Microglia Mediated Neuroinflammation: Focus on PI3K Modulation


 ,
, 
 , and
, and
Abstract
1. Introduction
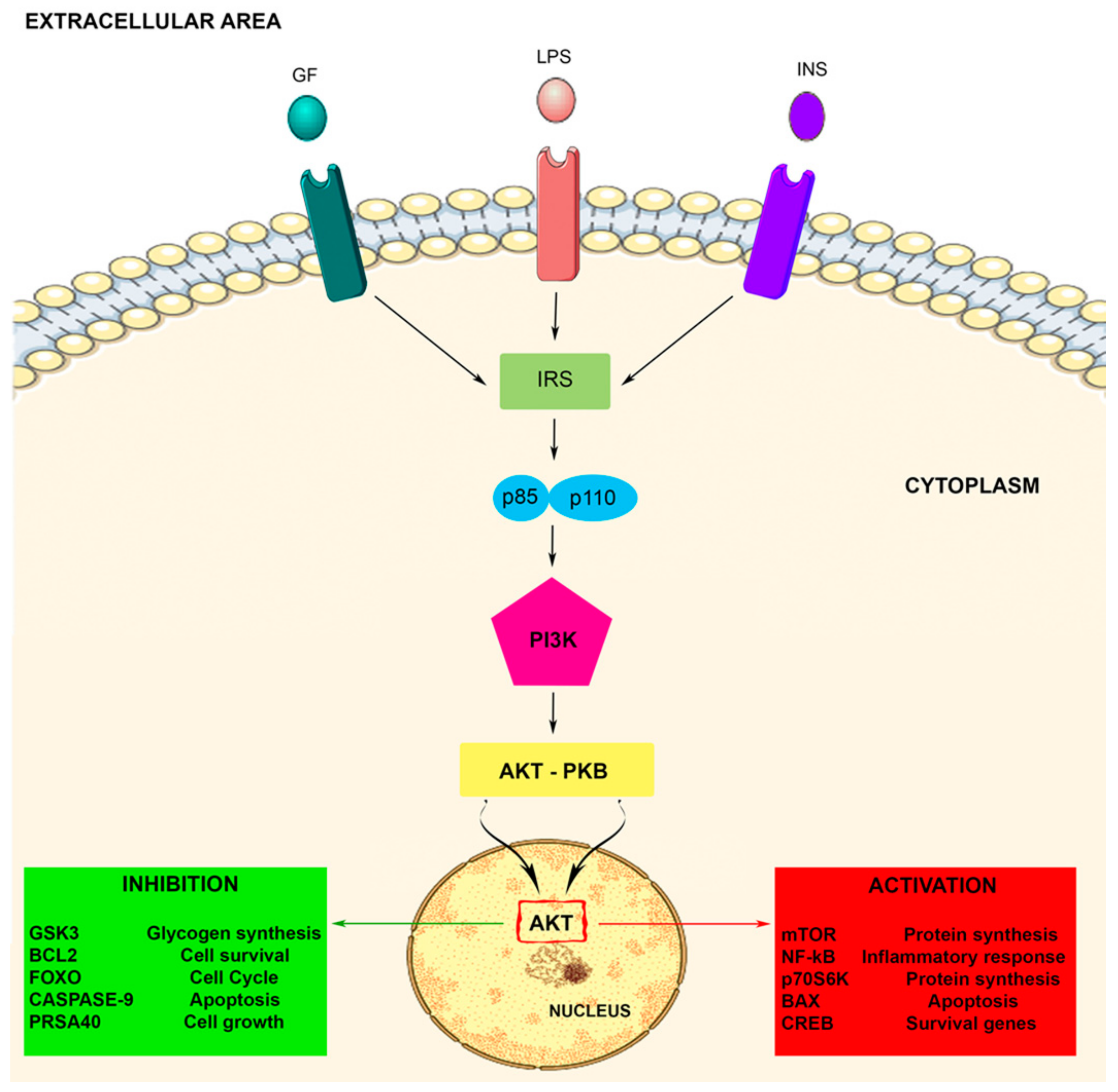
2. PI3K Signaling Pathway
2.1. Classes of PI3K Enzymes
2.2. PI3K Signaling Pathway Activation
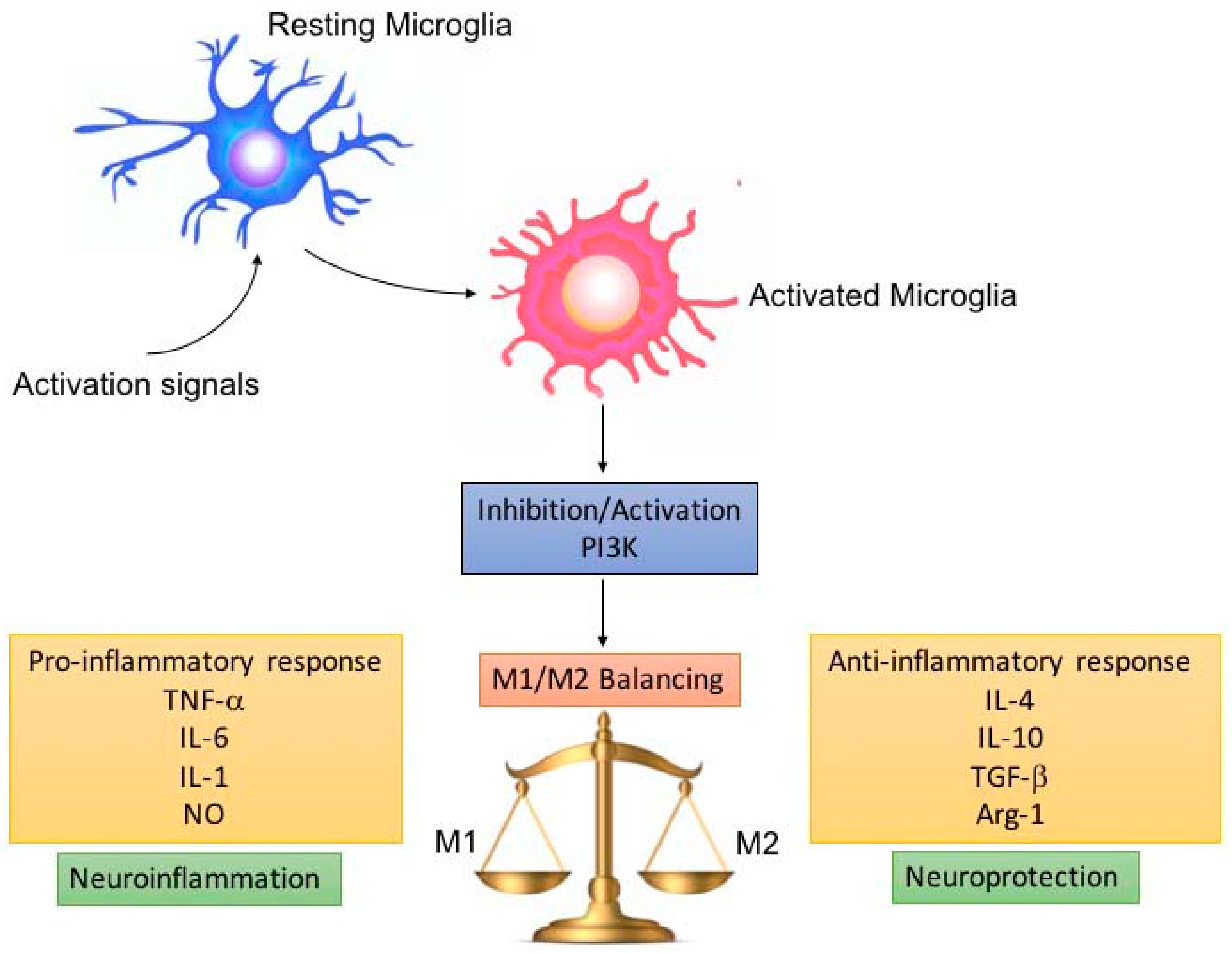
3. Role of PI3K in Microglia Activity
4. PI3K Role in Neuroinflammation
5. Therapeutic Strategies on the Modulation of PI3K Signaling
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell. Biol. 2000, 1, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Hoglund, K.; Salter, H. Molecular biomarkers of neurodegeneration. Expert Rev. Mol. Diagn. 2013, 13, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Puentes, F.; Baker, D.; Van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Gendelman, H. Neural immunity: Friend or foe? J. Neurovirol. 2002, 8, 474–479. [Google Scholar] [CrossRef] [PubMed]
- McManus, R.M.; Heneka, M.T. Role of neuroinflammation in neurodegeneration: New insights. Alzheimer Res. Ther. 2017, 9, 14. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Lehnardt, S. Innate immunity and neuroinflammation in the CNS: The role of microglia in Toll-like R in cancer. Nat. Rev. Drug Discov. 2010, 8, 627–644. [Google Scholar]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2015, 169, 1276–1290. [Google Scholar] [CrossRef]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging. 2014, 35, 2746–2760. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Whitehead, M.A.; Piñeiro, R. Molecules in medicine mini-review: Isoforms of PI3K in biology and disease. J. Mol. Med. (Berl) 2016, 94, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell. Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, C.; Cianciulli, A.; Calvello, R.; Dragone, T.; Iacobazzi, F.; Panaro, M.A. The PI3K/Akt pathway is required for LPS activation of microglial cells. Immunopharmacol. Immunotoxicol. 2012, 34, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Wang, H.; Ma, L.; Lei, P.; Zhang, Q. Ghrelin attenuates brain injury in septic mice via PI3K/Akt signalling activation. Brain. Res. Bull. 2016, 124, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.T.; Stephens, L.R. Emerging evidence of signalling roles for PI(3,4)P2 in Class I and II PI3K-regulated pathways. Biochem. Soc. Trans. 2016, 44, 307–314. [Google Scholar] [CrossRef]
- Deane, J.A.; Fruman, D.A. Phosphoinositide 3-kinase: Diverse roles in immune cell activation. Annu. Rev. Immunol. 2004, 22, 563–598. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Kobayashi, M.; Kikuta, K.; Matsuda, S. Roles of PI3K/AKT/GSK3/mTOR Pathway in Cell Signalling of Mental Illnesses. Depress. Res. Treat. 2012, 2012, 752563. [Google Scholar]
- Amzel, L.M.; Huang, C.H.; Mandelker, D.; Lengauer, C.; Gabelli, S.B.; Vogelstein, B. Structural comparisons of class I phosphoinositide 3-kinases. Nat. Rev. Cancer 2008, 8, 665–669. [Google Scholar] [CrossRef]
- Fruman, D.A.; Bismuth, G. Fine tuning the immune response with PI3K. Immunol. Rev. 2009, 228, 253–272. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, M.A.; Bombardieri, M.; Pitzalis, C.; Vanhaesebroeck, B. Isoform-selective induction of human p110δ PI3K expression by TNFα: Identification of a new and inducible PIK3CD promoter. Biochem. J. 2012, 443, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Leinders, M.; Koehrn, F.J.; Bartok, B.; Boyle, D.L.; Shubayev, V.; Kalcheva, I.; Yu, N.K.; Park, J.; Kaang, B.K.; Hefferan, M.P.; et al. Differential distribution of PI3K isoforms in spinal cord and dorsal root ganglia: Potential roles in acute inflammatory pain. Pain 2014, 155, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Geering, B.; Cutillas, P.R.; Nock, G.; Gharbi, S.I.; Vanhaesebroeck, B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc. Natl. Acad. Sci. USA 2007, 104, 7809–7814. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Kok, K.; Geering, B.; Vanhaesebroeck, B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends. Biochem. Sci. 2009, 34, 115–127. [Google Scholar] [CrossRef]
- Zhou, Z.; Tang, M.; Liu, Y.; Zhang, Z.; Lu, R.; Lu, J. Apigenin inhibits cell proliferation, migration, and invasion by targeting Akt in the A549 human lung cancer cell line. Anticancer Drugs 2017, 28, 446–456. [Google Scholar] [CrossRef]
- Suire, S.; Coadwell, J.; Ferguson, G.J.; Davidson, K.; Hawkins, P.; Stephens, L. p84, a new G beta gamma-activated regulatory subunit of the type IB phosphoinositide 3-kinase p110 gamma. Curr. Biol. 2005, 15, 566–570. [Google Scholar] [CrossRef]
- Hawkins, P.T.; Stephens, L.R. PI3K signalling in inflammation. Biochim. Biophys. Acta. 2015, 1851, 882–897. [Google Scholar] [CrossRef]
- Falasca, M.; Maffucci, T. Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem. J. 2012, 443, 587–601. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug. Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef]
- Valet, C.; Severin, S.; Chicanne, G.; Laurent, P.A.; Gaits-Iacovoni, F.; Gratacap, M.P.; Payrastre, B. The role of class I, II and III PI 3-kinases in platelet production and activation and their implication in thrombosis. Adv. Biol. Regul. 2016, 61, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, M.; Cao, B.; Hou, T.; Mao, X. Targeting the phosphatidylinositol 3-kinase/AKT pathway for the treatment of multiple myeloma. Curr. Med. Chem. 2014, 21, 3173–3187. [Google Scholar] [CrossRef] [PubMed]
- Popiolek-Barczyk, K.; Mika, J. Targeting the Microglial Signalling Pathways: New Insights in the Modulation of Neuropathic Pain. Curr. Med. Chem. 2016, 23, 2908–2928. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; Dorner, M.B.; Schaefer, M. Characterization of p87(PIKAP), a novel regulatory subunit of phosphoinositide 3-kinase gamma that is highly expressed in heart and interacts with PDE3B. J. Biol. Chem. 2006, 281, 9977–9986. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, R.; Downward, J. SnapShot: Class I PI3K isoform signalling. Cell 2013, 154, 940–940.e1. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Irie-Sasaki, J.; Jones, R.G.; Oliveira-dos-Santos, A.J.; Stanford, W.L.; Bolon, B.; Wakeham, A.; Itie, A.; Bouchard, D.; Kozieradzki, I.; et al. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science 2000, 287, 1040–1046. [Google Scholar] [CrossRef]
- Costa, C.; Martin-Conte, E.L.; Hirsch, E. Phosphoinositide 3-kinase p110gamma in immunity. IUBMB Life 2011, 63, 707–771. [Google Scholar]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, immunity, homeostasis, and cancer. Annu. Rev. Cell. Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef]
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schöneberg, J.; Ullrich, A.; Lampe, A.; Müller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 2013, 499, 233–237. [Google Scholar] [CrossRef]
- Brown, R.A.; Domin, J.; Arcaro, A.; Waterfield, M.D.; Shepherd, P.R. Insulin activates the α isoform of class II phosphoinositide 3-kinase. J. Biol. Chem. 1999, 274, 14529–14532. [Google Scholar] [CrossRef] [PubMed]
- Ktori, C.; Shepherd, P.R.; O’Rourke, L. TNF-alpha and leptin activate the a-isoform of class II phosphoinositide 3-kinase. Biochem. Biophys. Res. Commun. 2003, 306, 139–143. [Google Scholar] [CrossRef]
- Arcaro, A.; Khanzada, U.K.; Vanhaesebroeck, B.; Tetley, T.D.; Waterfield, M.D.; Seckl, M.J. Two distinct phosphoinositide 3-kinases mediate polypeptide growth factor-stimulated PKB activation. Embo J. 2002, 21, 5097–5108. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug. Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Visnjić, D.; Curić, J.; Crljen, V.; Batinić, D.; Volinia, S.; Banfić, H. Nuclear phosphoinositide 3-kinase C2beta activation during G2/M phase of the cell cycle in HL-60 cells. Biochim. Biophys. Acta. 2003, 631, 61–71. [Google Scholar] [CrossRef]
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Aβ-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta. 2016, 11, 2820–2834. [Google Scholar] [CrossRef]
- Backer, J.M. The regulation and function of Class III PI3Ks: Novel roles for Vps34. Biochem. J. 2008, 410, 1–17. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Ho, V.W.; Kuroda, E.; Ruschmann, J.; Antignano, F.; Lam, V.; Krystal, G. Role of SHIP in cancer. Exp. Hemat. 2011, 39, 2–13. [Google Scholar] [CrossRef]
- Blero, D.; Payrastre, B.; Schurmans, S.; Erneux, C. Phosphoinositide phosphatases in a network of signalling reactions. Pflugers. Archiv. 2007, 455, 31–44. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB signalling: Navigating the network. Cell 2017, 169, 281–405. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Ma, L.; Kaarela, T.; Li, Z. Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J. Neuroinflammation 2012, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef]
- Gertig, U.; Hanisch, U.K. Microglial diversity by responses and responders. Front. Cell. Neurosci. 2014, 8, 101. [Google Scholar] [CrossRef]
- Cianciulli, A.; Calvello, R.; Porro, C.; Trotta, T.; Salvatore, R.; Panaro, M.A. PI3k/Akt signalling pathway plays a crucial role in the anti-inflammatory effects of curcumin in LPS-activated microglia. Int. Immunopharmacol. 2016, 36, 282–290. [Google Scholar] [CrossRef]
- Elstner, M.; Morris, C.M.; Heim, K.; Bender, A.; Mehta, D.; Jaros, E.; Klopstock, T.; Meitinger, T.; Turnbull, D.M.; Prokisch, H. Expression analysis of dopaminergic neurons in Parkinson’s disease and aging links transcriptional dysregulation of energy metabolism to cell death. Acta Neuropathol. 2011, 122, 75–86. [Google Scholar] [CrossRef]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef]
- London, A.; Cohen, M.; Schwartz, M. Microglia and monocyte-derived macrophages: Functionally distinct populations that act in concert in CNS plasticity and repair. Front. Cell. Neurosci. 2013, 7, 34. [Google Scholar] [CrossRef]
- Murphy, S. Production of nitric oxide by glial cells: Regulation and potential roles in the CNS. Glia 2000, 29, 1–13. [Google Scholar] [CrossRef]
- Bal-Price, A.; Brown, G.C. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J. Neurosci. 2001, 21, 6480–6491. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Wada, Y.; Kitagishi, Y.; Matsuda, S. Link between PI3K/AKT/PTEN Pathway and NOX Proteinin Diseases. Aging Dis. 2014, 5, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.K.; Jha, N.K.; Kar, R.; Ambasta, R.K.; Kumar, P. p38 MAPK and PI3K/AKT Signalling Cascades in Parkinson’s Disease. Int. J. Mol. Cell. Med. 2015, 4, 67–86. [Google Scholar] [PubMed]
- Bozic, I.; Savic, D.; Laketa, D.; Bjelobaba, I.; Milenkovic, I.; Pekovic, S.; Nedeljkovic, N.; Lavrnja, I. Benfotiamine attenuates inflammatory response in LPS stimulated BV-2 microglia. PLoS ONE 2015, 10, e0118372. [Google Scholar] [CrossRef] [PubMed]
- Eggen, B.J.; Raj, D.; Hanisch, U.K.; Boddeke, H.W. Microglial phenotype and adaptation. J. Neuroimmune Pharmacol. 2013, 8, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, X.; Liu, Z.; Peng, Y.P.; Qiu, Y.H. Interleukin-10 Protection against Lipopolysaccharide-Induced Neuro-Inflammation and Neurotoxicity in Ventral Mesencephalic Cultures. Int. J. Mol. Sci. 2015, 17, E25. [Google Scholar] [CrossRef]
- Xing, B.; Xin, T.; Hunter, R.L.; Bing, G. Pioglitazone inhibition of lipopolysaccharide-induced nitric oxide synthase is associated with altered activity of p38 MAP kinase and PI3K/Akt. J. Neuroinflammation 2008, 5, 4. [Google Scholar] [CrossRef]
- Hu, X.; Zhou, H.; Zhang, D.; Yang, S.; Qian, L.; Wu, H.M.; Chen, P.S.; Wilson, B.; Gao, H.M.; Lu, R.B.; et al. Clozapine protects dopaminergic neurons from inflammation-induced damage by inhibiting microglial overactivation. J. Neuroimmune Pharmacol. 2012, 7, 187–201. [Google Scholar] [CrossRef]
- Tarassishin, L.; Suh, H.S.; Lee, S.C. Interferon regulatory factor 3 plays an anti-inflammatory role in microglia by activating the PI3K/Akt pathway. J. Neuroinflammation 2011, 8, 187. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, X.; Dai, X.; Lu, S.; Gui, B.; Jin, W.; Zhang, S.; Zhang, S.; Qian, Y. Lithium ameliorates lipopolysaccharide-induced microglial activation via inhibition of toll-like receptor 4 expression by activating the PI3K/Akt/FoxO1 pathway. J. Neuroinflammation 2014, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Chang, H.F.; Tsai, M.J.; Chen, J.S.; Wang, M.J. 6-Mercaptopurine attenuates tumor necrosis factor-α production in microglia through Nur77-mediated transrepression and PI3K/Akt/mTOR signalling-mediated translational regulation. J. Neuroinflammation 2016, 13, 78. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, R.; Burm, S.M.; Bajramovic, J.J. An Overview of in vitro Methods to Study Microglia. Front. Cell Neurosci. 2018, 12, 242. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yang, L.; Wan, C.X.; Xia, Y.Z.; Zhang, C.; Chen, M.H.; Wang, Z.D.; Li, Z.R.; Li, X.M.; Geng, Y.D.; et al. Anti-neuroinflammatory effect of Sophoraflavanone G from Sophora alopecuroides in LPS-activated BV2 microglia by MAPK, JAK/STAT and Nrf2/HO-1 signalling pathways. Phytomedicine 2016, 23, 1629–1637. [Google Scholar] [CrossRef]
- Jung, J.S.; Choi, M.J.; Lee, Y.Y.; Moon, B.I.; Park, J.S.; Kim, H.S. Suppression of Lipopolysaccharide-Induced Neuroinflammation by Morin via MAPK, PI3K/Akt, and PKA/HO-1 Signalling Pathway Modulation. J. Agric. Food. Chem. 2017, 65, 373–382. [Google Scholar] [CrossRef]
- Rivière, J.B.; Mirzaa, G.M.; O’Roak, B.J.; Beddaoui, M.; Alcantara, D.; Conway, R.L.; St-Onge, J.; Schwartzentruber, J.A.; Gripp, K.W.; Nikkel, S.M.; et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 2012, 44, 934–940. [Google Scholar] [CrossRef]
- Jansen, L.A.; Mirzaa, G.M.; Ishak, G.E.; O’Roak, B.J.; Hiatt, J.B.; Roden, W.H.; Gunter, S.A.; Christian, S.L.; Collins, S.; Adams, C.; et al. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 2015, 138, 1613–1628. [Google Scholar] [CrossRef]
- Brandt, C.; Hillmann, P.; Noack, A.; Römermann, K.; Öhler, L.A.; Rageot, D.; Beaufils, F.; Melone, A.; Sele, A.M.; Wymann, M.P.; et al. The novel, catalytic mTORC1/2 inhibitor PQR620 and the PI3K/mTORC1/2 inhibitor PQR530 effectively cross the blood-brain barrier and increase seizure threshold in a mouse model of chronic epilepsy. Neuropharmacology 2018, 140, 107–120. [Google Scholar] [CrossRef]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef]
- Li, S.; Chen, X.; Mao, L.; Zahid, K.R.; Wen, J.; Zhang, L.; Zhang, M.; Duan, J.; Duan, J.; Yin, X.; et al. Histone deacetylase 1 promotes glioblastoma cell proliferation and invasion via activation of PI3K/AKT and MEK/ERK signalling pathways. Brain. Res. 2018, 1692, 154–162. [Google Scholar] [CrossRef]
- Seitz, C.; Hugle, M.; Cristofanon, S.; Tchoghandjian, A.; Fulda, S. The dual PI3K/mTOR inhibitor NVP-BEZ235 and chloroquine synergize to trigger apoptosis via mitochondrial-lysosomal cross-talk. Int. J. Cancer 2013, 132, 2682–2693. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Qiu, J.; Liang, M.; Golinski, J.; Van Leyen, K.; Jung, J.E.; You, Z.; Lo, E.H.; Degterev, A.; Whalen, M.J. Akt and mTOR mediate programmed necrosis in neurons. Cell. Death Dis. 2014, 5, e1084. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira, C.R.; Rego, A.C. Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free Rad. Biol. Med. 2014, 74, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C.; Neel, B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. Usa 1999, 96, 4240–4245. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.L.; Xu, X.M. PTEN inhibitor bisperoxovanadium protects oligodendrocytes and myelin and prevents neuronal atrophy in adult rats following cervical hemicontusive spinal cord injury. Neurosci. Lett. 2014, 573, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Park, K.K.; Belin, S.; Wang, D.; Lu, T.; Chen, G.; Zhang, K.; Yeung, C.; Feng, G.; Yankner, B.A.; et al. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature 2011, 480, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, S.; Pan, R.; Li, G.; Tang, H.; Jiang, M.; Xing, Y.; Jin, F.; Lin, L.; Dong, J. Curcumin Attenuates gp120-Induced Microglial Inflammation by Inhibiting Autophagy via the PI3K Pathway. Cell. Mol. Neurobiol. 2018, 38, 1465–1477. [Google Scholar] [CrossRef]
- Ermak, G.; Hench, K.J.; Chang, K.T.; Sachdev, S.; Davies, K.J. Regulator of calcineurin (RCAN1-1L) is deficient in Huntington disease and protective against mutant huntingtin toxicity in vitro. J. Biol. Chem. 2009, 284, 11845–11853. [Google Scholar] [CrossRef]
- Liu, N.K.; Xu, X.M. Neuroprotection and its molecular mechanism following spinal cord injury. Neural. Regen. Res. 2012, 7, 2051–2062. [Google Scholar]
- Delgado-Esteban, M.; Martin-Zanca, D.; Andres-Martin, L.; Almeida, A.; Bolaños, J.P. Inhibition of PTEN by peroxynitrite activates the phosphoinositide-3-kinase/Akt neuroprotective signalling pathway. J. Neurochem. 2007, 102, 194–205. [Google Scholar] [CrossRef]
- Takeuchi, K.; Gertner, M.J.; Zhou, J.; Parada, L.F.; Bennett, M.V.; Zukin, R.S. Dysregulation of synaptic plasticity precedes appearance of morphological defects in a Pten conditional knockout mouse model of autism. Proc. Natl. Acad. Sci. USA 2013, 110, 4738–4743. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.; El Fissi, N.; Aouane, A.; Cabirol-Pol, M.J.; Rival, T.; Liévens, J.C. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell. Death Dis. 2015, 6, e1617. [Google Scholar] [CrossRef] [PubMed]
- Troutman, T.D.; Bazan, J.F.; Pasare, C. Toll-like receptors, signalling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle 2012, 11, 3559–3567. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, E.; Taboubi, S.; Torres, D.; Delbauve, S.; Hachani, A.; Whitehead, M.A.; Pearce, W.P.; Berenjeno, I.M.; Nock, G.; Filloux, A.; et al. The p110δ isoform of the kinase PI(3)K controls the subcellular compartmentalization of TLR4 signalling and protects from endotoxic shock. Nat. Immunol. 2012, 13, 1045–1054. [Google Scholar] [CrossRef]
- Deng, Q.; Huttenlocher, A. Leukocyte migration from a fish eye’s view. J. Cell. Sci. 2012, 125, 3949–3956. [Google Scholar] [CrossRef]
- Stephens, L.; Milne, L.; Hawkins, P. Moving towards a better understanding of chemotaxis. Curr. Biol. 2008, 18, 485–494. [Google Scholar] [CrossRef]
- Ferguson, G.J.; Milne, L.; Kulkarni, S.; Sasaki, T.; Walker, S.; Andrews, S.; Crabbe, T.; Finan, P.; Jones, G.; Jackson, S.; et al. PI(3)Kγ has an important context-dependent role in neutrophil chemokinesis. Nat. Cell. Biol. 2007, 9, 86–91. [Google Scholar] [CrossRef]
- Smith, D.F.; Deem, T.L.; Bruce, A.C.; Reutershan, J.; Wu, D.; Ley, K. Leukocyte phosphoinositide-3 kinase {gamma} is required for chemokine-induced, sustained adhesion under flow in vivo. J. Leukoc. Biol. 2006, 80, 1491–1499. [Google Scholar] [CrossRef]
- Bony, C.; Roche, S.; Shuichi, U.; Sasaki, T.; Crackower, M.A.; Penninger, J.; Mano, H.; Pucéat, M. A specific role of phosphatidylinositol 3-kinase gamma. A regulation of autonomic Ca2+ oscillations in cardiac cells. J. Cell. Biol. 2001, 152, 717–728. [Google Scholar] [CrossRef]
- Yanamandra, M.; Mitra, S.; Giri, A. Development and application of PI3K assays for novel drug discovery. Expert. Opin. Drug. Discov. 2015, 10, 171–186. [Google Scholar] [CrossRef]
- Sinagra, T.; Tamburella, A.; Urso, V.; Siarkos, I.; Drago, F.; Bucolo, C.; Salomone, S. Reversible inhibition of vasoconstriction by thiazolidinediones related to PI3K/Akt inhibition in vascular smooth muscle cells. Biochem. Pharmacol. 2013, 85, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; He, Y.; Li, D.; Han, R.; Liu, G.; Kong, D.; Hao, J. Class I PI3K inhibitor ZSTK474 mediates a shift in microglial/macrophage phenotype and inhibits inflammatory response in mice with cerebral ischemia/reperfusion injury. J. Neuroinflammation 2016, 13, 192. [Google Scholar] [CrossRef] [PubMed]
- Rice-Evans, C. Flavonoid antioxidants. Curr. Med. Chem. 2001, 8, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.E.; Rice-Evans, C.; Williams, R.J. Modulation of pro-survival Akt/PKB and ERK1/2 signalling cascades by quercetin and its in vivo metabolites underlie their action on neuronal viability. J. Biol. Chem. 2003, 278, 34783–34793. [Google Scholar] [CrossRef]
- Williams, R.J.; Spencer, J.P.E.; Rice-Evans, C. Flavonoids: Antioxidants or signalling molecules? Free Radic. Biol. Med. 2004, 36, 838–849. [Google Scholar] [CrossRef]
- Agullo, G.; Gamet-Payrastre, L.; Manenti, S.; Viala, C.; Rémésy, C.; Chap, H.; Payrastre, B. Relationship between flavonoid structure and inhibition of phosphatidylinositol 3-kinase: A comparison with tyrosine kinase and protein kinase C inhibition. Biochem. Pharmacol. 1997, 53, 1649–1657. [Google Scholar]
- Ferriola, P.C.; Cody, V.; Middleton, E., Jr. Protein kinase C inhibition by plant flavonoids. Kinetic mechanisms and structure-activity relationships. Biochem. Pharmacol. 1989, 38, 1617–1624. [Google Scholar] [CrossRef]
- Matter, W.F.; Brown, R.F.; Vlahos, C.J. The inhibition of phosphatidylinositol 3-kinase by quercetin and analogs. Biochem. Biophys. Res. Commun. 1992, 186, 624–631. [Google Scholar] [CrossRef]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell. 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Kong, A.N.; Yu, R.; Chen, C.; Mandlekar, S.; Primiano, T. Signal transduction events elicited by natural products: Role of MAPK and caspase pathways in homeostatic response and induction of apoptosis. Arch. Pharm. Res. 2000, 23, 1–16. [Google Scholar] [CrossRef]
- Cao, F.; Jin, T.Y.; Zhou, Y.F. Inhibitory effect of isoflavones on prostate cancer cells and PTEN gene. Biomed. Environ. Sci. 2006, 19, 35–41. [Google Scholar]
- Dave, B.; Eason, R.R.; Till, S.R.; Geng, Y.; Velarde, M.C.; Badger, T.M.; Simmen, R.C. The soy isoflavone genistein promotes apoptosis in mammary epithelial cells by inducing the tumor suppressor PTEN. Carcinogenesis 2005, 2, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Ye, X.; Bao, X.; Zhao, B.; Wang, X.; Zhang, D. Inhibition of Src tyrosine kinase activity by squamosamide derivative FLZ attenuates neuroinflammation in both in vivo and in vitro Parkinson’s disease models. Neuropharmacology 2013, 75, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.S.; Shi, H.L.; Huang, F.; Peterson, K.E.; Wu, H.; Lan, Y.Y.; Zhang, B.B.; He, Y.X.; Woods, T.; Du, M.; et al. Astragaloside IV inhibits microglia activation via glucocorticoid receptor mediated signalling pathway. Sci. Rep. 2016, 16, 19137. [Google Scholar] [CrossRef] [PubMed]
- Knafo, S.; Esteban, J.A. PTEN: Local and Global Modulation of Neuronal Function in Health and Disease. Trends Neurosci. 2017, 40, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.H.; Spinks, C.A.; Doleman, J.F.; Melchini, A.; Ball, R.Y.; Mills, R.D.; Mithen, R.F. The dietary isothiocyanate sulforaphane modulates gene expression and alternative gene splicing in a PTEN null preclinical murine model of prostate cancer. Mol. Cancer. 2010, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- De la Parra, C.; Castillo-Pichardo, L.; Cruz-Collazo, A.; Cubano, L.; Redis, R.; Calin, G.A.; Dharmawardhane, S. Soy isoflavone genistein-mediated downregulation of miR-155 contributes to the anticancer effects of genistein. Nutr. Cancer 2016, 68, 154–164. [Google Scholar] [CrossRef]
- Rovito, D.; Giordano, C.; Vizza, D.; Plastina, P.; Barone, I.; Casaburi, I.; Lanzino, M.; De Amicis, F.; Sisci, D.; Mauro, L.; et al. Omega-3 PUFA ethanolamides DhEA and EPEA induce autophagy through PPARγ activation in MCF-7 breast cancer cells. J. Cell. Physiol. 2013, 228, 1314–1322. [Google Scholar] [CrossRef]
- Ghosh-Choudhury, T.; Mandal, C.C.; Woodruff, K.; St Clair, P.; Fernandes, G.; Choudhury, G.G.; Ghosh-Choudhury, N. Fish oil targets PTEN to regulate NFkappaB for downregulation of anti-apoptotic genes in breast tumor growth. Breast Cancer Res. Treat. 2009, 118, 213–228. [Google Scholar] [CrossRef]
- Moreira, J.D.; Knorr, L.; Thomazi, A.P.; Simão, F.; Battú, C.; Oses, J.P.; Gottfried, C.; Wofchuk, S.; Salbego, C.; Souza, D.O.; et al. Dietary omega-3 fatty acids attenuate cellular damage after a hippocampal ischemic insult in adult rats. J. Nutr. Biochem. 2010, 21, 351–356. [Google Scholar] [CrossRef]
- Yu, H.; Deng, J.; Zuo, Z. High-fat diet reduces neuroprotection of isoflurane post-treatment: Role carboxyl-terminal modulator protein-Akt signalling. Obes. (Silver Spring) 2014, 22, 2396–2405. [Google Scholar] [CrossRef]
- Park, H.Y.; Kim, N.D.; Kim, G.Y.; Hwang, H.J.; Kim, B.W.; Kim, W.J.; Choi, Y.H. Inhibitory effects of diallyl disulfide on the production of inflammatory mediators and cytokines in lipopolysaccharide-activated BV2 microglia. Toxicol. Appl. Pharmacol. 2012, 262, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Herdegen, T.; Skene, P.; Bahr, M. The c-Jun transcription factor-bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997, 20, 227–231. [Google Scholar] [CrossRef]
- Zhao, M.; Zhou, A.; Xu, L.; Zhang, X. The role of TLR4-mediated PTEN/PI3K/AKT/NF-κB signalling pathway in neuroinflammation in hippocampal neurons. Neuroscience 2014, 269, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Mechlovich, D.; Amit, T.; Bar-Am, O.; Mandel, S.; Youdim, M.B.; Weinreb, O. The novel multi-target iron chelator, M30 modulates HIF-1α-related glycolytic genes and insulin signalling pathway in the frontal cortex of APP/PS1 Alzheimer’s disease mice. Curr. Alzheimer. Res. 2014, 11, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Qi, B.; Xiaoxiang, W.; Xu, J.; Liu, X. Baicalein increases cisplatin sensitivity of A549 lung adenocarcinoma cells via PI3K/Akt/NF-κB pathway. Biomed. Pharmacother. 2017, 90, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Milito, A.; Spagnuolo, C.; Carbone, V.; Rosén, A.; Minasi, P.; Lauria, F.; Russo, G.L. CK2 and PI3K are direct molecular targets of quercetin in chronic lymphocytic leukaemia. Oncotarget 2017, 8, 4257–42587. [Google Scholar] [CrossRef]
- Zhao, J.; Cheng, Y.Y.; Fan, W.; Yang, C.B.; Ye, S.F.; Cui, W.; Wei, W.; Lao, L.X.; Cai, J.; Han, Y.F.; et al. Botanical drug puerarin coordinates with nerve growth factor in the regulation of neuronal survival and neuritogenesis via activating ERK1/2 and PI3K/Akt signalling pathways in the neurite extension process. Cns Neurosci. Ther. 2015, 21, 61–70. [Google Scholar] [CrossRef]
- Nones, J.E.; Spohr, T.C.; Gomes, F.C. Hesperidin, a flavone glycoside, as mediator of neuronal survival. Neurochem. Res. 2011, 36, 1776–1784. [Google Scholar] [CrossRef]
- Landis-Piwowar, K.; Chen, D.; Foldes, R.; Chan, T.H.; Dou, Q.P. Novel epigallocatechin gallate analogs as potential anticancer agents: A patent review (2009–present). Expert Opin. Ther. Pat. 2013, 23, 189–202. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhu, Y.; He, T.; Li, W.; Li, Q.; Miao, Y. Brain-derived neurotrophic factor inhibits hyperglycemia-induced apoptosis and downregulation of synaptic plasticity-related proteins in hippocampal neurons via the PI3K/Akt pathway. Int. J. Mol. Med. 2019, 43, 294–304. [Google Scholar] [CrossRef]
- Liu, T.Y.; Shi, C.X.; Gao, R.; Sun, H.J.; Xiong, X.Q.; Ding, L.; Chen, Q.; Li, Y.H.; Wang, J.J.; Kang, Y.M.; et al. Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin. Sci. (Lond) 2015, 129, 839–850. [Google Scholar] [CrossRef]
- Niswender, K.D.; Morrison, C.D.; Clegg, D.J.; Olson, R.; Baskin, D.G.; Myers, M.G., Jr.; Seeley, R.J.; Schwartz, M.W. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: A key mediator of insulin-induced anorexia. Diabetes 2003, 52, 227–231. [Google Scholar] [CrossRef]
- Marchetti, L.; Klein, M.; Schlett, K.; Pfizenmaier, K.; Eisel, U.L. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J. Biol. Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef]
- Kim, W.K.; Hwang, S.Y.; Oh, E.S.; Piao, H.Z.; Kim, K.W.; Han, I.O. TGF-beta1 represses activation and resultant death of microglia via inhibition of phosphatidylinositol 3-kinase activity. J. Immunol. 2004, 172, 7015–7023. [Google Scholar] [CrossRef]
- Wang, C.; Chi, Y.; Li, J.; Miao, Y.; Li, S.; Su, W.; Jia, S.; Chen, Z.; Du, S.; Zhang, X.; et al. FAM3A activates PI3Kp110α/Akt signalling to ameliorate hepatic gluconeogenesis and lipogenesis. Hepatology 2014, 59, 1779–1790. [Google Scholar]
- Hafner, A.; Obermajer, N.; Kos, J. γ-Enolase C-terminal peptide promotes cell survival and neurite outgrowth by activation of the PI3K/Akt and MAPK/ERK signalling pathways. Biochem. J. 2012, 443, 439–450. [Google Scholar] [CrossRef]
- Molina-Holgado, E.; Vela, J.M.; Arévalo-Martín, A.; Almazán, G.; Molina-Holgado, F.; Borrell, J.; Guaza, C. Cannabinoids promote oligodendrocyte progenitor survival: Involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signalling. J. Neurosci. 2002, 22, 9742–9753. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| PI3k | Expression | Ref |
|---|---|---|
| p110 α | Ubiquitous | [14,23] |
| p110 β | Ubiquitous | [14,23,24] |
| p110 δ | Immune cells, Neurons and Microglia, Spleen, Platelets, Endothelial Cells | [25] |
| p85 α | Ubiquitous | [24] |
| p55 α | Brain, Muscle | [26] |
| p50 α | Liver, Kidney, Brain, T Cells | [25] |
| p85 β | Ubiquitous | [24,26] |
| p55 γ | Brain, Testis, Liver, Muscle, Fat, Spleen | [24] |
| p110 γ | Immune Cells, Heart, Pancreas, Liver, Skeletal Muscle | [25,27] |
| p101 | Immune Cells, Mast Cells | [26,28,29] |
| p84/p87 | Immune cells, Mast Cell, Heart | [26,28,29] |
| pI3k-C2 | Ubiquitous | [30] |
| pI3k-C2β | Ubiquitous | [30] |
| pI3k-C2 γ | Liver, Prostate, Breast, Salivary Gland | [31] |
| Vps34 | Ubiquitous | [32] |
| Vps15 | Ubiquitous | [26] |
| Compounds | Nature | Function | Ref. |
|---|---|---|---|
| Myricetin | Flavonoid | Inhibition | [106] |
| Baicalein | Flavonoid | Inhibition | [126] |
| Apigenin | Flavonoid | Inhibition | [27] |
| Morin | Flavonoid | Inhibition | [74] |
| Quercetin | Flavonoid | Inhibition | [127] |
| Puerarin | Flavonoid | Activation | [128] |
| Hesperidin | Flavonoid | Activation | [129] |
| EGCG | Polyphenol | Inhibition | [130] |
| Curcumin | Polyphenol | Inhibition | [57,88] |
| Wortmannin | A fungal metabolite | Inhibition | [131] |
| Irisin | Hormone | Activation | [132] |
| Insulin | Hormone | Activation | [133] |
| BDNF | Brain-derived neurotrophic factor | Activation | [131] |
| PDGF | Platelet-derived growth factor | Activation | [43] |
| EGF | Epidermal growth factor | Activation | [43] |
| TNFR2 | 75-kDa TNF receptor type II | Activation | [134] |
| TGF-1α | Pleiotropic cytokine | Inhibition | [135] |
| FAM3A | Cytokine-like gene family | Activation | [136] |
| NSE | Neuron specific enolase | Activation | [137] |
| 6-MP | Thiopurine | Inhibition | [137] |
| LY294002 | Inhibitor | Inhibition | [138] |
| ZSTK474 | Class I PI3K inhibitor | Inhibition | [132] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cianciulli, A.; Porro, C.; Calvello, R.; Trotta, T.; Lofrumento, D.D.; Panaro, M.A. Microglia Mediated Neuroinflammation: Focus on PI3K Modulation. Biomolecules 2020, 10, 137. https://doi.org/10.3390/biom10010137
Cianciulli A, Porro C, Calvello R, Trotta T, Lofrumento DD, Panaro MA. Microglia Mediated Neuroinflammation: Focus on PI3K Modulation. Biomolecules. 2020; 10(1):137. https://doi.org/10.3390/biom10010137
Chicago/Turabian StyleCianciulli, Antonia, Chiara Porro, Rosa Calvello, Teresa Trotta, Dario Domenico Lofrumento, and Maria Antonietta Panaro. 2020. "Microglia Mediated Neuroinflammation: Focus on PI3K Modulation" Biomolecules 10, no. 1: 137. https://doi.org/10.3390/biom10010137
APA StyleCianciulli, A., Porro, C., Calvello, R., Trotta, T., Lofrumento, D. D., & Panaro, M. A. (2020). Microglia Mediated Neuroinflammation: Focus on PI3K Modulation. Biomolecules, 10(1), 137. https://doi.org/10.3390/biom10010137