Vibrationally and Spin-Orbit-Resolved Inner-Shell X-ray Absorption Spectroscopy of the NH+ Molecular Ion: Measurements and ab Initio Calculations


 , ,
, , 
Abstract
1. Introduction
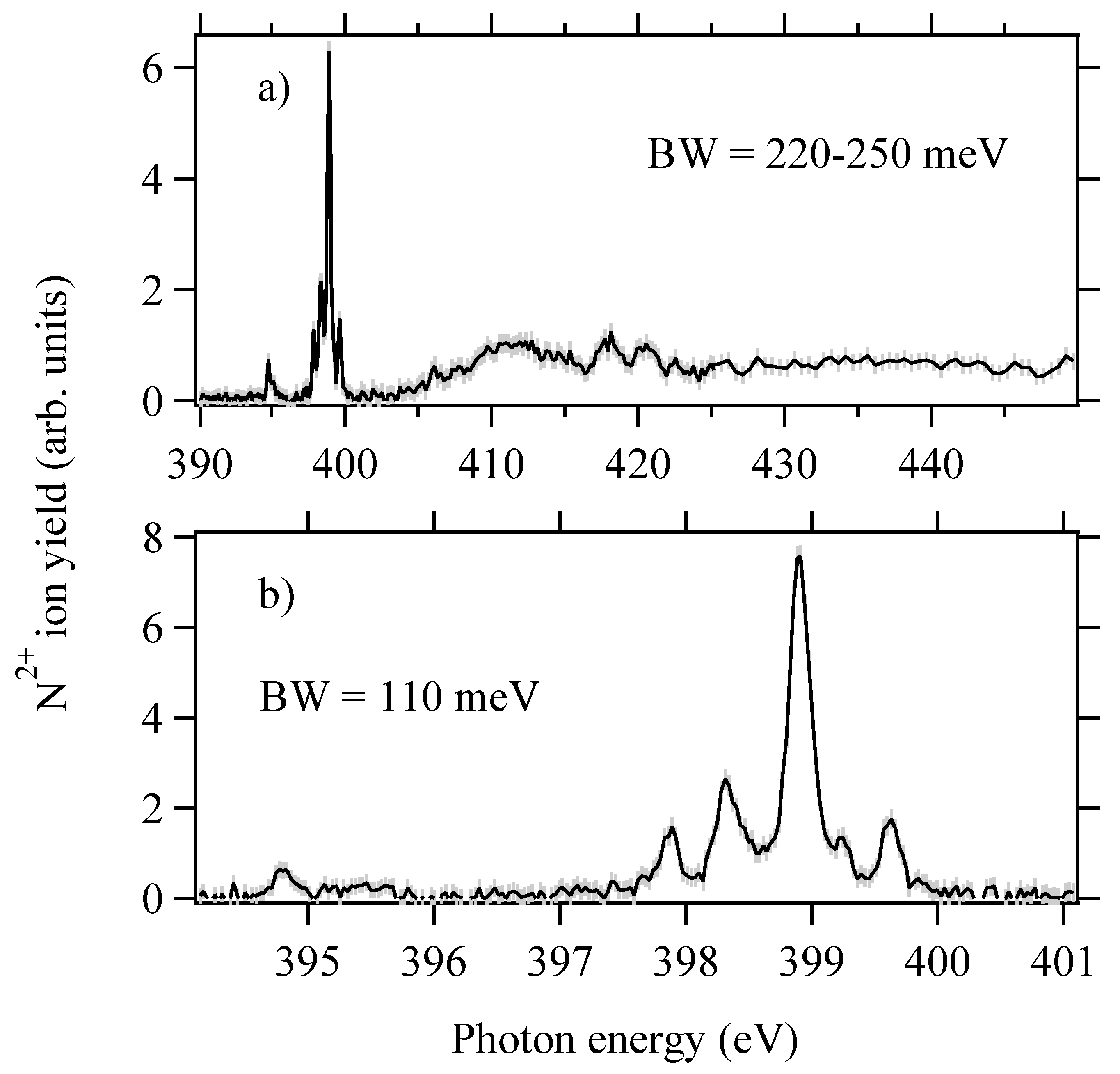
2. Experimental Details and Results
3. Theoretical Aspects
3.1. Electronic Energies
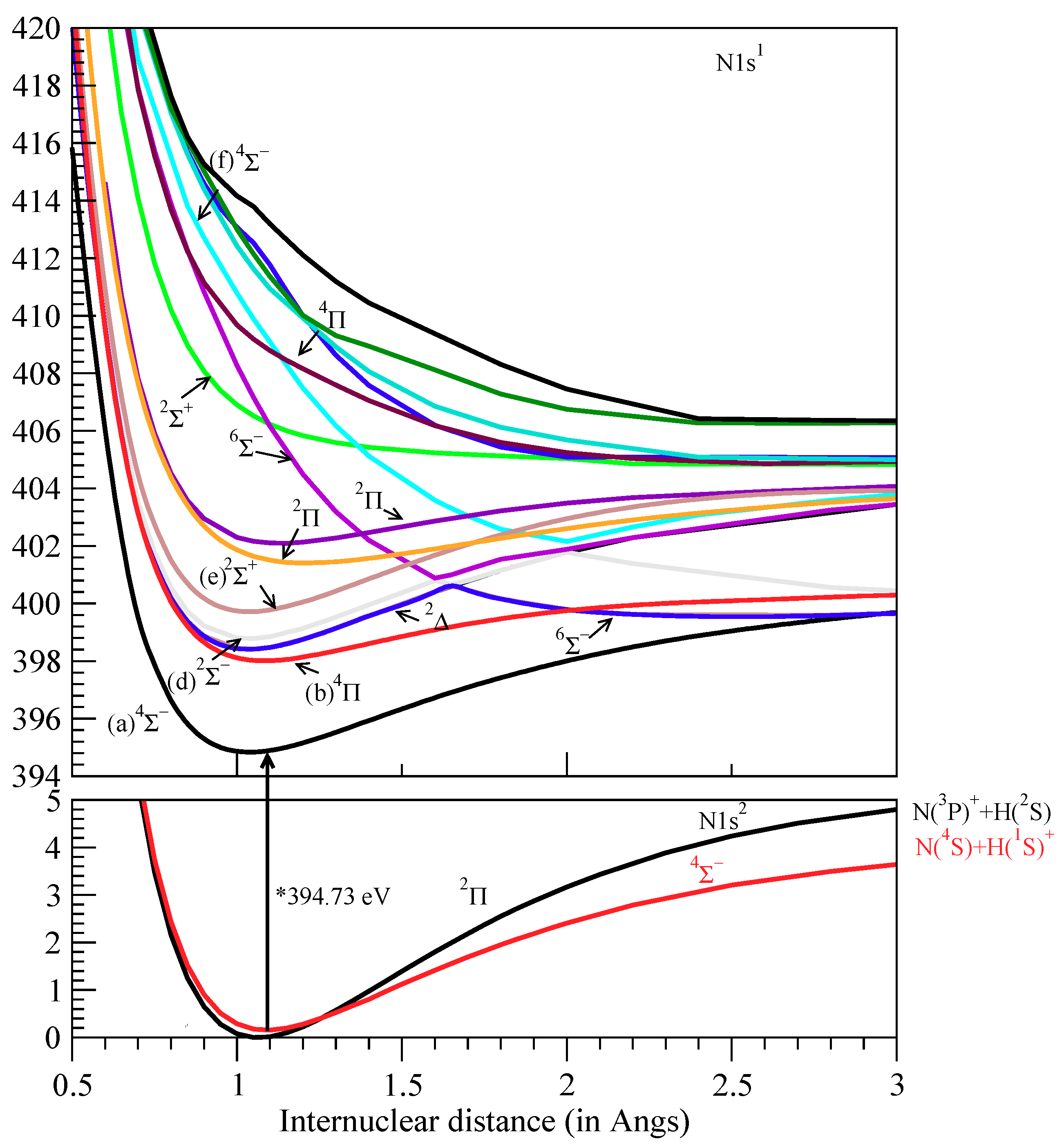
3.2. Vibrational Analysis, Potential Energy Curves
3.3. Absorption Cross-Section Spectrum
4. Results and Discussion
4.1. Ground States
4.2. Core-Excited States
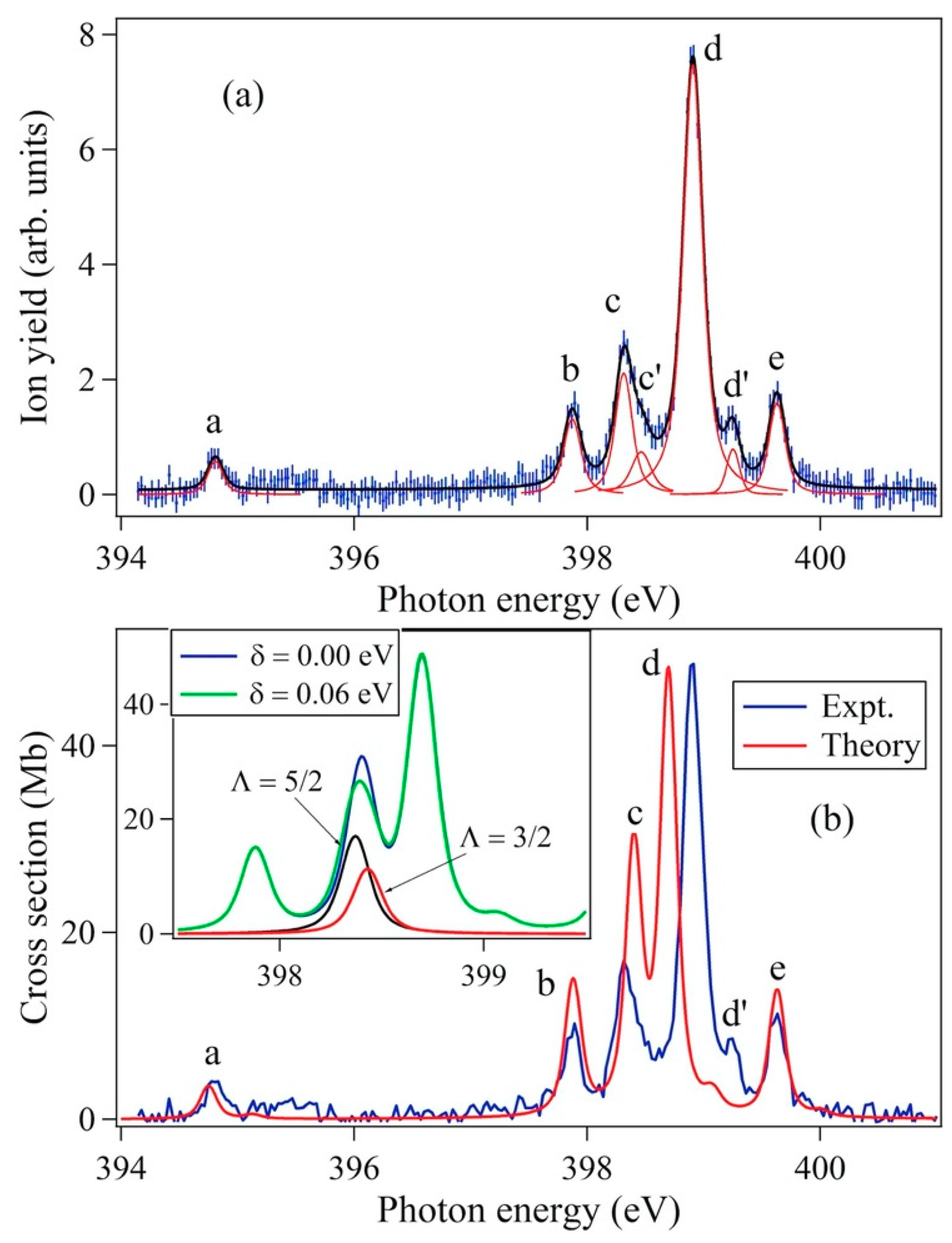
4.2.1. Region 394–396 eV (Peaks a 4Σ−)
4.2.2. Region 397–400 eV
Peak b 4Π
Peaks c 2Δ and e 2∑+
Peak d 2∑−
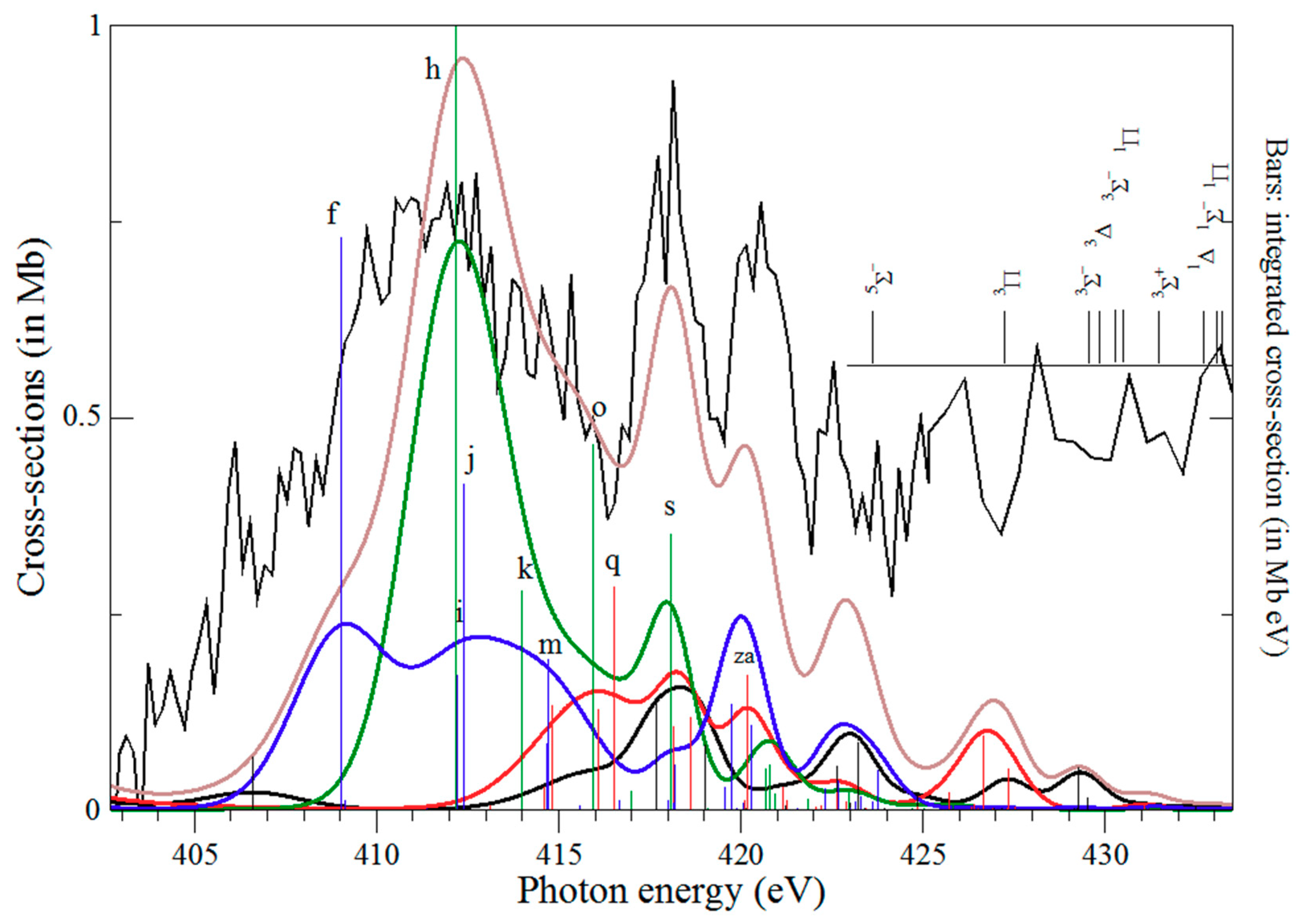
4.2.3. Region 400–430 eV
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gerin, M.; Neufeld, D.A.; Goicoechea, J.R. Interstellar Hydrides. Annu. Rev. Astron. Astrophys. 2016, 54, 181–225. [Google Scholar] [CrossRef]
- Rednyk, S.; Roucka, Š.; Kovalenko, A.; Tran, T.D.; Dohnal, P.; Plašil, R.; Glosík, J. Reaction of NH+, NH2+, and NH3+ ions with H2 at low temperatures. Astron. Astrophys. 2019, 625, A74. [Google Scholar] [CrossRef]
- Le Gal, R.; Hily-Blant, P.; Faure, A.; Pineau des Forêts, G.; Rist, C.; Maret, S. Interstellar chemistry of nitrogen hydrides in dark clouds. Astron. Astrophys. 2014, 562, A83. [Google Scholar] [CrossRef]
- Wagenblast, R.; Williams, D.A.; Millar, T.J.; Nejad, L.A.M. On the origin of NH in diffuse interstellar clouds. Mon. Not. R. Astron. Soc. 1993, 260, 420–424. [Google Scholar] [CrossRef]
- Amero, J.M.; Vazquez, G.J. Electronic Structure of NH+: An Ab Initio Study. Int. J. Quantum Chem. 2005, 101, 396–410. [Google Scholar] [CrossRef]
- Chen, C.T.; Ma, Y.; Sette, F. K-shell photoabsorption of the N2 molecule. Phys. Rev. A 1989, 40, 6737. [Google Scholar] [CrossRef]
- Mosnier, J.-P.; Kennedy, E.T.; Van Kampen, P.; Cubaynes, D.; Guilbaud, S.; Sisourat, N.; Puglisi, A.; Carniato, S.; Bizau, J.-M. Inner-shell photoexcitations as probes of the molecular ions CH+, OH+, and SiH+: Measurements and theory. Phys. Rev. A 2016, 93, 061401. [Google Scholar] [CrossRef]
- Kennedy, E.T.; Mosnier, J.-P.; Van Kampen, P.; Bizau, J.M.; Cubaynes, D.; Guilbaud, S.; Carniato, S.; Puglisi, A.; Sisourat, N. Evolution of L-shell photoabsorption of the molecular-ion series SiHn+ (n = 1,2,3): Experimental and theoretical studies. Phys. Rev. A 2018, 97, 043410. [Google Scholar] [CrossRef]
- Klumpp, S.; Guda, A.A.; Schubert, K.; Mertens, K.; Hellhund, J.; Müller, A.; Schippers, S.; Bari, S.; Martins, M. Photoabsorption of the molecular IH cation at the iodine 3d absorption edge. Phys. Rev. A 2018, 97, 033401. [Google Scholar] [CrossRef]
- Lindblad, R.; Kjellsson, L.; Couto, R.C.; Timm, M.; Bülow, C.; Zamudio-Bayer, V.; Lundberg, M.; Von Issendorff, B.; Lau, J.T.; Sorensen, S.L.; et al. X-ray Absorption Spectrum of the N2+ Molecular Ion. Phys. Rev. Lett. 2002, 124, 203001. [Google Scholar] [CrossRef]
- Couto, R.C.; Kjellsson, L.; Ågren, H.; Carravetta, V.; Sorensen, S.L.; Kubin, M.; Bülow, C.; Timm, M.; Zamudio-Bayer, V.; von Issendorff, B.; et al. The carbon and oxygen K-edge NEXAFS spectra of CO+. Phys. Chem. Chem. Phys. 2020, 22, 16215–16223. [Google Scholar] [CrossRef] [PubMed]
- Bari, S.; Inhester, L.; Schubert, K.; Mertens, K.; Schunck, J.O.; Dörner, S.; Deinert, S.; Schwob, L.; Schippers, S.; Müller, A.; et al. Inner-shell X-ray absorption spectra of the cationic series NHy+ (y = 0–3). Phys. Chem. Chem. Phys. 2019, 21, 16505–16514. [Google Scholar] [CrossRef] [PubMed]
- Bizau, J.M.; Cubaynes, D.; Guilbaud, S.; El Hassan, N.; Al Shorman, M.M.; Bouisset, E.; Guigand, J.; Moustier, O.; Marié, A.; Nadal, E.; et al. A merged-beam setup at SOLEIL dedicated to photoelectron–photoion coincidence studies on ionic species. J. Electron Spectrosc. Relat. Phenom. 2016, 210, 5–12. [Google Scholar] [CrossRef]
- Gharaibeh, M.F.; Bizau, J.M.; Cubaynes, D.; Guilbaud, S.; El Hassan, N.; Al Shorman, M.M.; Miron, C.; Nicolas, C.; Robert, E.; Blancard, C.; et al. K-shell photoionization of singly ionized atomic nitrogen: Experiment and theory. J. Phys. B At. Mol. Opt. Phys. 2011, 44, 175208. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. Montgomery, General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Prince, K.C.; Vondráček, M.; Karvonen, J.; Coreno, M.; Camilloni, R.; Avaldi, L.; de Simonel, M. A critical comparison of selected 1s and 2p core hole widths. J. Electron Spectrosc. Relat. Phenom. 1999, 101, 141–147. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Colin, R. Perturbations between the X2Π and a4Σ− states of the NH+ ion. J. Mol. Spec. 1989, 136, 387–401. [Google Scholar] [CrossRef]
- Feller, D.; Sordo, J.A. A CCSDT study of the effects of higher order correlation on spectroscopic constants. I. First row diatomic hydrides. J. Chem. Phys. 2000, 112, 5604–5610. [Google Scholar] [CrossRef]
- Wilson, D.L. Λ-type doubling in the molecules 14NH+, 15NH+, and 14ND+. Mol. Phys. 1978, 36, 597–610. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main Configuration | State | dNH (Å) | νNH (cm−1) | Type 1 |
|---|---|---|---|---|
| 1σ122σ23σ23px,y1 | 2Π | 1.0776 | 2974.3 | DFT/B3LYP |
| 1.065 | 3064.2 | MCSCF | ||
| 1.0692 | 3047.58 | Experimental value from Ref. [20] | ||
| 1σ122σ23σ13px13py1 | 4Σ− | 1.1135 | 2502.0 | DFT/B3LYP |
| 1.096 | 2716.0 | MCSCF | ||
| 1.093 | 2672.57 | Experimental value from Ref. [20] | ||
| 1σ11 2σ23σ23px13py1 | 4Σ− | 1.0341 | 3169.0 | DFT/B3LYP |
| 1.040 | 3182.0 | CI-SD | ||
| 1σ112σ23σ13px23py1/3px13py2 | 4Π | 1.0919 | 2517.0 | DFT/B3LYP |
| 1.082 | 2557.0 | CI-SD | ||
| 1σ112σ23σ23px23py0/3px03py2 | 2Δ | 1.0371 | 3140.0 | DFT/B3LYP |
| 1.035 | 3250 | CI-SD | ||
| 1σ112σ23σ23px13py1 | 2Δ | 1.035 | 3250.0 | CI-SD |
| Electronic Transition | Final State | Emea (eV) 1 | Ecal (eV) | W (%) | ftheo | Label |
|---|---|---|---|---|---|---|
| Region1 | ||||||
| 1σ22σ23σ11πx11πy1→1σ12σ23σ21πx11πy1 | 4Σ− | 394.81(6) | 394.73 2 | 98.0 | 0.0430 | a |
| Region2 | ||||||
| 1σ22σ23σ11πx11πy1→1σ12σ23σ11πx(y)11πy(x)2 | 4Π | 397.87(5) | 397.84 3 | 95.6 | 0.1560 | b |
| 1σ22σ23σ21πx(y)1→1σ13σ21πx(y)2 πy(x)0 | 2Δ | 398.31(6) | 398.40 4 | 98.7 | 0.0401 | c |
| 1σ22σ23σ21πx(y)1→1σ12σ23σ21πx1 πy1 | 2Δ | 398.46(11) | 398.40 | 98.7 | 0.0401 | c’ |
| 1σ22σ23σ21πx(y)1→1σ12σ23σ21πx1 πy1 | 2Σ− | 398.91(4) | 398.70 | 98.7 | 0.1208 | d |
| 1σ22σ23σ21πx(y)1→1σ12σ23σ21πx(y)2 πy(x)0 | 2Σ+ | 399.63(5) | 399.63 | 92.5 | 0.0385 | e |
| Region 3 | ||||||
| 1σ22σ23σ11πx11πy1→1σ12σ23σ11πx11πy1 4σ1 | 4Σ− | 409.04 | 88.0 | 0.0332 | f | |
| 1σ22σ23σ11πx11πy1→1σ12σ13σ21πx(y)21πy(x)1 | 4Π | 409.14 | 74.0 | 0.0012 | g | |
| 1σ22σ23σ21πx(y)1→1σ12σ23σ21πx(y)1 4σ1 | 2Π | 412.17 | 85.0 | 0.0261 | h | |
| 1σ22σ23σ11πx11πy1→1σ13σ11πx11πy1 4σ1 | 4Σ− | 412.20 | 77.8 | 0.0078 | i | |
| 1σ22σ23σ11πx11πy1→1σ13σ11πx11πy1 4σ1 | 4Σ− | 412.40 | 80.6 | 0.0189 | j | |
| 1σ22σ23σ21πx(y)1→1σ12σ23σ21πx(y)1 4σ1 | 2Π | 414.00 | 57.8 | 0.0032 | k | |
| 1σ22σ23σ11πx11πy1→1σ13σ11πx11πy1 5σ1 | 4Σ− | 414.67 | 89.2 | 0.0039 | l | |
| 1σ22σ23σ11πx11πy1→1σ13σ11πx11πy1 2πx(y)1 | 4Π | 414.70 | 59.6 | 0.0174 | m | |
| 1σ22σ23σ21πx1→1σ12σ23σ11πx1 πy14σ1 | 2Σ− | 414.80 | 82.0 | 0.0015 | n | |
| 1σ22σ23σ21πx(y)1→1σ12σ23σ21πx(y)1 4σ1 | 2Π | 415.95 | 57.8 | 0.0053 | o | |
| 1σ22σ23σ21πx1→1σ12σ23σ11πx1 πy14σ1 | 2Δ | 416.09 | 78.8 | 0.0015 | p | |
| 1σ22σ23σ21πx1→1σ12σ23σ11πx(y)2 πy(x)04σ1 | 2Δ | 416.09 | 78.8 | 0.0015 | p’ | |
| 1σ22σ23σ21πx1→1σ12σ23σ11πx1 πy14σ1 | 2Σ− | 416.51 | 76.9 | 0.0032 | q | |
| 1σ22σ23σ21πx1→1σ12σ23σ11πx(y)21πy(x)04σ1 | 2Σ+ | 417.68 | 50.1 | 0.0016 | r | |
| 1σ22σ23σ21πx(y)1→1σ12σ23σ21πx(y)1 4σ1 | 2Π | 418.07 | 77.4 | 0.0040 | s | |
| 1σ22σ23σ11πx11πy1→1σ13σ11πx11πy1 3πx(y)1 | 4Π | 418.01 | 94.5 | 0.0013 | t | |
| 1σ22σ23σ21πx1→1σ12σ23σ21πx1 2πy1 | 2Σ− | 418.14 | 76.9 | 0.0012 | u | |
| 1σ22σ23σ11πx11πy1→1σ13σ11πx11πy1 3πx(y)1 | 4Σ− | 418.19 | 53.5 | 0.0026 | v | |
| 1σ22σ23σ21πx1→1σ12σ23σ21πx1 2πy1 | 2Δ | 418.61 | 76.9 | 0.0013 | x | |
| 1σ22σ23σ21πx1→1σ12σ23σ21πx1 2πx1 | 2Δ | 418.61 | 76.9 | 0.0013 | x’ | |
| 1σ22σ23σ11πx11πy1→1σ13σ11πx11πy1 2πx(y)1 | 4Π | 419.55 | 84.5 | 0.0027 | y | |
| 1σ22σ23σ11πx11πy1→1σ13σ11πx11πy1 5σ1 | 4Σ− | 419.76 | 51.3 | 0.0062 | z | |
| 1σ22σ23σ21πx1→1σ12σ23σ11πx11πy15σ1 | 2Σ− | 420.18 | 69.0 | 0.0020 | za | |
| 1σ22σ23σ11πx11πy1→1σ13σ11πx11πy1 2πx(y)1 | 4Π | 420.27 | 81.7 | 0.0099 | zb |
| Line Label | Energy (eV) | Natural Width | |
|---|---|---|---|
| This Work | Reference [12] 1 | (meV) | |
| a | 394.81(6) | 394.9 | 84(6) |
| b | 397.87(5) | 397.8 | 108(3) |
| c | 398.31(6) | 398.8 | 108(5) |
| c’ | 398.46(11) | ||
| d | 398.91(4) | 398.8 | 142(20) |
| d’ | 399.25(5) | ||
| e | 399.63(5) | 399.6 | 94(29) |
| N1sσ1 | 1πx2 | 1πy2 | βαα | ααβ | αβα |
|---|---|---|---|---|---|
| 1πx2 | 0 | 0 | 0 | ||
| 1πy2 | = | 0 | 0 | 0 | |
| βαα | 0 | 0 | |||
| ααβ | 0 | 0 | |||
| αβα | 0 | 0 | = |
| Threshold Number | Absolute Energy (eV) | State | Main Configurations |
|---|---|---|---|
| 1 | 423.61 * | (5Σ−)Q | 1σ12σ23σ11πx11πx1 (97.4) |
| 2 | 426.93 * | (3Π)D | 1σ12σ23σ21πx,y1 |
| 3 | 429.39 | (3Σ−) Q | 1σ12σ23σ11πx11πy1 (75.6) |
| 4 | 429.52 | (3Δ) Q | 1σ12σ23σ11πx11πy1(8.1)/1σ12σ23σ11πx2(43.8)/1σ12σ23σ11πy2(43.8) 1σ12σ23σ11πx11πy1(87.6)/1σ12σ23σ11πx2(4.05)/1σ12σ23σ11πy2(4.05) |
| 5 | 429.95 | (3Σ−) Q | 1σ12σ23σ11πx11πx1 (96.2) |
| 6 | 430.22 | (1Π)D | 1σ12σ23σ21πx,y1 |
| 7 | 431.23 | (3Σ+) Q | 1σ12σ23σ11πx2(46.0) 1σ12σ23σ11πy2(46.0) |
| 8 | 432.43 | (1Δ) Q | 1σ12σ23σ11πx2(42.0) 1σ12σ23σ11πy2(42.0)/1σ12σ23σ11πx11πx1(8.6) 1σ12σ23σ11πx11πx1(84.0)/ 1σ12σ23σ11πx2(4.3) 1σ12σ23σ11πy2(4.3) |
| 9 | 432.79 | (1Σ−) Q | 1σ12σ23σ11πx11πy1 (91.1) |
| 10 | 432.87 | (1Π) Q | 1σ12σ23σ01πx1 1πy2(58.7)/ 1σ12σ23σ01πx21πy1(23.9) 1σ12σ23σ01πx2 1πy1(58.7)/ 1σ12σ23σ01πx11πy2(23.9) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carniato, S.; Bizau, J.-M.; Cubaynes, D.; Kennedy, E.T.; Guilbaud, S.; Sokell, E.; McLaughlin, B.; Mosnier, J.-P. Vibrationally and Spin-Orbit-Resolved Inner-Shell X-ray Absorption Spectroscopy of the NH+ Molecular Ion: Measurements and ab Initio Calculations. Atoms 2020, 8, 67. https://doi.org/10.3390/atoms8040067
Carniato S, Bizau J-M, Cubaynes D, Kennedy ET, Guilbaud S, Sokell E, McLaughlin B, Mosnier J-P. Vibrationally and Spin-Orbit-Resolved Inner-Shell X-ray Absorption Spectroscopy of the NH+ Molecular Ion: Measurements and ab Initio Calculations. Atoms. 2020; 8(4):67. https://doi.org/10.3390/atoms8040067
Chicago/Turabian StyleCarniato, Stéphane, Jean-Marc Bizau, Denis Cubaynes, Eugene T. Kennedy, Ségolène Guilbaud, Emma Sokell, Brendan McLaughlin, and Jean-Paul Mosnier. 2020. "Vibrationally and Spin-Orbit-Resolved Inner-Shell X-ray Absorption Spectroscopy of the NH+ Molecular Ion: Measurements and ab Initio Calculations" Atoms 8, no. 4: 67. https://doi.org/10.3390/atoms8040067
APA StyleCarniato, S., Bizau, J.-M., Cubaynes, D., Kennedy, E. T., Guilbaud, S., Sokell, E., McLaughlin, B., & Mosnier, J.-P. (2020). Vibrationally and Spin-Orbit-Resolved Inner-Shell X-ray Absorption Spectroscopy of the NH+ Molecular Ion: Measurements and ab Initio Calculations. Atoms, 8(4), 67. https://doi.org/10.3390/atoms8040067