Mercury and Alzheimer’s Disease: Hg(II) Ions Display Specific Binding to the Amyloid-β Peptide and Hinder Its Fibrillization

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Fluorescence Spectroscopy
2.2.1. ThT Fluorescence as a Probe for Aβ Aggregation Kinetics
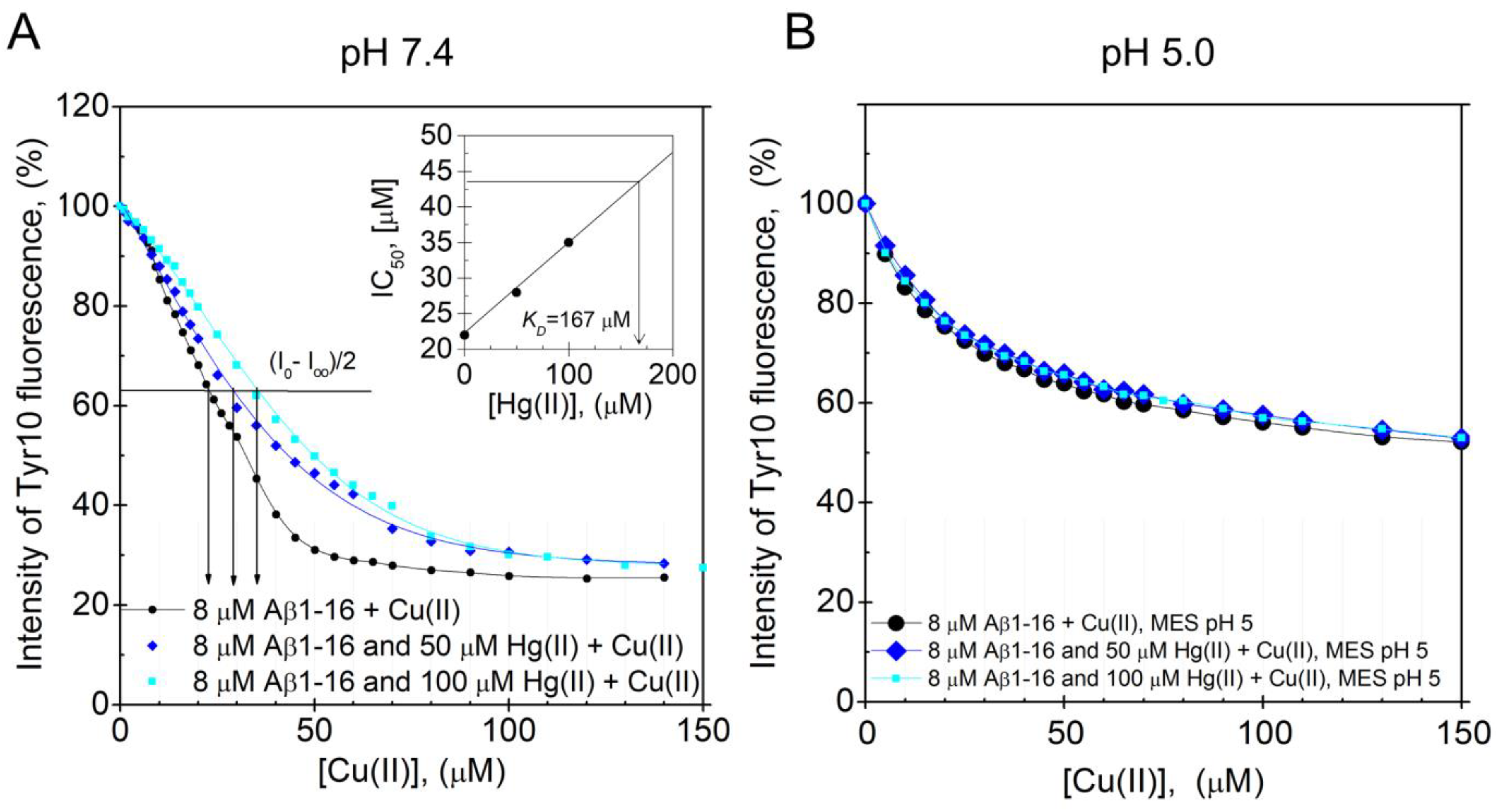
2.2.2. Tyrosine Fluorescence Quenching Reporting on Hg(II)·Aβ Binding Affinity
2.3. AFM Imaging
2.4. NMR Spectroscopy
3. Results
3.1. ThT Fluorescence: Kinetic Effects of Hg(II) Ions on the Aβ40 and Aβ42 Aggregation Processes
3.2. AFM Imaging: Effects of Hg(II) Ions on Aβ40 Aggregate Morphology
3.3. NMR Spectroscopy: Molecular Interactions Between Hg(II) Ions and Aβ40 Monomers
3.4. Fluorescence Spectroscopy: pH-Dependence of the Hg(II) Binding Affinity to the Aβ16 Monomer
4. Discussion
4.1. The In Vitro Analyses of the Hg(II)·Aβ Complexes and Their Aggregation
4.2. Mercury and AD: Clinical Studies and Sources of Exposure
4.3. Biological Relevance and Other Molecular Effects on AD Involving Hg Ions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015—The Global Impact of Dementia; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Alzheimer’s-Association. 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Bjørklund, G.; Aaseth, J.; Dadar, M.; Chirumbolo, S. Molecular Targets in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 7032–7044. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef]
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Miyata, M.; Smith, J.D. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat. Genet. 1996, 14, 55–61. [Google Scholar] [CrossRef]
- Mutter, J.; Naumann, J.; Sadaghiani, C.; Schneider, R.; Walach, H. Alzheimer disease: Mercury as pathogenetic factor and apolipoprotein E as a moderator. Neuroendocrinol. Lett. 2004, 25, 331–339. [Google Scholar]
- Godfrey, M.E.; Wojcik, D.P.; Krone, C.A. Apolipoprotein E genotyping as a potential biomarker for mercury neurotoxicity. J. Alzheimer’s Dis. 2003, 5, 189–195. [Google Scholar] [CrossRef]
- Gessel, M.M.; Bernstein, S.; Kemper, M.; Teplow, D.B.; Bowers, M.T. Familial Alzheimer’s disease mutations differentially alter amyloid beta-protein oligomerization. ACS Chem. Neurosci. 2012, 3, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Herring, A.; Munster, Y.; Metzdorf, J.; Bolczek, B.; Krussel, S.; Krieter, D.; Yavuz, I.; Karim, F.; Roggendorf, C.; Stang, A.; et al. Late running is not too late against Alzheimer’s pathology. Neurobiol. Dis. 2016, 94, 44–54. [Google Scholar] [CrossRef]
- Xu, W.; Tan, L.; Wang, H.F.; Jiang, T.; Tan, M.S.; Tan, L.; Zhao, Q.F.; Li, J.Q.; Wang, J.; Yu, J.T. Meta-analysis of modifiable risk factors for Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1299–1306. [Google Scholar] [CrossRef]
- Rolandi, E.; Frisoni, G.B.; Cavedo, E. Efficacy of lifestyle interventions on clinical and neuroimaging outcomes in elderly. Ageing Res. Rev. 2016, 25, 1–12. [Google Scholar] [CrossRef]
- Maher, B.A.; Ahmed, I.A.; Karloukovski, V.; MacLaren, D.A.; Foulds, P.G.; Allsop, D.; Mann, D.M.; Torres-Jardon, R.; Calderon-Garciduenas, L. Magnetite pollution nanoparticles in the human brain. Proc. Natl. Acad. Sci. USA 2016, 113, 10797–10801. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Kavanaugh, M.; Block, M.; D’Angiulli, A.; Delgado-Chavez, R.; Torres-Jardon, R.; Gonzalez-Maciel, A.; Reynoso-Robles, R.; Osnaya, N.; Villarreal-Calderon, R.; et al. Neuroinflammation, hyperphosphorylated tau, diffuse amyloid plaques, and down-regulation of the cellular prion protein in air pollution exposed children and young adults. J. Alzheimer’s Dis. 2012, 28, 93–107. [Google Scholar] [CrossRef]
- Durazzo, T.C.; Mattsson, N.; Weiner, M.W.; Alzheimer’s Disease Neuroimaging Initiative. Smoking and increased Alzheimer’s disease risk: A review of potential mechanisms. Alzheimer’s Dement. 2014, 10, S122–S145. [Google Scholar] [CrossRef]
- Cataldo, J.K.; Prochaska, J.J.; Glantz, S.A. Cigarette smoking is a risk factor for Alzheimer’s Disease: An analysis controlling for tobacco industry affiliation. J. Alzheimer’s Dis. 2010, 19, 465–480. [Google Scholar] [CrossRef]
- Wallin, C.; Sholts, S.B.; Österlund, N.; Luo, J.; Jarvet, J.; Roos, P.M.; Ilag, L.; Gräslund, A.; Wärmländer, S.K.T.S. Alzheimer’s disease and cigarette smoke components: Effects of nicotine, PAHs, and Cd(II), Cr(III), Pb(II), Pb(IV) ions on amyloid-beta peptide aggregation. Sci. Rep. 2017, 7, 14423. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.J.; Zhang, M.; Xu, Z.Q.; Gao, C.Y.; Fang, C.Q.; Yan, J.C.; Zhou, H.D.; Chongqing Ageing Study Group. Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology 2011, 76, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Reciprocal Molecular Interactions between the Abeta Peptide Linked to Alzheimer’s Disease and Insulin Linked to Diabetes Mellitus Type II. ACS Chem. Neurosci. 2016, 7, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Sivanandam, T.M.; Thakur, M.K. Traumatic brain injury: A risk factor for Alzheimer’s disease. Neurosci. Biobehav. Rev. 2012, 36, 1376–1381. [Google Scholar] [CrossRef]
- Wang, C.; Klechikov, A.G.; Gharibyan, A.L.; Wärmländer, S.K.; Jarvet, J.; Zhao, L.; Jia, X.; Narayana, V.K.; Shankar, S.K.; Olofsson, A.; et al. The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol. 2014, 127, 507–522. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, X.; Yang, J.; Suo, J.; Chen, J.; Liu, X.; Zhao, X. Chronic exposure to aluminum and risk of Alzheimer’s disease: A meta-analysis. Neurosci. Lett. 2016, 610, 200–206. [Google Scholar] [CrossRef]
- Modgil, S.; Lahiri, D.K.; Sharma, V.L.; Anand, A. Role of early life exposure and environment on neurodegeneration: Implications on brain disorders. Transl. Neurodegener. 2014, 3, 9. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Sunde, M.; Blake, C.C. From the globular to the fibrous state: Protein structure and structural conversion in amyloid formation. Q. Rev. Biophys. 1998, 31, 1–39. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Abelein, A.; Abrahams, J.P.; Danielsson, J.; Gräslund, A.; Jarvet, J.; Luo, J.; Tiiman, A.; Wärmländer, S.K. The hairpin conformation of the amyloid beta peptide is an important structural motif along the aggregation pathway. J. Biol. Inorg. Chem. 2014, 19, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Wärmländer, S.; Tiiman, A.; Abelein, A.; Luo, J.; Jarvet, J.; Söderberg, K.L.; Danielsson, J.; Gräslund, A. Biophysical studies of the amyloid beta-peptide: Interactions with metal ions and small molecules. Chembiochem 2013, 14, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yu, C.H.; Yu, H.; Borstnar, R.; Kamerlin, S.C.; Gräslund, A.; Abrahams, J.P.; Wärmländer, S.K. Cellular polyamines promote amyloid-beta (Abeta) peptide fibrillation and modulate the aggregation pathways. ACS Chem. Neurosci. 2013, 4, 454–462. [Google Scholar] [CrossRef]
- Luo, J.; Mohammed, I.; Wärmländer, S.K.; Hiruma, Y.; Gräslund, A.; Abrahams, J.P. Endogenous polyamines reduce the toxicity of soluble abeta peptide aggregates associated with Alzheimer’s disease. Biomacromolecules 2014, 15, 1985–1991. [Google Scholar] [CrossRef]
- Owen, M.C.; Gnutt, D.; Gao, M.; Wärmländer, S.K.T.S.; Jarvet, J.; Gräslund, A.; Winter, R.; Ebbinghaus, S.; Strodel, B. Effects of in vivo conditions on amyloid aggregation. Chem. Soc. Rev. 2019, 48, 3946–3996. [Google Scholar] [CrossRef]
- Faller, P.; Hureau, C.; Berthoumieu, O. Role of metal ions in the self-assembly of the Alzheimer’s amyloid-beta peptide. Inorg. Chem. 2013, 52, 12193–12206. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Sandberg, A.; Luheshi, L.M.; Sollvander, S.; Pereira de Barros, T.; Macao, B.; Knowles, T.P.; Biverstal, H.; Lendel, C.; Ekholm-Petterson, F.; Dubnovitsky, A.; et al. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc. Natl. Acad. Sci. USA 2010, 107, 15595–15600. [Google Scholar] [CrossRef]
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Alzheimer peptides aggregate into transient nanoglobules that nucleate fibrils. Biochemistry 2014, 53, 6302–6308. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an understanding of amyloid-beta oligomers: Characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-beta Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Agholme, L.; Kurudenkandy, F.R.; Granseth, B.; Marcusson, J.; Hallbeck, M. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of beta-amyloid. J. Neurosci. 2012, 32, 8767–8777. [Google Scholar] [CrossRef]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjo, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef]
- Lee, M.C.; Yu, W.C.; Shih, Y.H.; Chen, C.Y.; Guo, Z.H.; Huang, S.J.; Chan, J.C.C.; Chen, Y.R. Zinc ion rapidly induces toxic, off-pathway amyloid-beta oligomers distinct from amyloid-beta derived diffusible ligands in Alzheimer’s disease. Sci. Rep. 2018, 8, 4772. [Google Scholar] [CrossRef]
- Wärmländer, S.K.T.S.; Österlund, N.; Wallin, C.; Wu, J.; Luo, J.; Tiiman, A.; Jarvet, J.; Gräslund, A. Metal binding to the Amyloid-β peptides in the presence of biomembranes: Potential mechanisms of cell toxicity. J. Biol. Inorg. Chem. 2019, 24, 1189–1196, in press. [Google Scholar]
- Österlund, N.; Kulkarni, Y.S.; Misiaszek, A.D.; Wallin, C.; Krüger, D.M.; Liao, Q.; Mashayekhy Rad, F.; Jarvet, J.; Strodel, B.; Wärmländer, S.K.T.S.; et al. Amyloid-beta Peptide Interactions with Amphiphilic Surfactants: Electrostatic and Hydrophobic Effects. ACS Chem. Neurosci. 2018, 9, 1680–1692. [Google Scholar] [CrossRef]
- Österlund, N.; Moons, R.; Ilag, L.L.; Sobott, F.; Gräslund, A. Native Ion Mobility-Mass Spectrometry Reveals the Formation of beta-Barrel Shaped Amyloid-beta Hexamers in a Membrane-Mimicking Environment. J. Am. Chem. Soc. 2019, 141, 10440–10450. [Google Scholar] [CrossRef]
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Human lysozyme inhibits the in vitro aggregation of Abeta peptides, which in vivo are associated with Alzheimer’s disease. Chem. Commun. (Camb.) 2013, 49, 6507–6509. [Google Scholar] [CrossRef]
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Non-chaperone proteins can inhibit aggregation and cytotoxicity of Alzheimer amyloid beta peptide. J. Biol. Chem. 2014, 289, 27766–27775. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Cross-interactions between the Alzheimer Disease Amyloid-beta Peptide and Other Amyloid Proteins: A Further Aspect of the Amyloid Cascade Hypothesis. J. Biol. Chem. 2016, 291, 16485–16493. [Google Scholar] [CrossRef] [PubMed]
- Leshem, G.; Richman, M.; Lisniansky, E.; Antman-Passig, M.; Habashi, M.; Gräslund, A.; Wärmländer, S.K.T.S.; Rahimipour, S. Photoactive chlorin e6 is a multifunctional modulator of amyloid-beta aggregation and toxicity via specific interactions with its histidine residues. Chem. Sci. 2019, 10, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Richman, M.; Wilk, S.; Chemerovski, M.; Warmlander, S.K.; Wahlstrom, A.; Graslund, A.; Rahimipour, S. In vitro and mechanistic studies of an antiamyloidogenic self-assembled cyclic D,L-alpha-peptide architecture. J. Am. Chem. Soc. 2013, 135, 3474–3484. [Google Scholar] [CrossRef]
- Luo, J.; Otero, J.M.; Yu, C.H.; Wärmländer, S.K.; Gräslund, A.; Overhand, M.; Abrahams, J.P. Inhibiting and reversing amyloid-beta peptide (1–40) fibril formation with gramicidin S and engineered analogues. Chemistry 2013, 19, 17338–17348. [Google Scholar] [CrossRef]
- Goedert, M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018, 592, 2383–2391. [Google Scholar] [CrossRef]
- Gibbons, G.S.; Lee, V.M.Y.; Trojanowski, J.Q. Mechanisms of Cell-to-Cell Transmission of Pathological Tau: A Review. JAMA Neurol. 2019, 76, 101–108. [Google Scholar] [CrossRef]
- Wallin, C.; Hiruma, Y.; Wärmländer, S.K.T.S.; Huvent, I.; Jarvet, J.; Abrahams, J.P.; Gräslund, A.; Lippens, G.; Luo, J. The Neuronal Tau Protein Blocks in Vitro Fibrillation of the Amyloid-beta (Abeta) Peptide at the Oligomeric Stage. J. Am. Chem. Soc. 2018. [Google Scholar] [CrossRef]
- Regen, F.; Hellmann-Regen, J.; Costantini, E.; Reale, M. Neuroinflammation and Alzheimer’s Disease: Implications for Microglial Activation. Curr. Alzheimer Res. 2017, 14, 1140–1148. [Google Scholar] [CrossRef]
- Al-Hilaly, Y.K.; Williams, T.L.; Stewart-Parker, M.; Ford, L.; Skaria, E.; Cole, M.; Bucher, W.G.; Morris, K.L.; Sada, A.A.; Thorpe, J.R.; et al. A central role for dityrosine crosslinking of Amyloid-beta in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 83. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Duce, J.A.; Bush, A.I.; Adlard, P.A. Role of amyloid-β–metal interactions in Alzheimer’s disease. Future Neurol. 2011, 6, 641–659. [Google Scholar] [CrossRef]
- Wang, Z.X.; Tan, L.; Wang, H.F.; Ma, J.; Liu, J.; Tan, M.S.; Sun, J.H.; Zhu, X.C.; Jiang, T.; Yu, J.T. Serum Iron, Zinc, and Copper Levels in Patients with Alzheimer’s Disease: A Replication Study and Meta-Analyses. J. Alzheimer’s Dis. 2015, 47, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Tiiman, A.; Palumaa, P.; Tougu, V. The missing link in the amyloid cascade of Alzheimer’s disease—Metal ions. Neurochem. Int. 2013, 62, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Lei, P.; Bush, A.I. Metallostasis in Alzheimer’s disease. Free Radic. Biol. Med. 2013, 62, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R. Metals in Alzheimer’s disease: A systemic perspective. Front. Biosci. (Landmark Ed) 2012, 17, 451–472. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Miller, L.M.; Wang, Q.; Telivala, T.P.; Smith, R.J.; Lanzirotti, A.; Miklossy, J. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 2006, 155, 30–37. [Google Scholar] [CrossRef]
- Beauchemin, D.; Kisilevsky, R. A method based on ICP-MS for the analysis of Alzheimer’s amyloid plaques. Anal. Chem. 1998, 70, 1026–1029. [Google Scholar] [CrossRef]
- Sayre, L.M.; Perry, G.; Harris, P.L.; Liu, Y.; Schubert, K.A.; Smith, M.A. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: A central role for bound transition metals. J. Neurochem. 2000, 74, 270–279. [Google Scholar] [CrossRef]
- Wallin, C.; Kulkarni, Y.S.; Abelein, A.; Jarvet, J.; Liao, Q.; Strodel, B.; Olsson, L.; Luo, J.; Abrahams, J.P.; Sholts, S.B.; et al. Characterization of Mn(II) ion binding to the amyloid-beta peptide in Alzheimer’s disease. J. Trace Elem. Med. Biol. 2016, 38, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Faller, P. Copper and zinc binding to amyloid-beta: Coordination, dynamics, aggregation, reactivity and metal-ion transfer. ChemBioChem 2009, 10, 2837–2845. [Google Scholar] [CrossRef] [PubMed]
- Conte-Daban, A.; Borghesani, V.; Sayen, S.; Guillon, E.; Journaux, Y.; Gontard, G.; Lisnard, L.; Hureau, C. Link between Affinity and Cu(II) Binding Sites to Amyloid-beta Peptides Evaluated by a New Water-Soluble UV-Visible Ratiometric Dye with a Moderate Cu(II) Affinity. Anal. Chem. 2017, 89, 2155–2162. [Google Scholar] [CrossRef] [PubMed]
- Faller, P.; Hureau, C. Bioinorganic chemistry of copper and zinc ions coordinated to amyloid-beta peptide. Dalton Trans. 2009, 1080–1094. [Google Scholar] [CrossRef]
- Tõugu, V.; Karafin, A.; Palumaa, P. Binding of zinc(II) and copper(II) to the full-length Alzheimer’s amyloid-beta peptide. J. Neurochem. 2008, 104, 1249–1259. [Google Scholar] [CrossRef]
- Wild, K.; August, A.; Pietrzik, C.U.; Kins, S. Structure and Synaptic Function of Metal Binding to the Amyloid Precursor Protein and its Proteolytic Fragments. Front. Mol. Neurosci. 2017, 10, 21. [Google Scholar] [CrossRef]
- Branch, T.; Barahona, M.; Dodson, C.A.; Ying, L. Kinetic Analysis Reveals the Identity of Abeta-Metal Complex Responsible for the Initial Aggregation of Abeta in the Synapse. ACS Chem. Neurosci. 2017, 8, 1970–1979. [Google Scholar] [CrossRef]
- Basha, M.R.; Wei, W.; Bakheet, S.A.; Benitez, N.; Siddiqi, H.K.; Ge, Y.-W.; Lahiri, D.K.; Zawia, N.H. The Fetal Basis of Amyloidogenesis: Exposure to Lead and Latent Overexpression of Amyloid Precursor Protein and β-Amyloid in the Aging Brain. J. Neurosci. 2005, 25, 823. [Google Scholar] [CrossRef]
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776. [Google Scholar] [CrossRef]
- Huang, C.-L.; Hsiao, I.L.; Lin, H.-C.; Wang, C.-F.; Huang, Y.-J.; Chuang, C.-Y. Silver nanoparticles affect on gene expression of inflammatory and neurodegenerative responses in mouse brain neural cells. Environ. Res. 2015, 136, 253–263. [Google Scholar] [CrossRef]
- Ashok, A.; Rai, N.K.; Tripathi, S.; Bandyopadhyay, S. Exposure to As-, Cd-, and Pb-mixture induces Abeta, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol. Sci. 2015, 143, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Kato-Negishi, M. Link between aluminum and the pathogenesis of Alzheimer’s disease: The integration of the aluminum and amyloid cascade hypotheses. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Chen, X.; Li, W.; Han, Y.; Liu, P.; Pi, R. Exposure to metal ions regulates mRNA levels of APP and BACE1 in PC12 cells: Blockage by curcumin. Neurosci. Lett. 2008, 440, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Cheng, D.; Laferla, F.M. Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. J. Neurochem. 2009, 108, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lv, Y.; Yu, S.; Zhao, H.; Yao, L. The effect of cadmium on Abeta levels in APP/PS1 transgenic mice. Exp. Ther. Med. 2012, 4, 125–130. [Google Scholar] [CrossRef]
- Olivieri, G.; Brack, C.; Muller-Spahn, F.; Stahelin, H.B.; Herrmann, M.; Renard, P.; Brockhaus, M.; Hock, C. Mercury induces cell cytotoxicity and oxidative stress and increases beta-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells. J. Neurochem. 2000, 74, 231–236. [Google Scholar] [CrossRef]
- Monnet-Tschudi, F.; Zurich, M.G.; Boschat, C.; Corbaz, A.; Honegger, P. Involvement of environmental mercury and lead in the etiology of neurodegenerative diseases. Rev. Environ. Health 2006, 21, 105–117. [Google Scholar] [CrossRef]
- Song, J.W.; Choi, B.S. Mercury induced the Accumulation of Amyloid Beta (Abeta) in PC12 Cells: The Role of Production and Degradation of Abeta. Toxicol. Res. 2013, 29, 235–240. [Google Scholar] [CrossRef]
- Bjørklund, G.; Tinkov, A.A.; Dadar, M.; Rahman, M.M.; Chirumbolo, S.; Skalny, A.V.; Skalnaya, M.G.; Haley, B.E.; Ajsuvakova, O.P.; Aaseth, J. Insights into the Potential Role of Mercury in Alzheimer’s Disease. J. Mol. Neurosci. 2019, 67, 511–533. [Google Scholar] [CrossRef]
- Ehmann, W.D.; Markesbery, W.R.; Alauddin, M.; Hossain, T.I.; Brubaker, E.H. Brain trace elements in Alzheimer’s disease. Neurotoxicology 1986, 7, 195–206. [Google Scholar]
- Hock, C.; Drasch, G.; Golombowski, S.; Muller-Spahn, F.; Willershausen-Zonnchen, B.; Schwarz, P.; Hock, U.; Growdon, J.H.; Nitsch, R.M. Increased blood mercury levels in patients with Alzheimer’s disease. J. Neural Transm. (Vienna) 1998, 105, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Gerhardsson, L.; Lundh, T.; Minthon, L.; Londos, E. Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer’s disease. Dement. Geriatr. Cognit. Disord. 2008, 25, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Mutter, J.; Curth, A.; Naumann, J.; Deth, R.; Walach, H. Does inorganic mercury play a role in Alzheimer’s disease? A systematic review and an integrated molecular mechanism. J. Alzheimer’s Dis. 2010, 22, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.T.; Harry, G.J.; Hayden, K.M.; Szabo, D.T.; Birnbaum, L. Comparison of Metal Levels between Postmortem Brain and Ventricular Fluid in Alzheimer’s Disease and Nondemented Elderly Controls. Toxicol. Sci. 2016, 150, 292–300. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, D.W.; Park, K.S.; Joung, H. Serum trace metal levels in Alzheimer’s disease and normal control groups. Am. J. Alzheimer’s Dis. Other Dement. 2014, 29, 76–83. [Google Scholar] [CrossRef]
- Berlin, M.; Zalups, R.K.; Fowler, B.A. Chapter 46: Mercury. In Handbook on the Toxicology of Metals, 4th ed.; Nordberg, G.F., Fowler, B.A., Nordberg, M., Eds.; Elsevier/Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Carocci, A.; Rovito, N.; Sinicropi, M.S.; Genchi, G. Mercury toxicity and neurodegenerative effects. Rev. Environ. Contam. Toxicol. 2014, 229, 1–18. [Google Scholar] [CrossRef]
- Wu, X.; Cobbina, S.J.; Mao, G.; Xu, H.; Zhang, Z.; Yang, L. A review of toxicity and mechanisms of individual and mixtures of heavy metals in the environment. Environ. Sci. Pollut. Res. Int. 2016, 23, 8244–8259. [Google Scholar] [CrossRef]
- Aschner, M.; Aschner, J.L. Mercury neurotoxicity: Mechanisms of blood-brain barrier transport. Neurosci. Biobehav. Rev. 1990, 14, 169–176. [Google Scholar] [CrossRef]
- Yang, D.J.; Shi, S.; Zheng, L.F.; Yao, T.M.; Ji, L.N. Mercury(II) promotes the in vitro aggregation of tau fragment corresponding to the second repeat of microtubule-binding domain: Coordination and conformational transition. Biopolymers 2010, 93, 1100–1107. [Google Scholar] [CrossRef]
- Arnhold, F.; Guhrs, K.H.; von Mikecz, A. Amyloid domains in the cell nucleus controlled by nucleoskeletal protein lamin B1 reveal a new pathway of mercury neurotoxicity. PeerJ 2015, 3, e754. [Google Scholar] [CrossRef]
- Sharma, S.K.; Goloubinoff, P.; Christen, P. Heavy metal ions are potent inhibitors of protein folding. Biochem. Biophys. Res. Commun. 2008, 372, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Tamas, M.J.; Sharma, S.K.; Ibstedt, S.; Jacobson, T.; Christen, P. Heavy metals and metalloids as a cause for protein misfolding and aggregation. Biomolecules 2014, 4, 252–267. [Google Scholar] [CrossRef] [PubMed]
- Meleleo, D.; Notarachille, G.; Mangini, V.; Arnesano, F. Concentration-dependent effects of mercury and lead on Abeta42: Possible implications for Alzheimer’s disease. Eur. Biophys. J. 2019, 48, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Gade Malmos, K.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A primer on the use of thioflavin T to investigate amyloid formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hellstrand, E.; Boland, B.; Walsh, D.M.; Linse, S. Amyloid beta-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci. 2010, 1, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Alies, B.; Renaglia, E.; Rozga, M.; Bal, W.; Faller, P.; Hureau, C. Cu(II) affinity for the Alzheimer’s peptide: Tyrosine fluorescence studies revisited. Anal. Chem. 2013, 85, 1501–1508. [Google Scholar] [CrossRef]
- Österlund, N.; Luo, J.; Wärmländer, S.K.T.S.; Gräslund, A. Membrane-mimetic systems for biophysical studies of the amyloid-beta peptide. Biochim. Biophys. Acta Proteins Proteom. 2018. [Google Scholar] [CrossRef]
- Danielsson, J.; Andersson, A.; Jarvet, J.; Gräslund, A. 15N relaxation study of the amyloid beta-peptide: Structural propensities and persistence length. Magn. Reson. Chem. 2006, 44, S114–S121. [Google Scholar] [CrossRef]
- Roche, J.; Shen, Y.; Lee, J.H.; Ying, J.; Bax, A. Monomeric Abeta(1-40) and Abeta(1-42) Peptides in Solution Adopt Very Similar Ramachandran Map Distributions That Closely Resemble Random Coil. Biochemistry 2016, 55, 762–775. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Matsuzaki, K.; Hoshino, M. Transient formation of intermediate conformational states of amyloid-beta peptide revealed by heteronuclear magnetic resonance spectroscopy. FEBS Lett. 2011, 585, 1097–1102. [Google Scholar] [CrossRef]
- Jarvet, J.; Danielsson, J.; Damberg, P.; Oleszczuk, M.; Gräslund, A. Positioning of the Alzheimer Abeta(1–40) peptide in SDS micelles using NMR and paramagnetic probes. J. Biomol. NMR 2007, 39, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, J.; Wahlström, A.; Danielsson, J.; Markova, N.; Ekblad, C.; Gräslund, A.; Abrahmsen, L.; Karlström, A.E.; Wärmländer, S.K. N-terminal engineering of amyloid-beta-binding Affibody molecules yields improved chemical synthesis and higher binding affinity. Protein Sci. 2010, 19, 2319–2329. [Google Scholar] [CrossRef] [PubMed]
- Tiiman, A.; Luo, J.; Wallin, C.; Olsson, L.; Lindgren, J.; Jarvet, J.; Roos, P.M.; Sholts, S.B.; Rahimipour, S.; Abrahams, J.P.; et al. Specific Binding of Cu(II) Ions to Amyloid-Beta Peptides Bound to Aggregation-Inhibiting Molecules or SDS Micelles Creates Complexes that Generate Radical Oxygen Species. J. Alzheimer’s Dis. 2016, 54, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Serpell, L.C. Alzheimer’s amyloid fibrils: Structure and assembly. Biochim. Biophys. Acta 2000, 1502, 16–30. [Google Scholar] [CrossRef]
- Abelein, A.; Gräslund, A.; Danielsson, J. Zinc as chaperone-mimicking agent for retardation of amyloid beta peptide fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, 5407–5412. [Google Scholar] [CrossRef]
- Butterfield, S.M.; Lashuel, H.A. Amyloidogenic protein-membrane interactions: Mechanistic insight from model systems. Angew. Chem. Int. Ed. Engl. 2010, 49, 5628–5654. [Google Scholar] [CrossRef]
- Stewart, K.L.; Radford, S.E. Amyloid plaques beyond Abeta: A survey of the diverse modulators of amyloid aggregation. Biophys. Rev. 2017, 9, 405–419. [Google Scholar] [CrossRef]
- Zhang, S.; Casey, N.; Lee, J.P. Residual structure in the Alzheimer’s disease peptide: Probing the origin of a central hydrophobic cluster. Fold. Des. 1998, 3, 413–422. [Google Scholar] [CrossRef]
- Lindgren, J.; Segerfeldt, P.; Sholts, S.B.; Gräslund, A.; Karlström, A.E.; Wärmländer, S.K. Engineered non-fluorescent Affibody molecules facilitate studies of the amyloid-beta (Abeta) peptide in monomeric form: Low pH was found to reduce Abeta/Cu(II) binding affinity. J. Inorg. Biochem. 2013, 120, 18–23. [Google Scholar] [CrossRef]
- Ghalebani, L.; Wahlström, A.; Danielsson, J.; Wärmländer, S.K.; Gräslund, A. pH-dependence of the specific binding of Cu(II) and Zn(II) ions to the amyloid-beta peptide. Biochem. Biophys. Res. Commun. 2012, 421, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, J.; Pierattelli, R.; Banci, L.; Gräslund, A. High-resolution NMR studies of the zinc-binding site of the Alzheimer’s amyloid beta-peptide. FEBS J. 2007, 274, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Bousejra-ElGarah, F.; Bijani, C.; Coppel, Y.; Faller, P.; Hureau, C. Iron(II) binding to amyloid-beta, the Alzheimer’s peptide. Inorg. Chem. 2011, 50, 9024–9030. [Google Scholar] [CrossRef] [PubMed]
- Brännström, K.; Öhman, A.; Lindhagen-Persson, M.; Olofsson, A. Ca(2+) enhances Abeta polymerization rate and fibrillar stability in a dynamic manner. Biochem. J. 2013, 450, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, D.; Guo, H.-B.; Parks, J.M.; Gu, B.; Summers, A.O.; Miller, S.M.; Liang, L.; Smith, J.C. Why Mercury Prefers Soft Ligands. J. Phys. Chem. Lett. 2013, 4, 2317–2322. [Google Scholar] [CrossRef]
- Charlet, L.; Chapron, Y.; Faller, P.; Kirsch, R.; Stone, A.T.; Baveye, P.C. Neurodegenerative diseases and exposure to the environmental metals Mn, Pb, and Hg. Coord. Chem. Rev. 2012, 256, 2147–2163. [Google Scholar] [CrossRef]
- Lv, Z.; Condron, M.M.; Teplow, D.B.; Lyubchenko, Y.L. Nanoprobing of the effect of Cu(2+) cations on misfolding, interaction and aggregation of amyloid beta peptide. J. Neuroimmune Pharmacol. 2013, 8, 262–273. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Nussinov, R. Zinc ions promote Alzheimer Abeta aggregation via population shift of polymorphic states. Proc. Natl. Acad. Sci. USA 2010, 107, 9490–9495. [Google Scholar] [CrossRef]
- Wineman-Fisher, V.; Bloch, D.N.; Miller, Y. Challenges in studying the structures of metal-amyloid oligomers related to type 2 diabetes, Parkinson’s disease, and Alzheimer’s disease. Coord. Chem. Rev. 2016, 327, 20–26. [Google Scholar] [CrossRef]
- Tougu, V.; Karafin, A.; Zovo, K.; Chung, R.S.; Howells, C.; West, A.K.; Palumaa, P. Zn(II)- and Cu(II)-induced non-fibrillar aggregates of amyloid-beta (1-42) peptide are transformed to amyloid fibrils, both spontaneously and under the influence of metal chelators. J. Neurochem. 2009, 110, 1784–1795. [Google Scholar] [CrossRef]
- Serra-Batiste, M.; Ninot-Pedrosa, M.; Bayoumi, M.; Gairi, M.; Maglia, G.; Carulla, N. Abeta42 assembles into specific beta-barrel pore-forming oligomers in membrane-mimicking environments. Proc. Natl. Acad. Sci. USA 2016, 113, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Diaz, J.C.; Flora, M. Efficiency of histidine-associating compounds for blocking the alzheimer’s Abeta channel activity and cytotoxicity. Biophys. J. 2008, 95, 4879–4889. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.M.; Markesbery, W.R.; Ehmann, W.D.; Mao, Y.X.; Vance, D.E. Regional brain trace-element studies in Alzheimer’s disease. Neurotoxicology 1988, 9, 1–7. [Google Scholar] [PubMed]
- Basun, H.; Forssell, L.G.; Wetterberg, L.; Winblad, B. Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer’s disease. J. Neural Transm. Park Dis. Dement. Sect. 1991, 3, 231–258. [Google Scholar] [PubMed]
- Gerhardsson, L.; Lundh, T.; Londos, E.; Minthon, L. Cerebrospinal fluid/plasma quotients of essential and non-essential metals in patients with Alzheimer’s disease. J. Neural Transm. (Vienna) 2011, 118, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Ostertag, S.K.; Stern, G.A.; Wang, F.; Lemes, M.; Chan, H.M. Mercury distribution and speciation in different brain regions of beluga whales (Delphinapterus leucas). Sci. Total Environ. 2013, 456–457, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Pamphlett, R.; Kum Jew, S. Inorganic mercury in human astrocytes, oligodendrocytes, corticomotoneurons and the locus ceruleus: Implications for multiple sclerosis, neurodegenerative disorders and gliomas. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2018, 31, 807–819. [Google Scholar] [CrossRef]
- Pamphlett, R.; Kum Jew, S. Different Populations of Human Locus Ceruleus Neurons Contain Heavy Metals or Hyperphosphorylated Tau: Implications for Amyloid-beta and Tau Pathology in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 437–447. [Google Scholar] [CrossRef]
- Liu, A.K.; Chang, R.C.; Pearce, R.K.; Gentleman, S.M. Nucleus basalis of Meynert revisited: Anatomy, history and differential involvement in Alzheimer’s and Parkinson’s disease. Acta Neuropathol. 2015, 129, 527–540. [Google Scholar] [CrossRef]
- Roos, P.M. Ultraclean paired sampling for metal analysis in neurodegenerative disorders. J. Trace Elem. Med. Biol. 2019, 52, 48–52. [Google Scholar] [CrossRef]
- Nuttall, K.L. Interpreting mercury in blood and urine of individual patients. Ann. Clin. Lab. Sci. 2004, 34, 235–250. [Google Scholar] [PubMed]
- Frisoni, G.B.; Boccardi, M.; Barkhof, F.; Blennow, K.; Cappa, S.; Chiotis, K.; Demonet, J.F.; Garibotto, V.; Giannakopoulos, P.; Gietl, A.; et al. Strategic roadmap for an early diagnosis of Alzheimer’s disease based on biomarkers. Lancet Neurol. 2017, 16, 661–676. [Google Scholar] [CrossRef]
- Shim, Y.S.; Roe, C.M.; Buckles, V.D.; Morris, J.C. Clinicopathologic study of Alzheimer’s disease: Alzheimer mimics. J. Alzheimer’s Dis. 2013, 35, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P. Mercury exposure and Alzheimer’s disease in India—An imminent threat? Sci. Total Environ. 2017, 589, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Saxe, S.R.; Wekstein, M.W.; Kryscio, R.J.; Henry, R.G.; Cornett, C.R.; Snowdon, D.A.; Grant, F.T.; Schmitt, F.A.; Donegan, S.J.; Wekstein, D.R.; et al. Alzheimer’s disease, dental amalgam and mercury. J. Am. Dent. Assoc. 1999, 130, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Park, J.D.; Zheng, W. Human exposure and health effects of inorganic and elemental mercury. J. Prev. Med. Public Health 2012, 45, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Lin, H.; Zheng, W.; Tomanicek, S.J.; Johs, A.; Feng, X.; Elias, D.A.; Liang, L.; Gu, B. Oxidation and methylation of dissolved elemental mercury by anaerobic bacteria. Nat. Geosci. 2013, 6, 751–754. [Google Scholar] [CrossRef]
- UNEP. Global Mercury Assessment 2013: Sources, Emissions, Releases and Environmental Transport; UNEP Chemicals Branch: Geneva, Switzerland, 2013. [Google Scholar]
- Streets, D.G.; Devane, M.K.; Lu, Z.; Bond, T.C.; Sunderland, E.M.; Jacob, D.J. All-time releases of mercury to the atmosphere from human activities. Environ. Sci. Technol. 2011, 45, 10485–10491. [Google Scholar] [CrossRef]
- Roberts, K.F.; Elbert, D.L.; Kasten, T.P.; Patterson, B.W.; Sigurdson, W.C.; Connors, R.E.; Ovod, V.; Munsell, L.Y.; Mawuenyega, K.G.; Miller-Thomas, M.M.; et al. Amyloid-beta efflux from the central nervous system into the plasma. Ann. Neurol. 2014, 76, 837–844. [Google Scholar] [CrossRef]
- Leong, C.C.; Syed, N.I.; Lorscheider, F.L. Retrograde degeneration of neurite membrane structural integrity of nerve growth cones following in vitro exposure to mercury. Neuroreport 2001, 12, 733–737. [Google Scholar] [CrossRef]
- Fitsanakis, V.A.; Aschner, M. The importance of glutamate, glycine, and gamma-aminobutyric acid transport and regulation in manganese, mercury and lead neurotoxicity. Toxicol. Appl. Pharmacol. 2005, 204, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.S.; Kwok, K.M.; Chan, P.H.; So, H.K.; Li, A.M.; Ng, P.C.; Fok, T.F. Long term neurocognitive impact of low dose prenatal methylmercury exposure in Hong Kong. Environ. Int. 2013, 54, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Kerper, L.E.; Ballatori, N.; Clarkson, T.W. Methylmercury transport across the blood-brain barrier by an amino acid carrier. Am. J. Physiol. 1992, 262, R761–R765. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Zalups, R.K. Molecular and ionic mimicry and the transport of toxic metals. Toxicol. Appl. Pharmacol. 2005, 204, 274–308. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Farkas, S.; Kortbeek, S.; Zhang, F.X.; Chen, L.; Zamponi, G.W.; Syed, N.I. Mercury-induced toxicity of rat cortical neurons is mediated through N-Methyl-D-Aspartate receptors. Mol. Brain 2012, 5, 30. [Google Scholar] [CrossRef]
- Kim, D.K.; Park, J.D.; Choi, B.S. Mercury-induced amyloid-beta (Abeta) accumulation in the brain is mediated by disruption of Abeta transport. J. Toxicol. Sci. 2014, 39, 625–635. [Google Scholar] [CrossRef]
- Limson, J.; Nyokong, T.; Daya, S. The interaction of melatonin and its precursors with aluminium, cadmium, copper, iron, lead, and zinc: An adsorptive voltammetric study. J. Pineal Res. 1998, 24, 15–21. [Google Scholar] [CrossRef]
- Muche, A.; Arendt, T.; Schliebs, R. Oxidative stress affects processing of amyloid precursor protein in vascular endothelial cells. PLoS ONE 2017, 12, e0178127. [Google Scholar] [CrossRef]
- Suzuki, K.T.; Sasakura, C.; Yoneda, S. Binding sites for the (Hg-Se) complex on selenoprotein P. Biochim. Biophys. Acta 1998, 1429, 102–112. [Google Scholar] [CrossRef]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.H.; Luz, A.L.; Cao, X.; Maurer, L.L.; Blawas, A.M.; Aballay, A.; Pan, W.K.; Meyer, J.N. Effects of methyl and inorganic mercury exposure on genome homeostasis and mitochondrial function in Caenorhabditis elegans. DNA Repair (Amst) 2017, 52, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.C.; de Paula, E.S.; Pazin, M.; Carneiro, M.F.H.; Grotto, D.; Barbosa, F., Jr.; Dorta, D.J. Niacin prevents mitochondrial oxidative stress caused by sub-chronic exposure to methylmercury. Drug Chem. Toxicol. 2018, 1–7. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Ng, S.; Lin, C.-C.; Hwang, Y.-H.; Hsieh, W.-S.; Liao, H.-F.; Chen, P.-C. Mercury, APOE, and children’s neurodevelopment. Neurotoxicology 2013, 37, 85–92. [Google Scholar] [CrossRef]
- Snoj Tratnik, J.; Falnoga, I.; Trdin, A.; Mazej, D.; Fajon, V.; Miklavcic, A.; Kobal, A.B.; Osredkar, J.; Sesek Briski, A.; Krsnik, M.; et al. Prenatal mercury exposure, neurodevelopment and apolipoprotein E genetic polymorphism. Environ. Res. 2017, 152, 375–385. [Google Scholar] [CrossRef]
- Ng, S.; Lin, C.C.; Jeng, S.F.; Hwang, Y.H.; Hsieh, W.S.; Chen, P.C. Mercury, APOE, and child behavior. Chemosphere 2015, 120, 123–130. [Google Scholar] [CrossRef]
- Wojcik, D.P.; Godfrey, M.E.; Christie, D.; Haley, B.E. Mercury toxicity presenting as chronic fatigue, memory impairment and depression: Diagnosis, treatment, susceptibility, and outcomes in a New Zealand general practice setting (1994–2006). Neuroendocrinol Lett. 2006, 27, 415–423. [Google Scholar]
- Morris, M.C.; Brockman, J.; Schneider, J.A.; Wang, Y.; Bennett, D.A.; Tangney, C.C.; van de Rest, O. Association of Seafood Consumption, Brain Mercury Level, and APOE epsilon4 Status With Brain Neuropathology in Older Adults. JAMA 2016, 315, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Marechal, J.D.; Wärmländer, S.; Gräslund, A.; Peralvarez-Marin, A. In silico analysis of the apolipoprotein E and the amyloid beta peptide interaction: Misfolding induced by frustration of the salt bridge network. PLoS Comput. Biol. 2010, 6, e1000663. [Google Scholar] [CrossRef] [PubMed]
- Napier, M.D.; Poole, C.; Satten, G.A.; Ashley-Koch, A.; Marrie, R.A.; Williamson, D.M. Heavy metals, organic solvents, and multiple sclerosis: An exploratory look at gene-environment interactions. Arch. Environ. Occup. Health 2016, 71, 26–34. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| τ½ [h] | τlag [h] | rmax [h−1] | ThT End-Point [a.u] | |
|---|---|---|---|---|
| 15 μM Aβ40 | 10.4 ± 0.9 | 6.8 ± 0.4 | 0.6 ± 0.04 | 7800 ± 800 |
| 15 μM Aβ40 + 0.8 μM Hg(II) | 9.8 ± 1.1 | 7.1 ± 1.6 | 0.8 ± 0.06 | 6000 ± 2000 |
| 15 μM Aβ40 +1.5 μM Hg(II) | 11.4 ± 0.5 | 9.1 ± 1.0 | 0.9 ± 0.13 | 5500 ± 300 |
| 15 μM Aβ40 +3 μM Hg(II) | 19.9 ± 2.7 | 12.9 ± 2.2 | 0.3 ± 0.01 | 3500 ± 1100 |
| 15 μM Aβ40 +15 μM Hg(II) * | n/a * | n/a * | n/a * | n/a * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wallin, C.; Friedemann, M.; Sholts, S.B.; Noormägi, A.; Svantesson, T.; Jarvet, J.; Roos, P.M.; Palumaa, P.; Gräslund, A.; Wärmländer, S.K.T.S. Mercury and Alzheimer’s Disease: Hg(II) Ions Display Specific Binding to the Amyloid-β Peptide and Hinder Its Fibrillization. Biomolecules 2020, 10, 44. https://doi.org/10.3390/biom10010044
Wallin C, Friedemann M, Sholts SB, Noormägi A, Svantesson T, Jarvet J, Roos PM, Palumaa P, Gräslund A, Wärmländer SKTS. Mercury and Alzheimer’s Disease: Hg(II) Ions Display Specific Binding to the Amyloid-β Peptide and Hinder Its Fibrillization. Biomolecules. 2020; 10(1):44. https://doi.org/10.3390/biom10010044
Chicago/Turabian StyleWallin, Cecilia, Merlin Friedemann, Sabrina B. Sholts, Andra Noormägi, Teodor Svantesson, Jüri Jarvet, Per M. Roos, Peep Palumaa, Astrid Gräslund, and Sebastian K. T. S. Wärmländer. 2020. "Mercury and Alzheimer’s Disease: Hg(II) Ions Display Specific Binding to the Amyloid-β Peptide and Hinder Its Fibrillization" Biomolecules 10, no. 1: 44. https://doi.org/10.3390/biom10010044
APA StyleWallin, C., Friedemann, M., Sholts, S. B., Noormägi, A., Svantesson, T., Jarvet, J., Roos, P. M., Palumaa, P., Gräslund, A., & Wärmländer, S. K. T. S. (2020). Mercury and Alzheimer’s Disease: Hg(II) Ions Display Specific Binding to the Amyloid-β Peptide and Hinder Its Fibrillization. Biomolecules, 10(1), 44. https://doi.org/10.3390/biom10010044