The Search for Clinically Useful Biomarkers of Complex Disease: A Data Analysis Perspective

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Definition of a Complex Disease
3. Study Design and the Biomarker Discovery Pipeline: Considerations for Complex Disease
4. Data Pretreatment Step: Considerations for complex disease
5. The Data Analysis Step: Considerations for Complex Disease
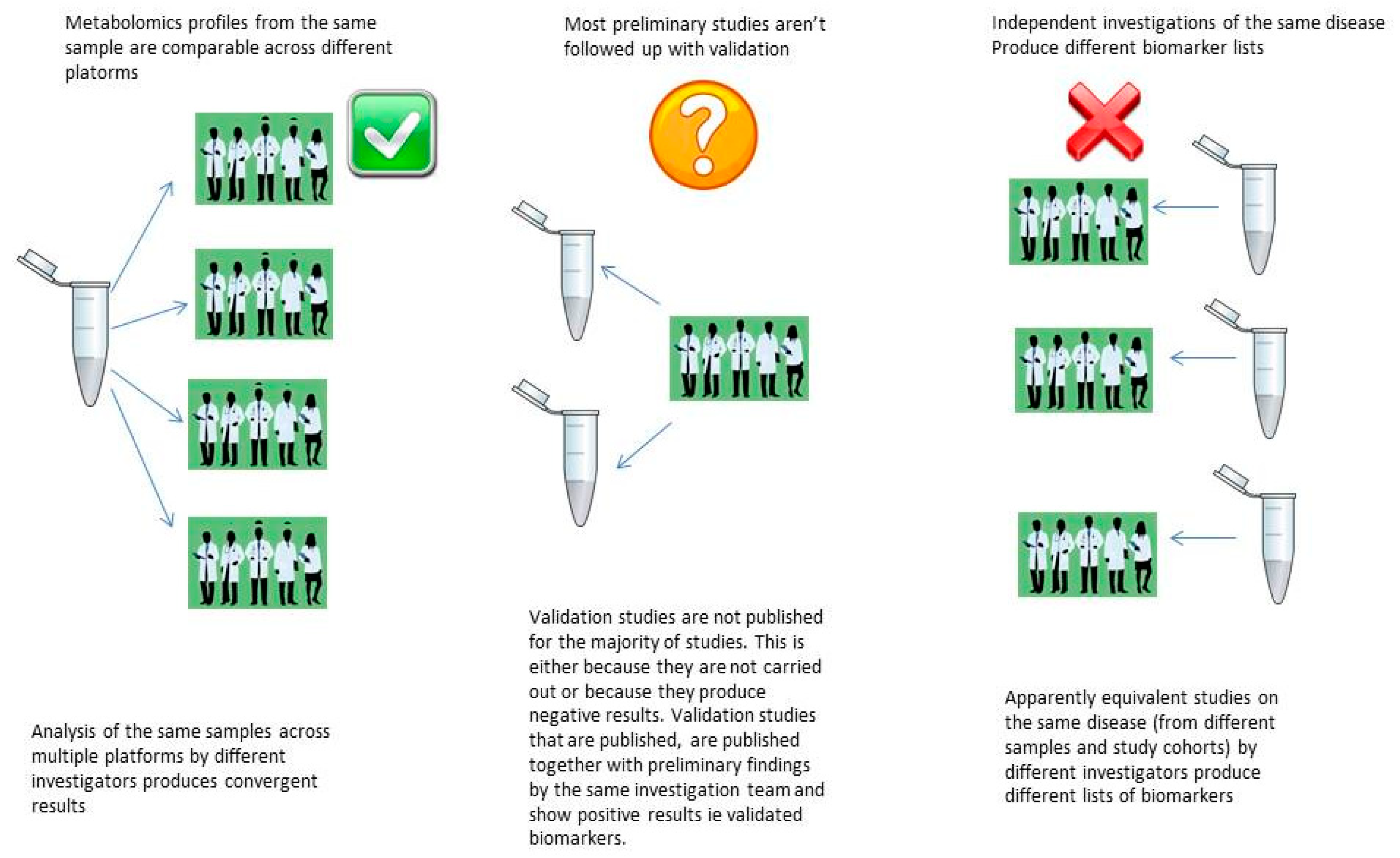
5.1. The Reproducibility Crisis in Metabolomics Biomarker Discovery
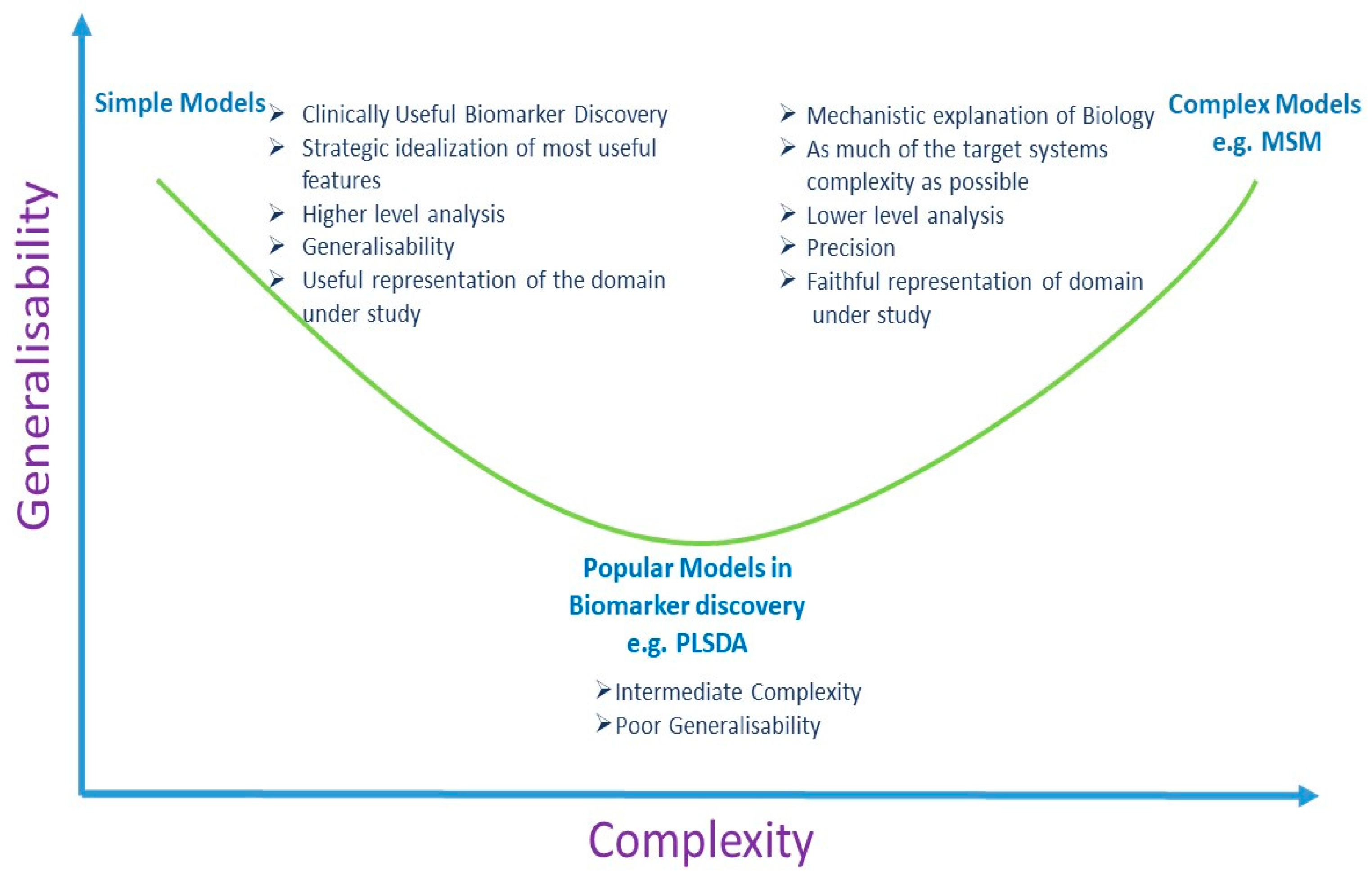
5.2. Multivariate Analysis and Univariate Analysis
6. Discussion
Funding
Conflicts of Interest
References
- Ioannidis, J.P.; Bossuyt, P.M. Waste, Leaks, and Failures in the Biomarker Pipeline. Clin. Chem. 2017, 63, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chan, D.W. The Road from Discovery to Clinical Diagnostics: Lessons Learned from the First FDA-Cleared In Vitro Diagnostic Multivariate Index Assay of Proteomic Biomarkers. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2995–2999. [Google Scholar] [CrossRef] [PubMed]
- Marchand, C.R.; Farshidfar, F.; Rattner, J.; Bathe, O.F. A Framework for Development of Useful Metabolomic Biomarkers and Their Effective Knowledge Translation. Metabolites 2018, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.E. Why Your New Cancer Biomarker May Never Work: Recurrent Patterns and Remarkable Diversity in Biomarker Failures. Cancer Res. 2012, 72, 6097–6101. [Google Scholar] [CrossRef] [PubMed]
- Barker, A.D.; Compton, C.C.; Poste, G. The National Biomarker Development Alliance accelerating the translation of biomarkers to the clinic. Biomark. Med. 2014, 8, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Spicer, R.; Salek, R.M.; Moreno, P.; Cañueto, D.; Steinbeck, C. Navigating freely-available software tools for metabolomics analysis. Metabolomics 2017, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Katajamaa, M.; Orešič, M. Data processing for mass spectrometry-based metabolomics. J. Chromatogr. A 2007, 1158, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Castillo, S.; Gopalacharyulu, P.; Yetukuri, L.; Orešič, M.; Peddinti, G. Algorithms and tools for the preprocessing of LC–MS metabolomics data. Chemom. Intell. Lab. Syst. 2011, 108, 23–32. [Google Scholar] [CrossRef]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D. Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef]
- Plomin, R.; Haworth, C.M.A.; Davis, O.S.P. Common disorders are quantitative traits. Nat. Rev. Genet. 2009, 10, 872–878. [Google Scholar] [CrossRef]
- Li, Y.; Vinckenbosch, N.; Tian, G.; Huerta-Sanchez, E.; Jiang, T.; Jiang, H.; Albrechtsen, A.; Andersen, G.; Cao, H.; Korneliussen, T.S.; et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 2010, 42, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Marth, G.T.; Yu, F.; Indap, A.R.; Garimella, K.; Gravel, S.; Leong, W.F.; Tyler-Smith, C.; Bainbridge, M.; Blackwell, T.; Zheng-Bradley, X.; et al. The functional spectrum of low-frequency coding variation. Genome Boil. 2011, 12, R84. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C. Beyond disease susceptibility-Leveraging genome-wide association studies for new insights into complex disease biology. HLA 2017, 90, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Yan, G.; Wang, P.; Wang, X. Metabolomics for Biomarker Discovery: Moving to the Clinic. BioMed Res. Int. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [PubMed]
- López-López, Á.; López-Gonzálvez, Á.; Barker-Tejeda, T.C.; Barbas, C.; Clive-Baker, T. A review of validated biomarkers obtained through metabolomics. Expert Rev. Mol. Diagn. 2018, 18, 557–575. [Google Scholar]
- Mitchell, K.J. What is complex about complex disorders? Genome Boil. 2012, 13, 237. [Google Scholar] [CrossRef] [PubMed]
- Elston, R.C.; Satagopan, J.M.; Sun, S. Genetic terminology. In Statistical Human Genetics; Humana Press: New York, NY, USA, 2012; pp. 1–9. [Google Scholar]
- Ransohoff, D.F. How to improve reliability and efficiency of research about molecular markers: roles of phases, guidelines, and study design. J. Clin. Epidemiol. 2007, 60, 1205–1219. [Google Scholar] [CrossRef]
- Fowke, J.H. Issues in the Design of Molecular and Genetic Epidemiologic Studies. J. Prev. Med. Public Heal. 2009, 42, 343. [Google Scholar] [CrossRef]
- Zheng, Y. Study Design Considerations for Cancer Biomarker Discoveries. J. Appl. Lab. Med. 2018, 3, 282–289. [Google Scholar] [CrossRef]
- Pepe, M.S.; Li, C.I.; Feng, Z. Improving the Quality of Biomarker Discovery Research: the Right Samples and Enough of Them. Cancer Epidemiol. Biomark. Prev. 2015, 24, 944–950. [Google Scholar] [CrossRef]
- Diamandis, E.P.; Diamandis, E.P.; Diamandis, E. Cancer Biomarkers: Can We Turn Recent Failures into Success? J. Natl. Cancer Inst. 2010, 102, 1462–1467. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.G.; Kramer, B.S.; Srivastava, S. Markers for early detection of cancer: Statistical guidelines for nested case-control studies. BMC Med Res. Methodol. 2002, 2, 4. [Google Scholar] [CrossRef]
- Baker, S.G.; Kramer, B.S.; McIntosh, M.; Patterson, B.H.; Shyr, Y.; Skates, S. Evaluating markers for the early detection of cancer: Overview of study designs and methods. Clin. Trials 2006, 3, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Pepe, M.S.; Feng, Z.; Janes, H.; Bossuyt, P.M.; Potter, J.D. Pivotal Evaluation of the Accuracy of a Biomarker Used for Classification or Prediction: Standards for Study Design. J. Natl. Cancer Inst. 2008, 100, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Rundle, A.G.; Vineis, P.; Ahsan, H. Design Options for Molecular Epidemiology Research within Cohort Studies. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Pesch, B.; Bruning, T.; Johnen, G.; Casjens, S.; Bonberg, N.; Taeger, D.; Müller, A.; Weber, D.; Behrens, T. Biomarker research with prospective study designs for the early detection of cancer. Biochim. et Biophys. Acta (BBA) - Proteins Proteom. 2014, 1844, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Wallstrom, G.; Anderson, K.S.; LaBaer, J. Biomarker Discovery for Heterogeneous Diseases. Cancer Epidemiology Biomarkers Prev. 2013, 22, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Manchia, M.; Cullis, J.; Turecki, G.; Rouleau, G.A.; Uher, R.; Alda, M. The Impact of Phenotypic and Genetic Heterogeneity on Results of Genome Wide Association Studies of Complex Diseases. PLOS ONE 2013, 8, e76295. [Google Scholar] [CrossRef]
- de Lemos, J.A.; Rohatgi, A.; Ayers, C.R. Applying a Big Data Approach to Biomarker Discovery: Running Before We Walk? Circulation 2015, 132, 2289–2292. [Google Scholar] [CrossRef][Green Version]
- Forshed, J. Experimental Design in Clinical ‘Omics Biomarker Discovery. J. Proteome Res. 2017, 16, 3954–3960. [Google Scholar] [CrossRef]
- Rothman, K.J.; Greenland, S.; Lash, T.L. Modern epidemiology, 3rd ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2008. [Google Scholar]
- Rundle, A.; Ahsan, H.; Vineis, P. Better Cancer Biomarker Discovery Through Better Study Design. Eur. J. Clin. Investig. 2012, 42, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Karpievitch, Y.V.; Dabney, A.R.; Smith, R.D. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinform. 2012, 13, S5. [Google Scholar] [CrossRef] [PubMed]
- Hrydziuszko, O.; Viant, M.R. Missing values in mass spectrometry based metabolomics: An undervalued step in the data processing pipeline. Metabolomics 2012, 8, 161. [Google Scholar] [CrossRef]
- Considine, E.C.; Thomas, G.; Boulesteix, A.L.; Khashan, A.S.; Kenny, L.C. Critical review of reporting of the data analysis step in metabolomics. Metabolomics 2018, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Hackstadt, A.J.; Hess, A.M. Filtering for increased power for microarray data analysis. BMC Bioinform. 2009, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Geman, D.; Ochs, M.; Price, N.D.; Tomasetti, C.; Younes, L. An argument for mechanism-based statistical inference in cancer. Hum. Genet. 2015, 134, 479–495. [Google Scholar] [CrossRef] [PubMed]
- Ejigu, B.A.; Valkenborg, D.; Baggerman, G.; Vanaerschot, M.; Witters, E.; Dujardin, J.-C.; Burzykowski, T.; Berg, M. Evaluation of Normalization Methods to Pave the Way Towards Large-Scale LC-MS-Based Metabolomics Profiling Experiments. OMICS: A J. Integr. Boil. 2013, 17, 473–485. [Google Scholar] [CrossRef]
- Kohl, S.M.; Klein, M.S.; Hochrein, J.; Oefner, P.J.; Spang, R.; Gronwald, W. State-of-the art data normalization methods improve NMR-based metabolomic analysis. Metabolomics 2012, 8, 146–160. [Google Scholar] [CrossRef]
- Berg, R.A.V.D.; Hoefsloot, H.C.J.; A Westerhuis, J.; Smilde, A.K.; Van Der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar]
- Shah, J.S.; Brock, G.N.; Rai, S.N. Metabolomics data analysis and missing value issues with application to infarcted mouse hearts. BMC Bioinform. 2015, 16, 16. [Google Scholar] [CrossRef][Green Version]
- Rubin, D.B. Inference and missing data. Biometrika 1976, 63, 581–592. [Google Scholar] [CrossRef]
- Webb-Robertson, B.-J.M.; Wiberg, H.K.; Matzke, M.M.; Brown, J.N.; Wang, J.; McDermott, J.E.; Smith, R.D.; Rodland, K.D.; Metz, T.O.; Pounds, J.G.; et al. Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry-based label-free global proteomics. J. Proteome Res. 2015, 14, 1993–2001. [Google Scholar] [CrossRef] [PubMed]
- Liew, A.W.-C.; Law, N.-F.; Yan, H. Missing value imputation for gene expression data: Computational techniques to recover missing data from available information. Briefings Bioinform. 2010, 12, 498–513. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, K.; Mohamad, M.; Deris, S. A Review on Missing Value Imputation Algorithms for Microarray Gene Expression Data. Curr. Bioinform. 2014, 9, 18–22. [Google Scholar] [CrossRef]
- Gromski, P.S.; Xu, Y.; Kotze, H.L.; Correa, E.; Ellis, D.I.; Armitage, E.G.; Turner, M.L.; Goodacre, R. Influence of Missing Values Substitutes on Multivariate Analysis of Metabolomics Data. Metabolites 2014, 4, 433–452. [Google Scholar] [CrossRef] [PubMed]
- Di Guida, R.; Engel, J.; Allwood, J.W.; Weber, R.J.M.; Jones, M.R.; Sommer, U.; Viant, M.R.; Dunn, W.B.; Guida, R. Non-targeted UHPLC-MS metabolomic data processing methods: A comparative investigation of normalisation, missing value imputation, transformation and scaling. Metabolomics 2016, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.L.; Ruhaak, L.R.; Kelly, K.; Weiss, R.H.; Kim, K. Effects of imputation on correlation: Implications for analysis of mass spectrometry data from multiple biological matrices. Brief. Bioinform. 2017, 18, 312–320. [Google Scholar] [CrossRef][Green Version]
- Lai, C.; Reinders, M.J.T.; Veer, L.J.V.; A Wessels, L.F. A comparison of univariate and multivariate gene selection techniques for classification of cancer datasets. BMC Bioinform. 2006, 7, 235. [Google Scholar] [CrossRef][Green Version]
- Ioannidis, J.P.A. Biomarker Failures. Clin. Chem. 2013, 59, 202. [Google Scholar] [CrossRef][Green Version]
- Jarvis, M.F.; Williams, M. Irreproducibility in Preclinical Biomedical Research: Perceptions, Uncertainties, and Knowledge Gaps. Trends Pharmacol. Sci. 2016, 37, 290–302. [Google Scholar] [CrossRef]
- Kaiser, J. Rigorous replication effort succeeds for just two of five cancer papers. Sci. 2017, 359, 6380. [Google Scholar] [CrossRef]
- Maniadis, Z.; Tufano, F.; List, J.A. How to make experimental economics research more reproducible: Lessons from other disciplines and a new proposal. In Replication in Experimental Economics; Emerald Publishing Ltd.: Bingley, UK, 2015; pp. 215–230. [Google Scholar]
- Baker, M. 1,500 scientists lift the lid on reproducibility. Nature 2016, 533, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.N.; Fanelli, D.; Ioannidis, J.P.A. What does research reproducibility mean? Sci. Transl. Med. 2016, 8, 341. [Google Scholar] [CrossRef]
- Peng, R. The reproducibility crisis in science: A statistical counterattack. Significance 2015, 12, 30–32. [Google Scholar] [CrossRef]
- Van Bavel, J.J.; Mende-Siedlecki, P.; Brady, W.J.; Reinero, D.A. Contextual sensitivity in scientific reproducibility. Proc. Natl. Acad. Sci. USA 2016, 113, 6454–6459. [Google Scholar] [CrossRef] [PubMed]
- Karp, N.A. Reproducible preclinical research—Is embracing variability the answer? PLoS Boil. 2018, 16, e2005413. [Google Scholar] [CrossRef]
- Voelkl, B.; Wuerbel, H. Reproducibility Crisis: Are We Ignoring Reaction Norms? Trends Pharmacol. Sci. 2016, 37, 509–510. [Google Scholar] [CrossRef] [PubMed]
- França, T.F.; Monserrat, J.M. Reproducibility crisis in science or unrealistic expectations? EMBO Rep. 2018, 19, e46008. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, B.; Vogt, L.; Sena, E.S.; Wurbel, H. Reproducibility of preclinical animal research improves with heterogeneity of study samples. PLoS Boil. 2018, 16, e2003693. [Google Scholar] [CrossRef]
- An, G. The Crisis of Reproducibility, the Denominator Problem and the Scientific Role of Multi-scale Modeling. Bull. Math. Boil. 2018, 80, 3071–3080. [Google Scholar] [CrossRef]
- Yoshino, M.; Kuhlmann, M.K.; Kotanko, P.; Greenwood, R.N.; Pisoni, R.L.; Port, F.K.; Jager, K.J.; Homel, P.; Augustijn, H.; De Charro, F.T.; et al. International Differences in Dialysis Mortality Reflect Background General Population Atherosclerotic Cardiovascular Mortality. J. Am. Soc. Nephrol. 2006, 17, 3510–3519. [Google Scholar] [CrossRef] [PubMed]
- Saccenti, E.; Hoefsloot, H.C.J.; Smilde, A.K.; Westerhuis, J.A.; Hendriks, M.M.W.B. Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics 2014, 10, 361–374. [Google Scholar] [CrossRef]
- Glazko, G.V.; Emmert-Streib, F. Unite and conquer: Univariate and multivariate approaches for finding differentially expressed gene sets. Bioinform. 2009, 25, 2348–2354. [Google Scholar] [CrossRef] [PubMed]
- Gromski, P.S.; Muhamadali, H.; Ellis, D.I.; Xu, Y.; Correa, E.; Turner, M.L.; Goodacre, R. A tutorial review: Metabolomics and partial least squares-discriminant analysis—A marriage of convenience or a shotgun wedding. Anal. Chim. Acta 2015, 879, 10–23. [Google Scholar] [CrossRef]
- Brereton, R.G.; Lloyd, G.R. Partial least squares discriminant analysis: Taking the magic away. J. Chemom. 2014, 28, 213–225. [Google Scholar] [CrossRef]
- Rodríguez-Pérez, R.; Fernández, L.; Marco, S. Overoptimism in cross-validation when using partial least squares-discriminant analysis for omics data: a systematic study. Anal. Bioanal. Chem. 2018, 410, 5981–5992. [Google Scholar] [CrossRef]
- Dupuy, A.; Simon, R.M. Critical Review of Published Microarray Studies for Cancer Outcome and Guidelines on Statistical Analysis and Reporting. J. Natl. Cancer Inst. 2007, 99, 147–157. [Google Scholar] [CrossRef]
- Shaffer, R.E. Multi- and Megavariate Data Analysis. Principles and Applications, I. Eriksson, E. Johansson, N. Kettaneh-Wold and S. Wold, Umetrics Academy, Umeå, 2001, ISBN 91-973730-1-X, 533pp. J. Chemom. 2002, 16, 261–262. [Google Scholar] [CrossRef]
- Levins, R.; Lewontin, R.C. The Dialectical Biologist; Harvard University Press: Cambridge, MA, USA, 1985. [Google Scholar]
- Levins, R. A Response to Orzack and Sober: Formal Analysis and the Fluidity of Science. Q. Rev. Boil. 1993, 68, 547–555. [Google Scholar] [CrossRef]


© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Considine, E.C. The Search for Clinically Useful Biomarkers of Complex Disease: A Data Analysis Perspective. Metabolites 2019, 9, 126. https://doi.org/10.3390/metabo9070126
Considine EC. The Search for Clinically Useful Biomarkers of Complex Disease: A Data Analysis Perspective. Metabolites. 2019; 9(7):126. https://doi.org/10.3390/metabo9070126
Chicago/Turabian StyleConsidine, Elizabeth C. 2019. "The Search for Clinically Useful Biomarkers of Complex Disease: A Data Analysis Perspective" Metabolites 9, no. 7: 126. https://doi.org/10.3390/metabo9070126
APA StyleConsidine, E. C. (2019). The Search for Clinically Useful Biomarkers of Complex Disease: A Data Analysis Perspective. Metabolites, 9(7), 126. https://doi.org/10.3390/metabo9070126