CrossRef]
- Nganga, R.; Oleinik, N.; Ogretmen, B. Mechanisms of Ceramide-Dependent Cancer Cell Death. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2018; Volume 140, pp. 1–25. [Google Scholar]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [CrossRef]
- Mizushima, N.; Koike, R.; Kohsaka, H.; Kushi, Y.; Handa, S.; Yagita, H.; Miyasaka, N. Ceramide induces apoptosis via CPP32 activation. FEBS Lett. 1996, 395, 267–271. [Google Scholar] [CrossRef]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef]
- Jung, J.S.; Ahn, Y.H.; Moon, B.I.; Kim, H.S. Exogenous C2 Ceramide Suppresses Matrix Metalloproteinase Gene Expression by Inhibiting ROS Production and MAPK Signaling Pathways in PMA-Stimulated Human Astroglioma Cells. Int. J. Mol. Sci. 2016, 17, 477. [Google Scholar] [CrossRef]
- Gatt, S. Enzymic hydrolysis of sphingolipids: Hydrolysis of ceramide glucoside by an enzyme from ox brain. Biochem. J. 1966, 101, 687–691. [Google Scholar] [PubMed]
- Momoi, T.; Ben-Yoseph, Y.; Nadler, H.L. Substrate-specificities of acid and alkaline ceramidases in fibroblasts from patients with Farber disease and controls. Biochem. J. 1982, 205, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Obeid, L.M. Ceramidases: Regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim. Biophys. Acta 2008, 1781, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Buehrer, B.M.; Bell, R.M. Inhibition of sphingosine kinase in vitro and in platelets. Implications for signal transduction pathways. J. Biol. Chem. 1992, 267, 3154–3159. [Google Scholar] [PubMed]
- Goetzl, E.J.; Kong, Y.; Mei, B. Lysophosphatidic acid and sphingosine 1-phosphate protection of T cells from apoptosis in association with suppression of Bax. J. Immunol. 1999, 162, 2049–2056. [Google Scholar] [PubMed]
- Augé, N.; Nikolova-Karakashian, M.; Carpentier, S.; Parthasarathy, S.; Nègre-Salvayre, A.; Salvayre, R.; Merrill, A.H., Jr.; Levade, T. Role of sphingosine 1-phosphate in the mitogenesis induced by oxidized low density lipoprotein in smooth muscle cells via activation of sphingomyelinase, ceramidase, and sphingosine kinase. J. Biol. Chem. 1999, 274, 21533–21538. [Google Scholar] [CrossRef]
- Clair, T.; Aoki, J.; Koh, E.; Bandle, R.W.; Nam, S.W.; Ptaszynska, M.M.; Mills, G.B.; Schiffmann, E.; Liotta, L.A.; Stracke, M.L. Autotaxin hydrolyzes sphingosylphosphorylcholine to produce the regulator of migration, sphingosine-1-phosphate. Cancer Res. 2003, 63, 5446–5453. [Google Scholar]
- Shaw, J.; Costa-Pinheiro, P.; Patterson, L.; Drews, K.; Spiegel, S.; Kester, M. Novel Sphingolipid-Based Cancer Therapeutics in the Personalized Medicine Era. Adv. Cancer Res. 2018, 140, 327–366. [Google Scholar] [CrossRef]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, J.S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef]
- Nagahashi, M.; Abe, M.; Sakimura, K.; Takabe, K.; Wakai, T. The role of sphingosine-1-phosphate in inflammation and cancer progression. Cancer Sci. 2018, 109, 3671–3678. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.E.; Milstien, S.; Spiegel, S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol. Metab. 2007, 18, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.R.; Becker, K.P.; Facchinetti, M.M.; Hannun, Y.A.; Obeid, L.M. PKC-dependent activation of sphingosine kinase 1 and translocation to the plasma membrane. Extracellular release of sphingosine-1-phosphate induced by phorbol 12-myristate 13-acetate (PMA). J. Biol. Chem. 2002, 277, 35257–35262. [Google Scholar] [CrossRef]
- Anelli, V.; Bassi, R.; Tettamanti, G.; Viani, P.; Riboni, L. Extracellular release of newly synthesized sphingosine-1-phosphate by cerebellar granule cells and astrocytes. J. Neurochem. 2005, 92, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Im, D.S.; Clemens, J.; Macdonald, T.L.; Lynch, K.R. Characterization of the human and mouse sphingosine 1-phosphate receptor, S1P5 (Edg-8): Structure-activity relationship of sphingosine1-phosphate receptors. Biochemistry 2001, 40, 14053–14060. [Google Scholar] [CrossRef] [PubMed]
- Hla, T.; Maciag, T. An abundant transcript induced in differentiating human endothelial cells encodes a polypeptide with structural similarities to G-protein-coupled receptors. J. Biol. Chem. 1990, 265, 9308–9313. [Google Scholar] [PubMed]
- Masana, M.I.; Brown, R.C.; Pu, H.; Gurney, M.E.; Dubocovich, M.L. Cloning and characterization of a new member of the G-protein coupled receptor EDG family. Recept. Channels 1995, 3, 255–262. [Google Scholar] [PubMed]
- Zondag, G.C.; Postma, F.R.; Etten, I.V.; Verlaan, I.; Moolenaar, W.H. Sphingosine 1-phosphate signalling through the G-protein-coupled receptor Edg-1. Biochem. J. 1998, 330 Pt 2, 605–609. [Google Scholar] [CrossRef]
- Siehler, S.; Manning, D.R. Pathways of transduction engaged by sphingosine 1-phosphate through G protein-coupled receptors. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2002, 1582, 94–99. [Google Scholar] [CrossRef]
- Sanchez, T.; Hla, T. Structural and functional characteristics of S1P receptors. J. Cell. Biochem. 2004, 92, 913–922. [Google Scholar] [CrossRef]
- Windh, R.T.; Lee, M.J.; Hla, T.; An, S.; Barr, A.J.; Manning, D.R. Differential coupling of the sphingosine 1-phosphate receptors Edg-1, Edg-3, and H218/Edg-5 to the G(i), G(q), and G(12) families of heterotrimeric G proteins. J. Biol. Chem. 1999, 274, 27351–27358. [Google Scholar] [CrossRef] [PubMed]
- Aloulou, A.; Rahier, R.; Arhab, Y.; Noiriel, A.; Abousalham, A. Phospholipases: An Overview. In Lipases and Phospholipases: Methods and Protocols; Sandoval, G., Ed.; Springer: New York, NY, USA, 2018. [Google Scholar]
- Nishizuka, Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 1992, 258, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Delon, C.; Manifava, M.; Wood, E.; Thompson, D.; Krugmann, S.; Pyne, S.; Ktistakis, N.T. Sphingosine kinase 1 is an intracellular effector of phosphatidic acid. J. Biol. Chem. 2004, 279, 44763–44774. [Google Scholar] [CrossRef] [PubMed]
- Mitra, P.; Oskeritzian, C.A.; Payne, S.G.; Beaven, M.A.; Milstien, S.; Spiegel, S. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc. Natl. Acad. Sci. USA 2006, 103, 16394–16399. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Simmons, S.; Kawamura, S.; Inoue, A.; Orba, Y.; Tokudome, T.; Sunden, Y.; Arai, Y.; Moriwaki, K.; Ishida, J.; et al. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J. Clin. Investig. 2012, 122, 1416–1426. [Google Scholar] [CrossRef]
- Zhou, J.; Saba, J.D. Identification of the first mammalian sphingosine phosphate lyase gene and its functional expression in yeast. Biochem. Biophys. Res. Commun. 1998, 242, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.R.; Perry, A. Glioblastoma. Arch. Pathol. Lab. Med. 2007, 131, 397–406. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef]
- Libby, C.J.; Tran, A.N.; Scott, S.E.; Griguer, C.; Hjelmeland, A.B. The pro-tumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Gallagher, J.; Myers, J.T.; Li, M.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; Huang, A.Y.; Rich, J.N. Direct in vivo evidence for tumor propagation by glioblastoma cancer stem cells. PLoS ONE 2011, 6, e24807. [Google Scholar] [CrossRef] [PubMed]
- Marfia, G.; Campanella, R.; Navone, S.E.; Di Vito, C.; Riccitelli, E.; Hadi, L.A.; Bornati, A.; de Rezende, G.; Giussani, P.; Tringali, C.; et al. Autocrine/paracrine sphingosine-1-phosphate fuels proliferative and stemness qualities of glioblastoma stem cells. Glia 2014, 62, 1968–1981. [Google Scholar] [CrossRef]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.D.; Young, B. Demographics of brain metastasis. Neurosurg. Clin. N. Am. 1996, 7, 337–344. [Google Scholar] [CrossRef]
- Nussbaum, E.S.; Djalilian, H.R.; Cho, K.H.; Hall, W.A. Brain metastases. Histology, multiplicity, surgery, and survival. Cancer 1996, 78, 1781–1788. [Google Scholar] [CrossRef]
- Graf, A.H.; Buchberger, W.; Langmayr, H.; Schmid, K.W. Site preference of metastatic tumours of the brain. Virchows Arch. A 1988, 412, 493–498. [Google Scholar] [CrossRef]
- Nayak, L.; Lee, E.Q.; Wen, P.Y. Epidemiology of Brain Metastases. Curr. Oncol. Rep. 2012, 14, 48–54. [Google Scholar] [CrossRef]
- Lowery, F.J.; Yu, D. Brain metastasis: Unique challenges and open opportunities. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 49–57. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Kotecha, R.; Gondi, V.; Ahluwalia, M.S.; Brastianos, P.K.; Mehta, M.P. Recent advances in managing brain metastasis. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Hubbs, J.L.; Boyd, J.A.; Hollis, D.; Chino, J.P.; Saynak, M.; Kelsey, C.R. Factors associated with the development of brain metastases: Analysis of 975 patients with early stage nonsmall cell lung cancer. Cancer 2010, 116, 5038–5046. [Google Scholar] [CrossRef]
- Dawood, S.; Broglio, K.; Esteva, F.J.; Yang, W.; Kau, S.W.; Islam, R.; Albarracin, C.; Yu, T.K.; Green, M.; Hortobagyi, G.N.; et al. Survival among women with triple receptor-negative breast cancer and brain metastases. Ann. Oncol. 2009, 20, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, J.; Geraghty, L.N.; Rose, A.E.; Christos, P.J.; Mazumdar, M.; Polsky, D.; Shapiro, R.; Berman, R.; Darvishian, F.; Hernando, E.; et al. Clinical variables and primary tumor characteristics predictive of the development of melanoma brain metastases and post-brain metastases survival. Cancer 2011, 117, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F. The brain metastatic niche. J. Mol. Med. 2015, 93, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Santana, P.; Peña, L.A.; Haimovitz-Friedman, A.; Martin, S.; Green, D.; McLoughlin, M.; Cordon-Cardo, C.; Schuchman, E.H.; Fuks, Z.; Kolesnick, R. Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell 1996, 86, 189–199. [Google Scholar] [CrossRef]
- Dbaibo, G.S.; Pushkareva, M.Y.; Rachid, R.A.; Alter, N.; Smyth, M.J.; Obeid, L.M.; Hannun, Y.A. p53-dependent ceramide response to genotoxic stress. J. Clin. Investig. 1998, 102, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Grammatikos, G.; Teichgräber, V.; Carpinteiro, A.; Trarbach, T.; Weller, M.; Hengge, U.R.; Gulbins, E. Overexpression of acid sphingomyelinase sensitizes glioma cells to chemotherapy. Antioxid. Redox Signal. 2007, 9, 1449–1456. [Google Scholar] [CrossRef]
- Gramatzki, D.; Herrmann, C.; Happold, C.; Becker, K.A.; Gulbins, E.; Weller, M.; Tabatabai, G. Glioma cell death induced by irradiation or alkylating agent chemotherapy is independent of the intrinsic ceramide pathway. PLoS ONE 2013, 8, e63527. [Google Scholar] [CrossRef] [PubMed]
- Van Meir, E.G.; Kikuchi, T.; Tada, M.; Li, H.; Diserens, A.C.; Wojcik, B.E.; Huang, H.J.; Friedmann, T.; de Tribolet, N.; Cavenee, W.K. Analysis of the p53 gene and its expression in human glioblastoma cells. Cancer Res. 1994, 54, 649–652. [Google Scholar] [PubMed]
- Wang, X.; Chen, J.X.; Liu, Y.H.; You, C.; Mao, Q. Mutant TP53 enhances the resistance of glioblastoma cells to temozolomide by up-regulating O(6)-methylguanine DNA-methyltransferase. Neurol. Sci. 2013, 34, 1421–1428. [Google Scholar] [CrossRef]
- Hara, S.; Nakashima, S.; Kiyono, T.; Sawada, M.; Yoshimura, S.; Iwama, T.; Banno, Y.; Shinoda, J.; Sakai, N. p53-Independent ceramide formation in human glioma cells during gamma-radiation-induced apoptosis. Cell Death Differ. 2004, 11, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Nakashima, S.; Kiyono, T.; Nakagawa, M.; Yamada, J.; Yamakawa, H.; Banno, Y.; Shinoda, J.; Nishimura, Y.; Nozawa, Y.; et al. p53 regulates ceramide formation by neutral sphingomyelinase through reactive oxygen species in human glioma cells. Oncogene 2001, 20, 1368–1378. [Google Scholar] [CrossRef]
- Xu, R.; Garcia-Barros, M.; Wen, S.; Li, F.; Lin, C.-L.; Hannun, Y.A.; Obeid, L.M.; Mao, C. Tumor suppressor p53 links ceramide metabolism to DNA damage response through alkaline ceramidase 2. Cell Death Differ. 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tseng, T.H.; Shen, C.H.; Huang, W.S.; Chen, C.N.; Liang, W.H.; Lin, T.H.; Kuo, H.C. Activation of neutral-sphingomyelinase, MAPKs, and p75 NTR-mediating caffeic acid phenethyl ester-induced apoptosis in C6 glioma cells. J. Biomed. Sci. 2014, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Kachler, K.; Bailer, M.; Heim, L.; Schumacher, F.; Reichel, M.; Holzinger, C.D.; Trump, S.; Mittler, S.; Monti, J.; Trufa, D.I.; et al. Enhanced Acid Sphingomyelinase Activity Drives Immune Evasion and Tumor Growth in Non-Small Cell Lung Carcinoma. Cancer Res. 2017, 77, 5963–5976. [Google Scholar] [CrossRef] [PubMed]
- Carpinteiro, A.; Beckmann, N.; Seitz, A.; Hessler, G.; Wilker, B.; Soddemann, M.; Helfrich, I.; Edelmann, B.; Gulbins, E.; Becker, K.A. Role of Acid Sphingomyelinase-Induced Signaling in Melanoma Cells for Hematogenous Tumor Metastasis. Cell Physiol. Biochem. 2016, 38, 1–14. [Google Scholar] [CrossRef]
- Dahiya, R.; Boyle, B.; Goldberg, B.C.; Yoon, W.H.; Konety, B.; Chen, K.; Yen, T.S.; Blumenfeld, W.; Narayan, P. Metastasis-associated alterations in phospholipids and fatty acids of human prostatic adenocarcinoma cell lines. Biochem. Cell Biol. 1992, 70, 548–554. [Google Scholar] [CrossRef]
- Cervia, D.; Assi, E.; De Palma, C.; Giovarelli, M.; Bizzozero, L.; Pambianco, S.; Di Renzo, I.; Zecchini, S.; Moscheni, C.; Vantaggiato, C.; et al. Essential role for acid sphingomyelinase-inhibited autophagy in melanoma response to cisplatin. Oncotarget 2016, 7, 24995–25009. [Google Scholar] [CrossRef]
- Smith, E.L.; Schuchman, E.H. Acid Sphingomyelinase Overexpression Enhances the Antineoplastic Effects of Irradiation In Vitro and In Vivo. Mol. Ther. 2008, 16, 1565–1571. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R.; Young, N.; Roof, R. Sphingosine-1-phosphate stimulates motility and invasiveness of human glioblastoma multiforme cells. Cancer Lett. 2003, 199, 53–60. [Google Scholar] [CrossRef]
- Young, N.; Pearl, D.K.; Van Brocklyn, J.R. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol. Cancer Res. 2009, 7, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.B.; Nguyen, H.S.; Al-Gizawiy, M.M.; Mueller, W.M.; Sabbadini, R.A.; Rand, S.D.; Connelly, J.M.; Chitambar, C.R.; Schmainda, K.M.; Mirza, S.P. Acid ceramidase confers radioresistance to glioblastoma cells. Oncol. Rep. 2017, 38, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Bien-Möller, S.; Lange, S.; Holm, T.; Böhm, A.; Paland, H.; Küpper, J.; Herzog, S.; Weitmann, K.; Havemann, C.; Vogelgesang, S.; et al. Expression of S1P metabolizing enzymes and receptors correlate with survival time and regulate cell migration in glioblastoma multiforme. Oncotarget 2016, 7, 13031–13046. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.B.; Alhajala, H.; Al-Gizawiy, M.M.; Mueller, W.M.; Rand, S.D.; Connelly, J.M.; Cochran, E.J.; Chitambar, C.R.; Clark, P.; Kuo, J.; et al. Acid ceramidase and its inhibitors: A de novo drug target and a new class of drugs for killing glioblastoma cancer stem cells with high efficiency. Oncotarget 2017, 8, 112662–112674. [Google Scholar] [CrossRef] [PubMed]
- Riccitelli, E.; Giussani, P.; Di Vito, C.; Condomitti, G.; Tringali, C.; Caroli, M.; Galli, R.; Viani, P.; Riboni, L. Extracellular sphingosine-1-phosphate: A novel actor in human glioblastoma stem cell survival. PLoS ONE 2013, 8, e68229. [Google Scholar] [CrossRef] [PubMed]
- Abdel Hadi, L.; Anelli, V.; Guarnaccia, L.; Navone, S.; Beretta, M.; Moccia, F.; Tringali, C.; Urechie, V.; Campanella, R.; Marfia, G.; et al. A bidirectional crosstalk between glioblastoma and brain endothelial cells potentiates the angiogenic and proliferative signaling of sphingosine-1-phosphate in the glioblastoma microenvironment. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.S.; Yu, Z.Q.; Zhang, S.M.; Sun, G.; Zhu, J.; Xu, J.; Guo, J.; Fu, L.S. The short chain cell-permeable ceramide (C6) restores cell apoptosis and perifosine sensitivity in cultured glioblastoma cells. Mol. Biol. Rep. 2013, 40, 5645–5655. [Google Scholar] [CrossRef]
- Yoshida, Y.; Nakada, M.; Sugimoto, N.; Harada, T.; Hayashi, Y.; Kita, D.; Uchiyama, N.; Hayashi, Y.; Yachie, A.; Takuwa, Y.; et al. Sphingosine-1-phosphate receptor type 1 regulates glioma cell proliferation and correlates with patient survival. Int. J. Cancer 2010, 126, 2341–2352. [Google Scholar] [CrossRef]
- Young, N.; Van Brocklyn, J.R. Roles of sphingosine-1-phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp. Cell Res. 2007, 313, 1615–1627. [Google Scholar] [CrossRef]
- Lepley, D.; Paik, J.H.; Hla, T.; Ferrer, F. The G protein-coupled receptor S1P2 regulates Rho/Rho kinase pathway to inhibit tumor cell migration. Cancer Res. 2005, 65, 3788–3795. [Google Scholar] [CrossRef]
- Malchinkhuu, E.; Sato, K.; Maehama, T.; Mogi, C.; Tomura, H.; Ishiuchi, S.; Yoshimoto, Y.; Kurose, H.; Okajima, F. S1P(2) receptors mediate inhibition of glioma cell migration through Rho signaling pathways independent of PTEN. Biochem. Biophys. Res. Commun. 2008, 366, 963–968. [Google Scholar] [CrossRef]
- Quint, K.; Stiel, N.; Neureiter, D.; Schlicker, H.U.; Nimsky, C.; Ocker, M.; Strik, H.; Kolodziej, M.A. The role of sphingosine kinase isoforms and receptors S1P1, S1P2, S1P3, and S1P5 in primary, secondary, and recurrent glioblastomas. Tumour Biol. 2014, 35, 8979–8989. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Jaillard, C.; Harrison, S.; Stankoff, B.; Aigrot, M.S.; Calver, A.R.; Duddy, G.; Walsh, F.S.; Pangalos, M.N.; Arimura, N.; Kaibuchi, K.; et al. Edg8/S1P5: An oligodendroglial receptor with dual function on process retraction and cell survival. J. Neurosci. 2005, 25, 1459–1469. [Google Scholar] [CrossRef]
- Rothhammer, V.; Kenison, J.E.; Tjon, E.; Takenaka, M.C.; de Lima, K.A.; Borucki, D.M.; Chao, C.C.; Wilz, A.; Blain, M.; Healy, L.; et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, 2012–2017. [Google Scholar] [CrossRef] [PubMed]
- O’sullivan, S.A.; O’Sullivan, C.; Healy, L.M.; Dev, K.K.; Sheridan, G.K. Sphingosine 1-phosphate receptors regulate TLR4-induced CXCL5 release from astrocytes and microglia. J. Neurochem. 2018, 144, 736–747. [Google Scholar] [CrossRef]
- Dai, Z.; Wu, J.; Chen, F.; Cheng, Q.; Zhang, M.; Wang, Y.; Guo, Y.; Song, T. CXCL5 promotes the proliferation and migration of glioma cells in autocrine- and paracrine-dependent manners. Oncol. Rep. 2016, 36, 3303–3310. [Google Scholar] [CrossRef]
- Rostami, N.; Nikkhoo, A.; Ajjoolabady, A.; Azizi, G.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Yousefi, B.; Yousefi, M.; Jadidi-Niaragh, F. S1PR1 as a Novel Promising Therapeutic Target in Cancer Therapy. Mol. Diagn. Ther. 2019, 23, 467–487. [Google Scholar] [CrossRef] [PubMed]
- Beider, K.; Rosenberg, E.; Bitner, H.; Shimoni, A.; Leiba, M.; Koren-Michowitz, M.; Ribakovsky, E.; Klein, S.; Olam, D.; Weiss, L.; et al. The Sphingosine-1-Phosphate Modulator FTY720 Targets Multiple Myeloma via the CXCR4/CXCL12 Pathway. Clin. Cancer Res. 2017, 23, 1733–1747. [Google Scholar] [CrossRef] [PubMed]
- Azizidoost, S.; Asnafi, A.A.; Saki, N. Signaling-chemokine axis network in brain as a sanctuary site for metastasis. J. Cell Physiol. 2019, 234, 3376–3382. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Takabe, K.; Hait, N.C. Metastatic triple-negative breast cancer is dependent on SphKs/S1P signaling for growth and survival. Cell Signal. 2017, 32, 85–92. [Google Scholar] [CrossRef]
- Wang, S.; Liang, Y.; Chang, W.; Hu, B.; Zhang, Y. Triple Negative Breast Cancer Depends on Sphingosine Kinase 1 (SphK1)/Sphingosine-1-Phosphate (S1P)/Sphingosine 1-Phosphate Receptor 3 (S1PR3)/Notch Signaling for Metastasis. Med. Sci. Monit. 2018, 24, 1912–1923. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Yamada, A.; Katsuta, E.; Aoyagi, T.; Huang, W.C.; Terracina, K.P.; Hait, N.C.; Allegood, J.C.; Tsuchida, J.; Yuza, K.; et al. Targeting the SphK1/S1P/S1PR1 Axis That Links Obesity, Chronic Inflammation, and Breast Cancer Metastasis. Cancer Res. 2018, 78, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, Y.; Sun, X.; Hong, T.L.; Huang, L.S.; Zhong, M. Prognostic roles of the expression of sphingosine-1-phosphate metabolism enzymes in non-small cell lung cancer. Transl. Lung Cancer Res. 2019, 8, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, J.; Lee, J.F.; Zhang, W.; Kandouz, M.; VanHecke, G.C.; Chen, S.; Ahn, Y.H.; Lonardo, F.; Lee, M.J. TGF-β/SMAD3 Pathway Stimulates Sphingosine-1 Phosphate Receptor 3 Expression: IMPLICATION OF SPHINGOSINE-1 PHOSPHATE RECEPTOR 3 IN LUNG ADENOCARCINOMA PROGRESSION. J. Biol. Chem. 2016, 291, 27343–27353. [Google Scholar] [CrossRef]
- Filipenko, I.; Schwalm, S.; Reali, L.; Pfeilschifter, J.; Fabbro, D.; Huwiler, A.; Zangemeister-Wittke, U. Upregulation of the S1P(3) receptor in metastatic breast cancer cells increases migration and invasion by induction of PGE(2) and EP(2)/EP(4) activation. Biochim. Biophys. Acta 2016, 1861, 1840–1851. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Condeelis, J.S.; Oktay, M.H. Chemotherapy-induced metastasis: Mechanisms and translational opportunities. Clin. Exp. Metastasis 2018, 35, 269–284. [Google Scholar] [CrossRef]
- Liu, G.; Chen, Y.; Qi, F.; Jia, L.; Lu, X.A.; He, T.; Fu, Y.; Li, L.; Luo, Y. Specific chemotherapeutic agents induce metastatic behaviour through stromal- and tumour-derived cytokine and angiogenic factor signalling. J. Pathol. 2015, 237, 190–202. [Google Scholar] [CrossRef]
- Arikawa, K.; Takuwa, N.; Yamaguchi, H.; Sugimoto, N.; Kitayama, J.; Nagawa, H.; Takehara, K.; Takuwa, Y. Ligand-dependent inhibition of B16 melanoma cell migration and invasion via endogenous S1P2 G protein-coupled receptor. Requirement of inhibition of cellular RAC activity. J. Biol. Chem. 2003, 278, 32841–32851. [Google Scholar] [CrossRef] [PubMed]
- Ohotski, J.; Long, J.S.; Orange, C.; Elsberger, B.; Mallon, E.; Doughty, J.; Pyne, S.; Pyne, N.J.; Edwards, J. Expression of sphingosine 1-phosphate receptor 4 and sphingosine kinase 1 is associated with outcome in oestrogen receptor-negative breast cancer. Br. J. Cancer 2012, 106, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Bedia, C.; Casas, J.; Andrieu-Abadie, N.; Fabriàs, G.; Levade, T. Acid ceramidase expression modulates the sensitivity of A375 melanoma cells to dacarbazine. J. Biol. Chem. 2011, 286, 28200–28209. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Realini, N.; La Ferla, M.; Passalacqua, I.; Matteoli, G.; Ganesan, A.; Pistello, M.; Mazzanti, C.M.; Piomelli, D. Complete Acid Ceramidase ablation prevents cancer-initiating cell formation in melanoma cells. Sci. Rep. 2017, 7, 7411. [Google Scholar] [CrossRef]
- Pyne, N.J.; Pyne, S. Sphingosine kinase 1 enables communication between melanoma cells and fibroblasts that provides a new link to metastasis. Oncogene 2014, 33, 3361–3363. [Google Scholar] [CrossRef]
- Van der Weyden, L.; Arends, M.J.; Campbell, A.D.; Bald, T.; Wardle-Jones, H.; Griggs, N.; Velasco-Herrera, M.D.; Tüting, T.; Sansom, O.J.; Karp, N.A.; et al. Genome-wide in vivo screen identifies novel host regulators of metastatic colonization. Nature 2017, 541, 233–236. [Google Scholar] [CrossRef]
- Kang, D.W.; Hwang, W.C.; Noh, Y.N.; Park, K.S.; Min, D.S. Phospholipase D1 inhibition sensitizes glioblastoma to temozolomide and suppresses its tumorigenicity. J. Pathol. 2020. [Google Scholar] [CrossRef]
- Walker, K.; Boyd, N.H.; Anderson, J.C.; Willey, C.D.; Hjelmeland, A.B. Kinomic profiling of glioblastoma cells reveals PLCG1 as a target in restricted glucose. Biomark. Res. 2018, 6, 22. [Google Scholar] [CrossRef]
- Mercurio, L.; Cecchetti, S.; Ricci, A.; Pacella, A.; Cigliana, G.; Bozzuto, G.; Podo, F.; Iorio, E.; Carpinelli, G. Phosphatidylcholine-specific phospholipase C inhibition down- regulates CXCR4 expression and interferes with proliferation, invasion and glycolysis in glioma cells. PLoS ONE 2017, 12, e0176108. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, X.; Li, H.; Fan, J.; Qian, X.; Li, H.; Xu, Y. Phospholipase D as a key modulator of cancer progression. Biol. Rev. Camb. Philos. Soc. 2020, 95, 911–935. [Google Scholar] [CrossRef]
- Kim, E.S.; Kim, J.S.; Kim, S.G.; Hwang, S.; Lee, C.H.; Moon, A. Sphingosine 1-phosphate regulates matrix metalloproteinase-9 expression and breast cell invasion through S1P3-Gαq coupling. J. Cell Sci. 2011, 124, 2220–2230. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Nakayama, Y.; Umeda, M.; Miyazaki, K. Induction of 103-kDa gelatinase/type IV collagenase by acidic culture conditions in mouse metastatic melanoma cell lines. J. Biol. Chem. 1992, 267, 11424–11430. [Google Scholar] [PubMed]
- Kato, Y.; Ozawa, S.; Tsukuda, M.; Kubota, E.; Miyazaki, K.; St-Pierre, Y.; Hata, R. Acidic extracellular pH increases calcium influx-triggered phospholipase D activity along with acidic sphingomyelinase activation to induce matrix metalloproteinase-9 expression in mouse metastatic melanoma. FEBS J. 2007, 274, 3171–3183. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Ui, M.; Okajima, F. Differential roles of Edg-1 and Edg-5, sphingosine 1-phosphate receptors, in the signaling pathways in C6 glioma cells. Mol. Brain Res. 2000, 85, 151–160. [Google Scholar] [CrossRef]
- Desai, N.N.; Zhang, H.; Olivera, A.; Mattie, M.E.; Spiegel, S. Sphingosine-1-phosphate, a metabolite of sphingosine, increases phosphatidic acid levels by phospholipase D activation. J. Biol. Chem. 1992, 267, 23122–23128. [Google Scholar]
- Yoon, C.M.; Hong, B.S.; Moon, H.G.; Lim, S.; Suh, P.G.; Kim, Y.K.; Chae, C.B.; Gho, Y.S. Sphingosine-1-phosphate promotes lymphangiogenesis by stimulating S1P1/Gi/PLC/Ca2+ signaling pathways. Blood 2008, 112, 1129–1138. [Google Scholar] [CrossRef]
- Stacker, S.A.; Achen, M.G.; Jussila, L.; Baldwin, M.E.; Alitalo, K. Lymphangiogenesis and cancer metastasis. Nature Rev. Cancer 2002, 2, 573–583. [Google Scholar] [CrossRef]
- Paduch, R. The role of lymphangiogenesis and angiogenesis in tumor metastasis. Cell Oncol. 2016, 39, 397–410. [Google Scholar] [CrossRef]
- Meacci, E.; Nuti, F.; Catarzi, S.; Vasta, V.; Donati, C.; Bourgoin, S.; Bruni, P.; Moss, J.; Vaughan, M. Activation of Phospholipase D by Bradykinin and Sphingosine 1-Phosphate in A549 Human Lung Adenocarcinoma Cells via Different GTP-Binding Proteins and Protein Kinase C Delta Signaling Pathways. Biochemistry 2003, 42, 284–292. [Google Scholar] [CrossRef]
- Dusaban, S.S.; Purcell, N.H.; Rockenstein, E.; Masliah, E.; Cho, M.K.; Smrcka, A.V.; Brown, J.H. Phospholipase C epsilon links G protein-coupled receptor activation to inflammatory astrocytic responses. Proc. Natl. Acad. Sci. USA 2013, 110, 3609–3614. [Google Scholar] [CrossRef]
- Obermeier, B.; Verma, A.; Ransohoff, R.M. The blood-brain barrier. Handb. Clin. Neurol. 2016, 133, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Park, M.H.; Lee, J.Y.; Park, K.H.; Jung, I.K.; Kim, K.T.; Lee, Y.S.; Ryu, H.H.; Jeong, Y.; Kang, M.; Schwaninger, M.; et al. Vascular and Neurogenic Rejuvenation in Aging Mice by Modulation of ASM. Neuron 2018, 100, 167–182.e169. [Google Scholar] [CrossRef]
- Li, Y.Q.; Chen, P.; Haimovitz-Friedman, A.; Reilly, R.M.; Wong, C.S. Endothelial apoptosis initiates acute blood-brain barrier disruption after ionizing radiation. Cancer Res. 2003, 63, 5950–5956. [Google Scholar]
- Prager, B.; Spampinato, S.F.; Ransohoff, R.M. Sphingosine 1-phosphate signaling at the blood-brain barrier. Trends Mol. Med. 2015, 21, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Aruga, J. Sphingosine 1-Phosphate Signaling Is Involved in Impaired Blood-Brain Barrier Function in Ischemia-Reperfusion Injury. Mol. Neurobiol. 2020, 57, 1594–1606. [Google Scholar] [CrossRef] [PubMed]
- Stepanovska, B.; Lange, A.I.; Schwalm, S.; Pfeilschifter, J.; Coldewey, S.M.; Huwiler, A. Downregulation of S1P Lyase Improves Barrier Function in Human Cerebral Microvascular Endothelial Cells Following an Inflammatory Challenge. Int. J. Mol. Sci. 2020, 21, 1240. [Google Scholar] [CrossRef]
- Li, Q.; Chen, B.; Zeng, C.; Fan, A.; Yuan, Y.; Guo, X.; Huang, X.; Huang, Q. Differential activation of receptors and signal pathways upon stimulation by different doses of sphingosine-1-phosphate in endothelial cells. Exp. Physiol. 2015, 100, 95–107. [Google Scholar] [CrossRef]
- Van Doorn, R.; Lopes Pinheiro, M.A.; Kooij, G.; Lakeman, K.; van het Hof, B.; van der Pol, S.M.; Geerts, D.; van Horssen, J.; van der Valk, P.; van der Kam, E.; et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J. Neuroinflammation 2012, 9, 133. [Google Scholar] [CrossRef]
- Spampinato, S.F.; Obermeier, B.; Cotleur, A.; Love, A.; Takeshita, Y.; Sano, Y.; Kanda, T.; Ransohoff, R.M. Sphingosine 1 Phosphate at the Blood Brain Barrier: Can the Modulation of S1P Receptor 1 Influence the Response of Endothelial Cells and Astrocytes to Inflammatory Stimuli? PLoS ONE 2015, 10, e0133392. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, M.J.; Jung, H.H.; Chang, W.S.; Choi, H.S.; Rachmilevitch, I.; Zadicario, E.; Chang, J.W. Safety and feasibility of multiple blood-brain barrier disruptions for the treatment of glioblastoma in patients undergoing standard adjuvant chemotherapy. J. Neurosurg. 2020, 1–9. [Google Scholar] [CrossRef]
- Yanagida, K.; Liu, C.H.; Faraco, G.; Galvani, S.; Smith, H.K.; Burg, N.; Anrather, J.; Sanchez, T.; Iadecola, C.; Hla, T. Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc. Natl. Acad. Sci. USA 2017, 114, 4531–4536. [Google Scholar] [CrossRef]
- Fecci, P.E.; Heimberger, A.B.; Sampson, J.H. Immunotherapy for Primary Brain Tumors: No Longer a Matter of Privilege. Clin. Cancer Res. 2014, 20, 5620. [Google Scholar] [CrossRef] [PubMed]
- Prionisti, I.; Bühler, L.H.; Walker, P.R.; Jolivet, R.B. Harnessing Microglia and Macrophages for the Treatment of Glioblastoma. Front. Pharmacol. 2019, 10, 506. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e1617. [Google Scholar] [CrossRef]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E., 2nd; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.A.; Dey, M.; Chang, A.; Lesniak, M.S. Targeting Tregs in Malignant Brain Cancer: Overcoming IDO. Front. Immunol. 2013, 4, 116. [Google Scholar] [CrossRef]
- Bai, A.; Guo, Y. Acid sphingomyelinase mediates human CD4(+) T-cell signaling: Potential roles in T-cell responses and diseases. Cell Death Dis. 2017, 8, e2963. [Google Scholar] [CrossRef]
- Zhou, Y.; Salker, M.S.; Walker, B.; Münzer, P.; Borst, O.; Gawaz, M.; Gulbins, E.; Singh, Y.; Lang, F. Acid Sphingomyelinase (ASM) is a Negative Regulator of Regulatory T Cell (Treg) Development. Cell Physiol. Biochem. 2016, 39, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef]
- Volpi, C.; Orabona, C.; Macchiarulo, A.; Bianchi, R.; Puccetti, P.; Grohmann, U. Preclinical discovery and development of fingolimod for the treatment of multiple sclerosis. Expert Opin. Drug Discov. 2019, 14, 1199–1212. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Perrotta, C.; Cervia, D.; Di Renzo, I.; Moscheni, C.; Bassi, M.T.; Campana, L.; Martelli, C.; Catalani, E.; Giovarelli, M.; Zecchini, S.; et al. Nitric Oxide Generated by Tumor-Associated Macrophages Is Responsible for Cancer Resistance to Cisplatin and Correlated With Syntaxin 4 and Acid Sphingomyelinase Inhibition. Front. Immunol. 2018, 9, 1186. [Google Scholar] [CrossRef]
- Assi, E.; Cervia, D.; Bizzozero, L.; Capobianco, A.; Pambianco, S.; Morisi, F.; De Palma, C.; Moscheni, C.; Pellegrino, P.; Clementi, E.; et al. Modulation of Acid Sphingomyelinase in Melanoma Reprogrammes the Tumour Immune Microenvironment. Mediat. Inflamm. 2015, 2015, 370482. [Google Scholar] [CrossRef]
- Gude, D.R.; Alvarez, S.E.; Paugh, S.W.; Mitra, P.; Yu, J.; Griffiths, R.; Barbour, S.E.; Milstien, S.; Spiegel, S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008, 22, 2629–2638. [Google Scholar] [CrossRef]
- Weigert, A.; Tzieply, N.; von Knethen, A.; Johann, A.M.; Schmidt, H.; Geisslinger, G.; Brüne, B. Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol. Biol. Cell 2007, 18, 3810–3819. [Google Scholar] [CrossRef]
- Mrad, M.; Imbert, C.; Garcia, V.; Rambow, F.; Therville, N.; Carpentier, S.; Ségui, B.; Levade, T.; Azar, R.; Marine, J.C.; et al. Downregulation of sphingosine kinase-1 induces protective tumor immunity by promoting M1 macrophage response in melanoma. Oncotarget 2016, 7, 71873–71886. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; von Bernstorff, W.; Heidecke, C.D.; Schulze, T. Differential S1P Receptor Profiles on M1- and M2-Polarized Macrophages Affect Macrophage Cytokine Production and Migration. Biomed. Res. Int. 2017, 2017, 7584621. [Google Scholar] [CrossRef] [PubMed]
- Weichand, B.; Popp, R.; Dziumbla, S.; Mora, J.; Strack, E.; Elwakeel, E.; Frank, A.C.; Scholich, K.; Pierre, S.; Syed, S.N.; et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1β. J. Exp. Med. 2017, 214, 2695–2713. [Google Scholar] [CrossRef]
- Kim, E.Y.; Choi, B.; Kim, J.E.; Park, S.O.; Kim, S.M.; Chang, E.J. Interleukin-22 Mediates the Chemotactic Migration of Breast Cancer Cells and Macrophage Infiltration of the Bone Microenvironment by Potentiating S1P/SIPR Signaling. Cells 2020, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, T.; Wang, Y.; Ning, C.; Lv, Z.; Han, G.; Morris, J.C.; Taylor, E.N.; Wang, R.; Xiao, H.; et al. The protumorigenic potential of FTY720 by promoting extramedullary hematopoiesis and MDSC accumulation. Oncogene 2017, 36, 3760–3771. [Google Scholar] [CrossRef]
- Guo, X.D.; Ji, J.; Xue, T.F.; Sun, Y.Q.; Guo, R.B.; Cheng, H.; Sun, X.L. FTY720 Exerts Anti-Glioma Effects by Regulating the Glioma Microenvironment Through Increased CXCR4 Internalization by Glioma-Associated Microglia. Front. Immunol. 2020, 11, 178. [Google Scholar] [CrossRef]
- Karunakaran, I.; Alam, S.; Jayagopi, S.; Frohberger, S.J.; Hansen, J.N.; Kuehlwein, J.; Hölbling, B.V.; Schumak, B.; Hübner, M.P.; Gräler, M.H.; et al. Neural sphingosine 1-phosphate accumulation activates microglia and links impaired autophagy and inflammation. Glia 2019, 67, 1859–1872. [Google Scholar] [CrossRef]
- Tea, M.N.; Poonnoose, S.I.; Pitson, S.M. Targeting the Sphingolipid System as a Therapeutic Direction for Glioblastoma. Cancers 2020, 12, 111. [Google Scholar] [CrossRef]
- Naser, E.; Kadow, S.; Schumacher, F.; Mohamed, Z.H.; Kappe, C.; Hessler, G.; Pollmeier, B.; Kleuser, B.; Arenz, C.; Becker, K.A.; et al. Characterization of the small molecule ARC39, a direct and specific inhibitor of acid sphingomyelinase in vitro. J. Lipid Res. 2020, 61, 896–910. [Google Scholar] [CrossRef]
- Magrassi, L.; Adorni, L.; Montorfano, G.; Rapelli, S.; Butti, G.; Berra, B.; Milanesi, G. Vitamin D metabolites activate the sphingomyelin pathway and induce death of glioblastoma cells. Acta Neurochir. 1998, 140, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Oancea-Castillo, L.R.; Klein, C.; Abdollahi, A.; Weber, K.J.; Régnier-Vigouroux, A.; Dokic, I. Comparative analysis of the effects of a sphingosine kinase inhibitor to temozolomide and radiation treatment on glioblastoma cell lines. Cancer Biol. Ther. 2017, 18, 400–406. [Google Scholar] [CrossRef] [PubMed]
- French, K.J.; Schrecengost, R.S.; Lee, B.D.; Zhuang, Y.; Smith, S.N.; Eberly, J.L.; Yun, J.K.; Smith, C.D. Discovery and Evaluation of Inhibitors of Human Sphingosine Kinase. Cancer Res. 2003, 63, 5962. [Google Scholar] [PubMed]
- Bektas, M.; Johnson, S.P.; Poe, W.E.; Bigner, D.D.; Friedman, H.S. A sphingosine kinase inhibitor induces cell death in temozolomide resistant glioblastoma cells. Cancer Chemother. Pharmacol. 2009, 64, 1053–1058. [Google Scholar] [CrossRef]
- Sordillo, L.A.; Sordillo, P.P.; Helson, L. Sphingosine Kinase Inhibitors as Maintenance Therapy of Glioblastoma After Ceramide-Induced Response. Anticancer Res. 2016, 36, 2085–2095. [Google Scholar]
- Cattaneo, M.G.; Vanetti, C.; Samarani, M.; Aureli, M.; Bassi, R.; Sonnino, S.; Giussani, P. Cross-talk between sphingosine-1-phosphate and EGFR signaling pathways enhances human glioblastoma cell invasiveness. FEBS Lett. 2018, 592, 949–961. [Google Scholar] [CrossRef]
- Dementiev, A.; Joachimiak, A.; Nguyen, H.; Gorelik, A.; Illes, K.; Shabani, S.; Gelsomino, M.; Ahn, E.-Y.E.; Nagar, B.; Doan, N. Molecular Mechanism of Inhibition of Acid Ceramidase by Carmofur. J. Med. Chem. 2019, 62, 987–992. [Google Scholar] [CrossRef]
- White-Gilbertson, S.; Lu, P.; Jones, C.M.; Chiodini, S.; Hurley, D.; Das, A.; Delaney, J.R.; Norris, J.S.; Voelkel-Johnson, C. Tamoxifen is a candidate first-in-class inhibitor of acid ceramidase that reduces amitotic division in polyploid giant cancer cells—Unrecognized players in tumorigenesis. Cancer Med. 2020, 9, 3142–3152. [Google Scholar] [CrossRef]
- Mahdy, A.E.; Cheng, J.C.; Li, J.; Elojeimy, S.; Meacham, W.D.; Turner, L.S.; Bai, A.; Gault, C.R.; McPherson, A.S.; Garcia, N.; et al. Acid ceramidase upregulation in prostate cancer cells confers resistance to radiation: AC inhibition, a potential radiosensitizer. Mol. Ther. 2009, 17, 430–438. [Google Scholar] [CrossRef]
- Proksch, D.; Klein, J.J.; Arenz, C. Potent inhibition of Acid ceramidase by novel B-13 analogues. J. Lipids 2011, 2011, 971618. [Google Scholar] [CrossRef]
- Reynolds, C.P.; Maurer, B.J.; Kolesnick, R.N. Ceramide synthesis and metabolism as a target for cancer therapy. Cancer Lett. 2004, 206, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Lovat, P.E.; Oliverio, S.; Di Sano, F.; Donnorso, R.P.; Redfern, C.P.; Piacentini, M. Fenretinide: A p53-independent way to kill cancer cells. Biochem. Biophys. Res. Commun. 2005, 331, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Orienti, I.; Salvati, V.; Sette, G.; Zucchetti, M.; Bongiorno-Borbone, L.; Peschiaroli, A.; Zolla, L.; Francescangeli, F.; Ferrari, M.; Matteo, C.; et al. A novel oral micellar fenretinide formulation with enhanced bioavailability and antitumour activity against multiple tumours from cancer stem cells. J. Exp. Clin. Cancer Res. 2019, 38, 373. [Google Scholar] [CrossRef] [PubMed]
- Song, M.M.; Makena, M.R.; Hindle, A.; Koneru, B.; Nguyen, T.H.; Verlekar, D.U.; Cho, H.; Maurer, B.J.; Kang, M.H.; Reynolds, C.P. Cytotoxicity and molecular activity of fenretinide and metabolites in T-cell lymphoid malignancy, neuroblastoma, and ovarian cancer cell lines in physiological hypoxia. Anti Cancer Drugs 2019, 30, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Puduvalli, V.K.; Yung, W.K.; Hess, K.R.; Kuhn, J.G.; Groves, M.D.; Levin, V.A.; Zwiebel, J.; Chang, S.M.; Cloughesy, T.F.; Junck, L.; et al. Phase II study of fenretinide (NSC 374551) in adults with recurrent malignant gliomas: A North American Brain Tumor Consortium study. J. Clin. Oncol. 2004, 22, 4282–4289. [Google Scholar] [CrossRef]
- Erbay, M.F.; Kamisli, O. A case of high grade glioma following treatment of relapsing-remitting multiple sclerosis with fingolimod. Neurol. India 2020, 68, 478–480. [Google Scholar] [CrossRef]
- Schuhmann, M.K.; Bittner, S.; Meuth, S.G.; Kleinschnitz, C.; Fluri, F. Fingolimod (FTY720-P) Does Not Stabilize the Blood-Brain Barrier under Inflammatory Conditions in an in Vitro Model. Int. J. Mol. Sci. 2015, 16, 29454–29466. [Google Scholar] [CrossRef]
- Cartier, A.; Leigh, T.; Liu, C.H.; Hla, T. Endothelial sphingosine 1-phosphate receptors promote vascular normalization and antitumor therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 3157–3166. [Google Scholar] [CrossRef]
- Pchejetski, D.; Bohler, T.; Brizuela, L.; Sauer, L.; Doumerc, N.; Golzio, M.; Salunkhe, V.; Teissié, J.; Malavaud, B.; Waxman, J.; et al. FTY720 (fingolimod) sensitizes prostate cancer cells to radiotherapy by inhibition of sphingosine kinase-1. Cancer Res. 2010, 70, 8651–8661. [Google Scholar] [CrossRef]
- Paugh, S.W.; Payne, S.G.; Barbour, S.E.; Milstien, S.; Spiegel, S. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 2003, 554, 189–193. [Google Scholar] [CrossRef]
- Alshaker, H.; Wang, Q.; Srivats, S.; Chao, Y.; Cooper, C.; Pchejetski, D. New FTY720-docetaxel nanoparticle therapy overcomes FTY720-induced lymphopenia and inhibits metastatic breast tumour growth. Breast Cancer Res. Treat. 2017, 165, 531–543. [Google Scholar] [CrossRef]
- Ito, K.; Yamaguchi, A.; Miura, K.; Kato, T.; Baba, S.; Matsumoto, S.; Ishii, M.; Takagi, H. Effect of oral adjuvant therapy with Carmofur (HCFU) for distant metastasis of colorectal cancer. Int. J. Clin. Oncol. 2000, 5, 29–35. [Google Scholar] [CrossRef]
- Realini, N.; Solorzano, C.; Pagliuca, C.; Pizzirani, D.; Armirotti, A.; Luciani, R.; Costi, M.P.; Bandiera, T.; Piomelli, D. Discovery of highly potent acid ceramidase inhibitors with in vitro tumor chemosensitizing activity. Sci. Rep. 2013, 3, 1035. [Google Scholar] [CrossRef]
- Matsumoto, S.; Nishizawa, S.; Murakami, M.; Noma, S.; Sano, A.; Kuroda, Y. Carmofur-induced leukoencephalopathy: MRI. Neuroradiology 1995, 37, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.Q.; Gao, H.; Han, M.; Liu, K.L.; Peng, J.J.; Han, Y.T. Hispidulin suppresses tumor growth and metastasis in renal cell carcinoma by modulating ceramide-sphingosine 1-phosphate rheostat. Am. J. Cancer Res. 2017, 7, 1501–1514. [Google Scholar] [PubMed]
- Lewis, C.S.; Voelkel-Johnson, C.; Smith, C.D. Targeting Sphingosine Kinases for the Treatment of Cancer. Adv. Cancer Res. 2018, 140, 295–325. [Google Scholar] [CrossRef] [PubMed]
- Antoon, J.W.; White, M.D.; Driver, J.L.; Burow, M.E.; Beckman, B.S. Sphingosine kinase isoforms as a therapeutic target in endocrine therapy resistant luminal and basal-A breast cancer. Exp. Biol. Med. 2012, 237, 832–844. [Google Scholar] [CrossRef] [PubMed]



| Cancer Type | NCT Number | Dates | Drug Name | Primary Target | Phase | Results | FDA Approval? |
|---|---|---|---|---|---|---|---|
| GBM | NCT02490930 | July 2015–September 2017 | Fingolimod | S1PR1 antagonist | Early Phase 1 | Not Published | Yes |
| Recurrent Glioma | NCT00006080 | September 2000–September 2004 | Fenretinide (Single Agent) | Ceramide | Phase 2 | Ineffective at the doses given | Yes |
| Recurrent GBM | NCT00075491 | December 2003–March 2005 | Fenretinide (Combination treatment) | Ceramide | Phase 2 | Not Published due to termination | Yes |
| Advanced solid tumors | NCT01488513 | August 2014–August 2015 | ABC294640 | SK inhibitor | Phase 1 | Tolerated up to 500mg bid | No |
| Advanced solid tumors | NCT02834611 | March 2017–August 2019 | Ceramide NanoLiposome | Ceramide | Phase 1 | Not Published | No |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hawkins, C.C.; Ali, T.; Ramanadham, S.; Hjelmeland, A.B. Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules 2020, 10, 1357. https://doi.org/10.3390/biom10101357
Hawkins CC, Ali T, Ramanadham S, Hjelmeland AB. Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules. 2020; 10(10):1357. https://doi.org/10.3390/biom10101357
Chicago/Turabian StyleHawkins, Cyntanna C., Tomader Ali, Sasanka Ramanadham, and Anita B. Hjelmeland. 2020. "Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate" Biomolecules 10, no. 10: 1357. https://doi.org/10.3390/biom10101357
APA StyleHawkins, C. C., Ali, T., Ramanadham, S., & Hjelmeland, A. B. (2020). Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules, 10(10), 1357. https://doi.org/10.3390/biom10101357