Screening for Preterm Birth: Potential for a Metabolomics Biomarker Panel


Abstract
1. Introduction
1.1. Aim
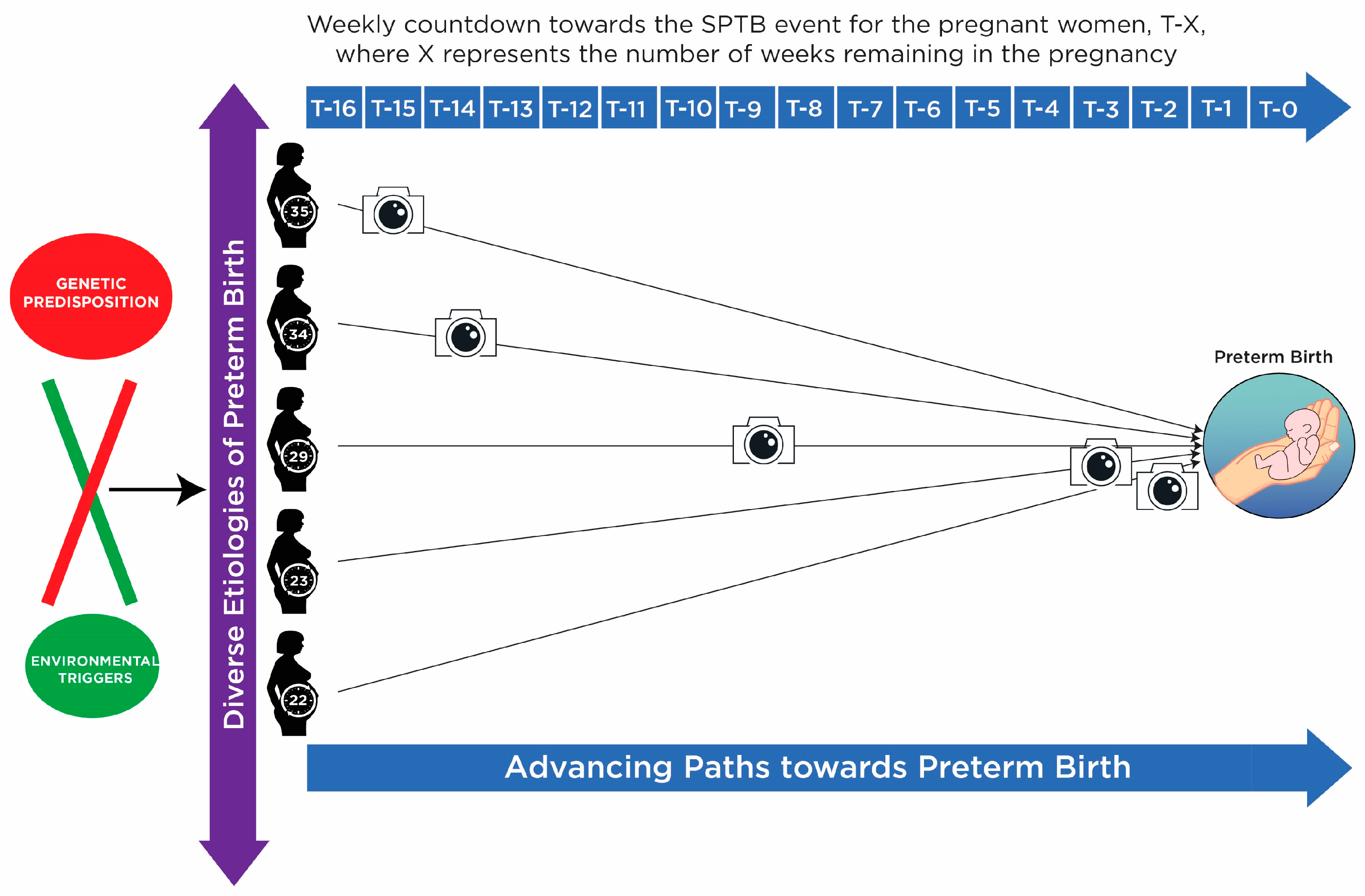
1.2. Background of SPTB
1.3. Biomarker Discovery from Heterogeneous Disease
2. Methods
2.1. Patient Recruitment
2.2. Study Design and Patient Demographics (Table 1)
2.3. Ethics
2.4. Sample Collection and Bio-Banking Procedures
2.5. LCMS Analysis
2.6. Feature Annotation and Identification
2.7. Data Preprocessing
2.8. Data Analysis
- (1)
- There was a risk of over matching due to BMI’s relationship with the outcome (SPTB) being controversial despite many investigations not established as a true confounder as opposed to a mediator.
- (2)
- Matching can lead to bias that is offset by appropriate analysis, ie conditional logistic regression. However in this study the low stringency analysis used here is unlikely to be appropriate for matching.
- (3)
- If the control group is too similar to the case group, the study may fail to detect the difference even if one exists. Since over 40% of the cases give birth very close to term from 36 weeks on, metabolomically speaking it is likely that they are very similar to controls. Matching to controls on age and BMI in this situation leads to overmatching as a risk.
2.8.1. Incorporating Biological Knowledge—Explanation and Rationale
- Before data analysis begins obvious xenobiotics and exogenous metabolites (drugs, plant metabolites and others) and unidentified metabolites (Identified compounds were defined as compounds where the biological group was putatively identified at least even if the compound was not absolutely and uniquely identified) were removed from the unbiased and untargeted metabolomics dataset as these features would not form part of the biomarker panel.
- After data analysis domain knowledge was employed to guide the selection of plausible biomarker candidates from the top ranked features by:
- ○
- Selecting those features that have supporting literature of their biological relevance and involvement in the disease process;
- ○
- Selecting only those top ranked features with low or zero missing values for assessment in PanelomiX to avoid the caveats of the imputation method (identification of false positives);
- ○
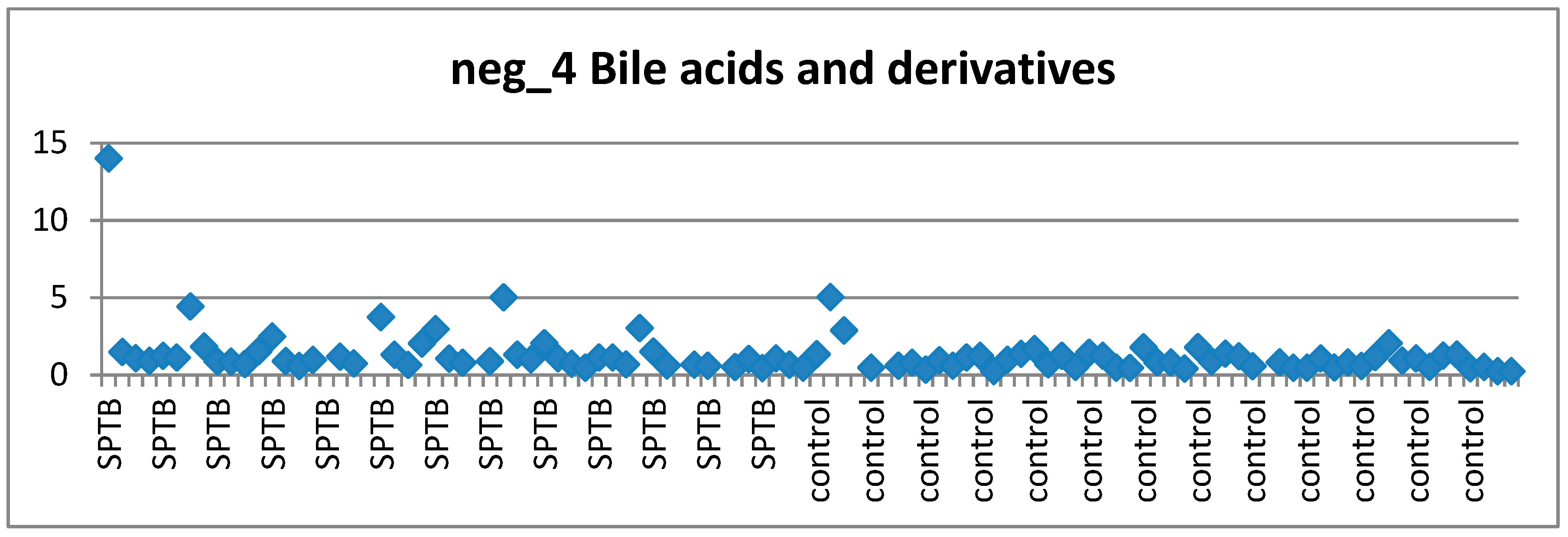
- Visually investigating the behaviour of each of the top ranked features across cases and controls according to the clinical requirements of a suitable biomarker candidate (stable in controls, perturbed in at least some cases) via scatter plots.
2.8.2. Pretreatment—Explanation and Rationale
2.8.3. Univariate Analysis (Fold Change)-Explanation and Rationale
2.8.4. Multivariate Analysis—Explanation and Rationale
2.8.5. Knowledge-Based Approach in Selecting Plausible Candidates
2.8.6. Missing Value Assessment for Avoiding Potential False Positives
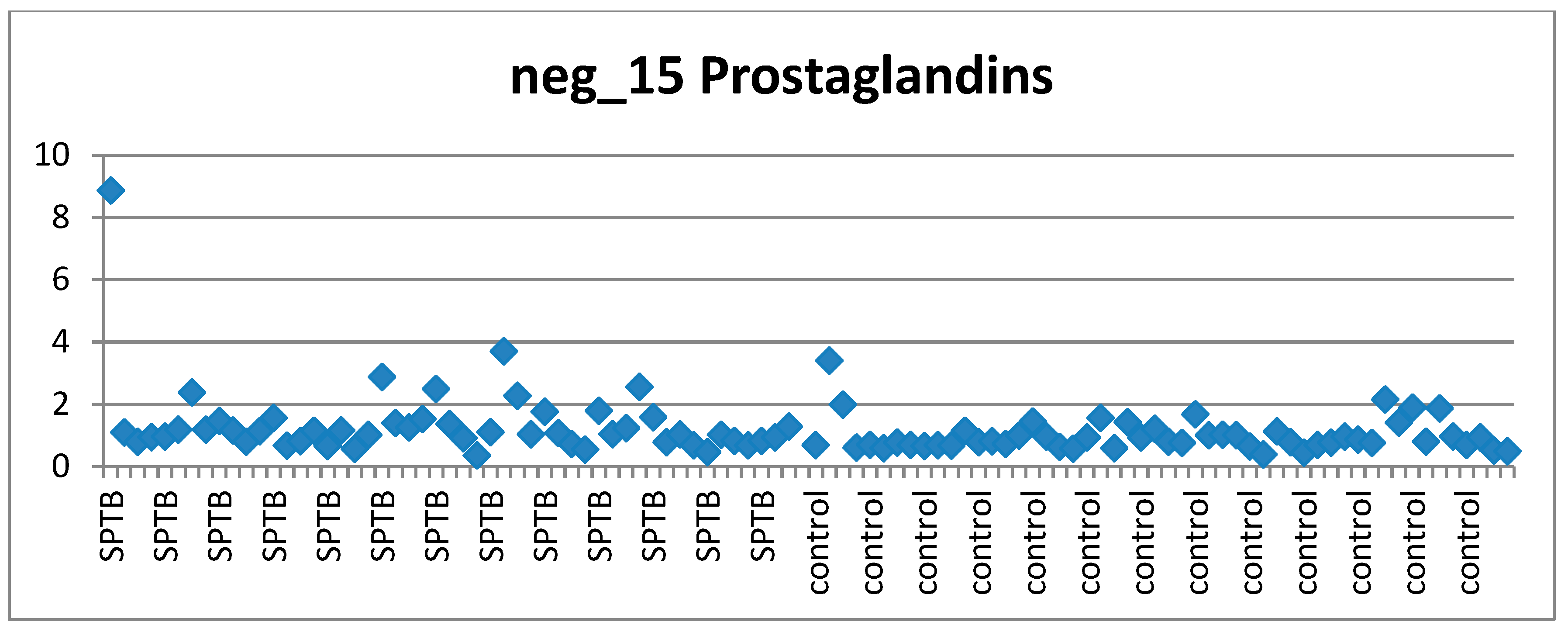
2.8.7. Visual Assessment of Features of Interest Distribution across Cases and Controls
2.8.8. Panel Development
2.8.9. Alternative Biomarker Panels
3. Results
3.1. Pretreatment: QC, Filtering, Normalisation, Scaling and Imputation
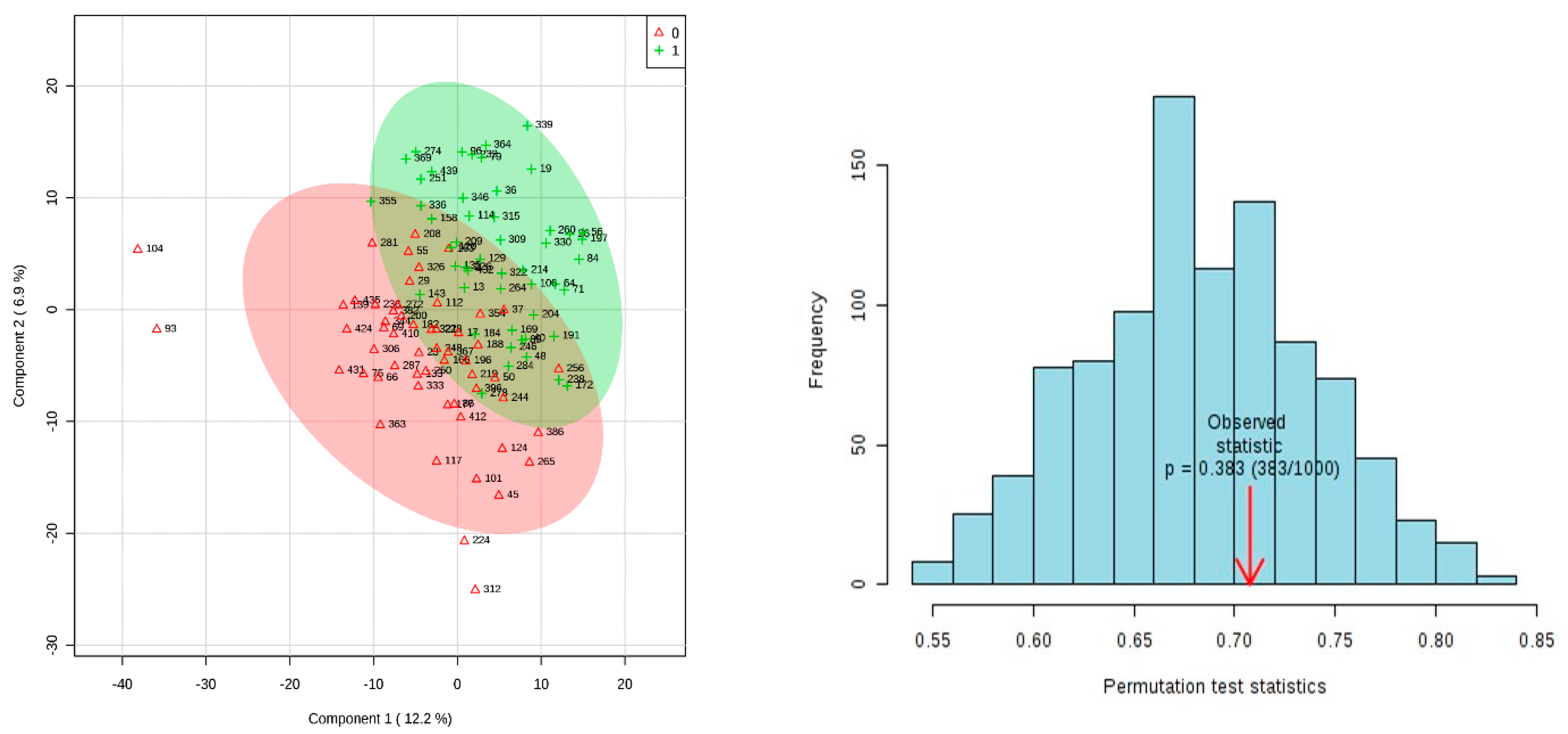
3.2. Results of Univariate and Multivariate Analysis on 15 Week Dataset Matched On Age Only
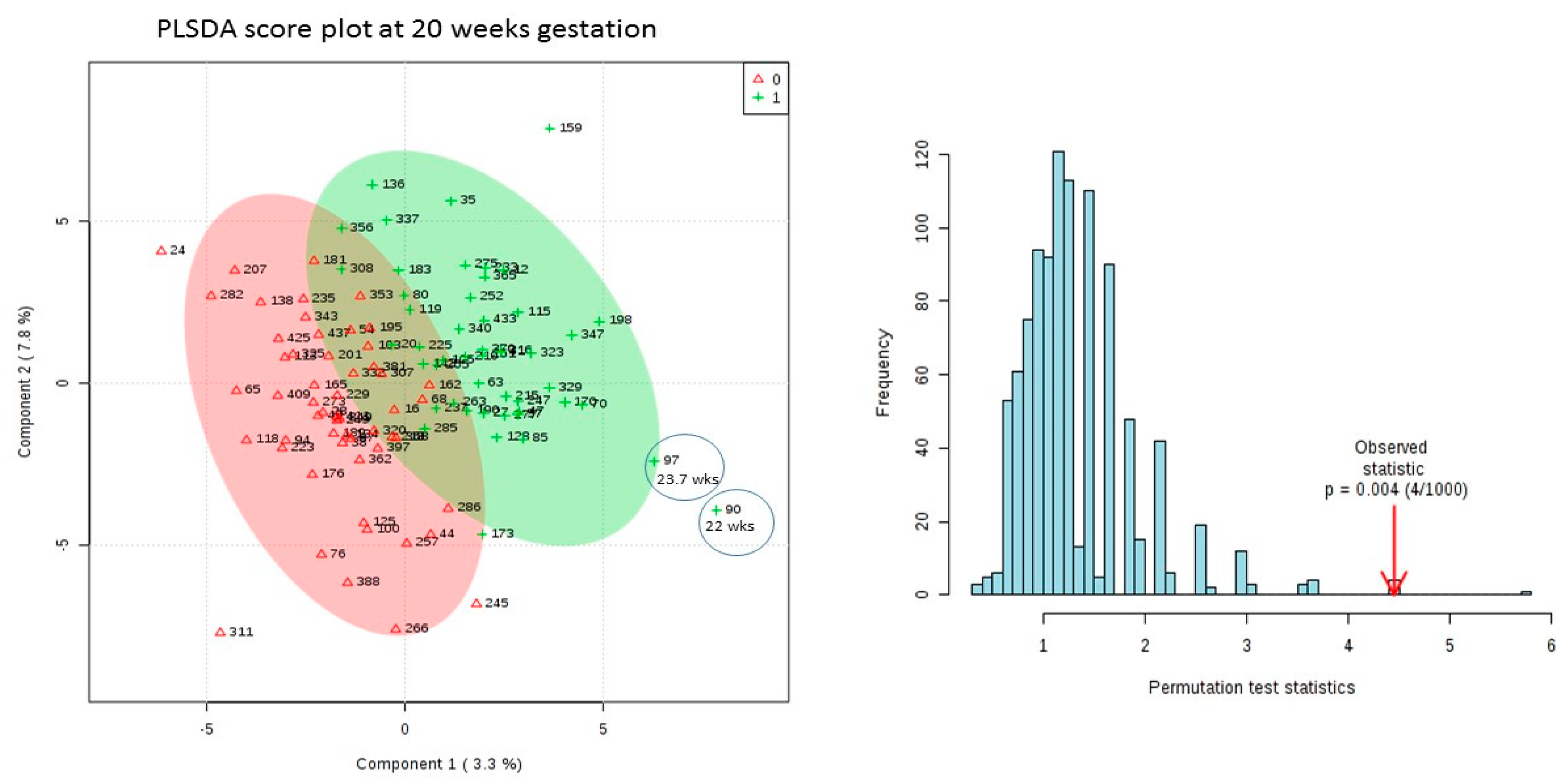
3.3. Results of Univariate and Multivariate Analysis on 20 Week Dataset Matched by Age Only
3.4. Domain Knowledge to Guide Feature Selection for Panel Assessment
3.4.1. Bile Acids and Preterm Birth
3.4.2. Prostaglandins and Preterm Birth
3.4.3. Vitamin D and Preterm Birth
3.4.4. Fatty Acids and Preterm Birth
3.5. Missing Value Assessment to Guide Feature Selection for Panel Assessment
3.6. Visual Examination to Guide Feature Selection for Panel Assesment
3.7. Biomarker Panel Assessment with PanelomiX
3.7.1. Panel A: 4 Features from 20 Week Dataset Ranked Highly by Fold Change/PLSDA and VIP Preceded by Separate Imputation
3.7.2. Panel B: 6 Features from 20 Week Dataset Ranked by Mann–Whitney U test
3.7.3. Panel C: 4 Features from 15-Week Dataset Ranked in Top 20 by Fold Change
3.7.4. Panel D: 4 Features from 15-Week Dataset Ranked by Mann–Whitney U test
3.7.5. PanelomiX Results Summary
4. Discussion
- (1)
- This imputation method is biased towards identifying features with high missing values,
- (2)
- This imputation method is biased towards artificial separation of cases and controls by increasing inter group similarity and decreasing intra group similarity so that good performance of PLSDA upon permutation is expected,
- (3)
- Lack of an independent validation cohort, and
- (4)
- This imputation method could lead to the identification of potential of false positives.
- (1)
- Selecting at the end of our analysis those features with low or zero missing values for assessment in biomarker panels using the PanelomiX program;
- (2)
- Showing that for the 15-week dataset despite the exact same imputation and analysis method being used as for the 20 week dataset this does not lead to a PLSDA model showing separation of cases and controls that stands up to permutation.
- (3)
- Although at this point we do not validate in an independent cohort the features are assessed in PanelomiX by 10-fold cross validation and perform well with a p-values of 0.0013. Also the assessment occurred on the dataset with both sets of controls so an extra 49 healthy subjects who weren’t involved in the discovery of the candidate features.
- (4)
- The fact that MWU test top ranked features at 20 weeks are tested by PanelomiX and create a panel that performs not as well upon cross validation as those found through fold change and imputation method is an argument against the method being more susceptible to false positives.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Enquobahrie, D.A.; Denis, M.; Tadesse, M.G.; Gelaye, B.; Ressom, H.W.; Williams, M.A. Maternal Early Pregnancy Serum Metabolites and Risk of Gestational Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2015, 100, 4348–4356. [Google Scholar] [CrossRef]
- Romero, R.; Espinoza, J.; Kusanovic, J.P.; Gotsch, F.; Hassan, S.; Erez, O.; Chaiworapongsa, T.; Mazor, M. The preterm parturition syndrome. BJOG 2006, 113 (Suppl. 3), 17–42. [Google Scholar] [CrossRef] [PubMed]
- Ward, K.; Argyle, V.; Meade, M.; Nelson, L. The Heritability of Preterm Delivery. Obstet. Gynecol. 2005, 106, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.G.; Jungner, G. Principles and Practice of Screening; WHO: Geneva, Switzerland, 1968; Available online: https://apps.who.int/iris/handle/10665/37650 (accessed on 1 May 2019).
- Heazell, A.E.P.; Bernatavicius, G.; Warrander, L.; Brown, M.C.; Dunn, W.B. A Metabolomic Approach Identifies Differences in Maternal Serum in Third Trimester Pregnancies That End in Poor Perinatal Outcome. Reprod. Sci. 2012, 19, 863–875. [Google Scholar] [CrossRef]
- Lizewska, B.; Teul, J.; Kuc, P.; Lemancewicz, A.; Charkiewicz, K.; Goscik, J.; Kacerovsky, M.; Menon, R.; Miltyk, W.; Laudański, P. Maternal Plasma Metabolomic Profiles in Spontaneous Preterm Birth: Preliminary Results. Mediat. Inflamm. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, A.; Forshed, J.; Nordström, A. Overlap in serum metabolic profiles between non-related diseases: Implications for LC-MS metabolomics biomarker discovery. Biochem. Biophys. Commun. 2016, 478, 1472–1477. [Google Scholar] [CrossRef]
- Wallstrom, G.; Anderson, K.S.; LaBaer, J. Biomarker Discovery for Heterogeneous Diseases. Cancer Epidemiol. Biomark. Prev. 2013, 22, 747–755. [Google Scholar] [CrossRef]
- North, R.A.; McCowan, L.M.E.; Dekker, G.A.; Poston, L.; Chan, E.H.Y.; Stewart, A.W.; Black, M.A.; Taylor, R.S.; Walker, J.J.; Baker, P.N.; et al. Clinical risk prediction for pre-eclampsia in nulliparous women: Development of model in international prospective cohort. BMJ 2011, 342, d1875. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelená, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Brown, M.; Wedge, D.C.; Goodacre, R.; Kell, D.B.; Baker, P.N.; Kenny, L.C.; Mamas, M.A.; Neyses, L.; Dunn, W.B. Automated workflows for accurate mass-based putative metabolite identification in LC/MS-derived metabolomic datasets. Bioinformatics 2011, 27, 1108–1112. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Biomarkers: Hopes and Challenges in the Path from Discovery to Clinical Practice. Transl. Res. 2012, 159, 197–204. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.E.; Wang, J.; Mitchell, H.; Webb-Robertson, B.J.; Hafen, R.; Ramey, J.; Rodland, K.D. Challenges in Biomarker Discovery: Combining Expert Insights with Statistical Analysis of Complex Omics Data. Expert. Opin. Med. Diagn. 2013, 7, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Karpievitch, Y.V.; Dabney, A.R.; Smith, R.D. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinform. 2012, 13 (Suppl. 16), S5. [Google Scholar] [CrossRef]
- Witten, D.; Tibshirani, R. A comparison of fold-change and the t-statistic for microarray data analysis. Analysis 2007, 58–85. [Google Scholar]
- Jeffery, I.B.; Higgins, D.G.; Culhane, A.C. Comparison and evaluation of methods for generating differentially expressed gene lists from microarray data. BMC Bioinform. 2006, 7, 359. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Jones, W.D.; Jensen, R.V.; Harris, S.C.; Perkins, R.G.; Goodsaid, F.M.; Guo, L.; Croner, L.J.; Boysen, C.; Fang, H.; et al. The balance of reproducibility, sensitivity, and specificity of lists of differentially expressed genes in microarray studies. BMC Bioinform. 2008, 9 (Suppl. 9), S10. [Google Scholar] [CrossRef]
- Guo, L.; Lobenhofer, E.K.; Wang, C.; Shippy, R.; Harris, S.C.; Zhang, L.; Mei, N.; Chen, T.; Herman, D.; Goodsaid, F.M.; et al. Rat toxicogenomic study reveals analytical consistency across microarray platforms. Nat. Biotechnol. 2006, 24, 1162–1169. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. PanelomiX: A threshold-based algorithm to create panels of biomarkers. Transl. Proteom. 2013, 1, 57–64. [Google Scholar] [CrossRef]
- Calderón-Santiago, M.; Priego-Capote, F.; Turck, N.; Robin, X.; Jurado-Gámez, B.; Sanchez, J.C.; De Castro, M.D.L. Human sweat metabolomics for lung cancer screening. Anal. Bioanal. Chem. 2015, 407, 5381–5392. [Google Scholar] [CrossRef] [PubMed]
- Peralbo-Molina, A.; Calderón-Santiago, M.; Priego-Capote, F.; Gamez, B.J.; De Castro, M.D.L. Identification of metabolomics panels for potential lung cancer screening by analysis of exhaled breath condensate. J. Breath 2016, 10, 26002. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Povedano, M.M.; Calderón-Santiago, M.; Priego-Capote, F.; Jurado-Gámez, B.; de Castro, M.D.L. Recent advances in human sweat metabolomics for lung cancer screening. Metabolomics 2016, 12, 166. [Google Scholar] [CrossRef]
- Hogan, S.R.; Alvarado-Velez, M.; Wang, M.D.; LaPlaca, M.C.; Phan, J.H.; Bellamkonda, R.V.; Fernandez, F.M. Discovery of Lipidome Alterations Following Traumatic Brain Injury via High-Resolution Metabolomics. J. Proteome 2018, 17, 2131–2143. [Google Scholar] [CrossRef]
- De Paoli, M.; Perco, P.; Mühlberger, I.; Lukas, A.; Pandha, H.; Morgan, R.; Feng, G.J.; Marquette, C. Disease map-based biomarker selection and pre-validation for bladder cancer diagnostic. Biomarkers 2015, 20, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Lagerstedt, L.; Egea-Guerrero, J.J.; Bustamante, A.; Rodríguez-Rodríguez, A.; El Rahal, A.; Quintana-Diaz, M.; García-Armengol, R.; Prica, C.M.; Andereggen, E.; Rinaldi, L.; et al. Combining H-FABP and GFAP increases the capacity to differentiate between CT-positive and CT-negative patients with mild traumatic brain injury. PLoS ONE 2018, 13, e0200394. [Google Scholar] [CrossRef]
- Pata, O.; Vardareli, E.; Ozcan, A.; Serteser, M.; Ünsal, I.; Saruç, M.; Unlu, C.; Tözün, N. Intrahepatic cholestasis of pregnancy: Correlation of preterm delivery with bile acids. Turk. J. Gastroenterol. 2011, 22, 602–605. [Google Scholar] [CrossRef]
- Mawson, A.R. A Role for the Liver in Parturition and Preterm Birth. J. Transl. Sci. 2016, 2, 154–159. [Google Scholar] [CrossRef]
- Challis, J.R.G.; Sloboda, D.M.; Alfaidy, N.; Lye, S.J.; Gibb, W.; Patel, F.A.; Whittle, W.L.; Newnham, J.P. Prostaglandins and mechanisms of preterm birth. Reproduction 2002, 124, 1–17. [Google Scholar] [CrossRef]
- O’Brien, W.F. The Role of Prostaglandins in Labor and Delivery. Clin. Perinatol. 1995, 22, 973–984. [Google Scholar] [CrossRef]
- Bodnar, L.M.; Platt, R.W.; Simhan, H.N. Early-Pregnancy Vitamin D Deficiency and Risk of Preterm Birth Subtypes. Obstet. Gynecol. 2015, 125, 439–447. [Google Scholar] [CrossRef]
- Tabatabaei, N.; Auger, N.; Herba, C.M.; Wei, S.; Allard, C.; Fink, G.D.; Fraser, W.D. Maternal Vitamin D Insufficiency Early in Pregnancy Is Associated with Increased Risk of Preterm Birth in Ethnic Minority Women in Canada. J. Nutr. 2017, 147, 1145–1151. [Google Scholar] [CrossRef]
- Wei, S.-Q.; Qi, H.-P.; Luo, Z.-C.; Fraser, W.D. Maternal vitamin D status and adverse pregnancy outcomes: A systematic review and meta-analysis. J. Matern. Neonatal Med. 2013, 26, 889–899. [Google Scholar] [CrossRef]
- Qin, L.-L.; Lu, F.-G.; Yang, S.-H.; Xu, H.-L.; Luo, B.-A. Does Maternal Vitamin D Deficiency Increase the Risk of Preterm Birth: A Meta-Analysis of Observational Studies. Nutrients 2016, 8, 301. [Google Scholar] [CrossRef]
- Menon, R.; Jones, J.; Gunst, P.R.; Kacerovsky, M.; Fortunato, S.J.; Saade, G.R.; Basraon, S. Amniotic fluid metabolomic analysis in spontaneous preterm birth. Reprod. Sci. 2014, 21, 791–803. [Google Scholar] [CrossRef]
- Catov, J.M.; Bertolet, M.; Chen, Y.-F.; Evans, R.W.; Hubel, C.A. Nonesterified fatty acids and spontaneous preterm birth: A factor analysis for identification of risk patterns. Am. J. Epidemiol. 2014, 179, 1208–1215. [Google Scholar] [CrossRef]
- Olsen, S.F.; Halldorsson, T.I.; Thorne-Lyman, A.L.; Strøm, M.; Gørtz, S.; Granstrøm, C.; Nielsen, P.H.; Wohlfahrt, J.; Lykke, J.A.; Langhoff-Roos, J. Plasma Concentrations of Long Chain N-3 Fatty Acids in Early and Mid-Pregnancy and Risk of Early Preterm Birth. EBioMedicine 2018, 35, 325–333. [Google Scholar] [CrossRef]
- Kim, Y.S.; Maruvada, P.; Milner, J.A. Metabolomics in biomarker discovery: Future uses for cancer prevention. Futur. Oncol. 2008, 4, 93–102. [Google Scholar] [CrossRef]
- Ostrow, J.D. Metabolism of bile salts in cholestasis in humans. In Hepatic Transport and Bile Secretion: Physiology and Pathophysiology; Raven Press: New York, NY, USA, 1993; pp. 673–712. [Google Scholar]
- Mesaros, C.; Lee, S.H.; Blair, I.A. Targeted quantitative analysis of eicosanoid lipids in biological samples using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2009, 877, 2736–2745. [Google Scholar] [CrossRef]
- Milne, S.B.; Ivanova, P.T.; DeCamp, D.; Hsueh, R.C.; Brown, H.A. A targeted mass spectrometric analysis of phosphatidylinositol phosphate species. J. Lipid. Res. 2005, 46, 1796–1802. [Google Scholar] [CrossRef]
- Müller, M.J.; Volmer, D.A. Mass Spectrometric Profiling of Vitamin D Metabolites beyond 25-Hydroxyvitamin D. Clin. Chem. 2015, 61, 1033–1048. [Google Scholar] [CrossRef]
- Bodnar, L.M.; Catov, J.M.; Roberts, J.M.; Simhan, H.N. Prepregnancy Obesity Predicts Poor Vitamin D Status in Mothers and Their Neonates. J. Nutr. 2007, 137, 2437–2442. [Google Scholar] [CrossRef]
- Forrest, K.Y.; Stuhldreher, W.L. Prevalence and correlates of vitamin D deficiency in US adults. Nutr. Res. 2011, 31, 48–54. [Google Scholar] [CrossRef]
- Quinkler, M.; Bujalska, I.J.; Tomlinson, J.W.; Smith, D.M.; Stewart, P.M. Depot-specific prostaglandin synthesis in human adipose tissue: A novel possible mechanism of adipogenesis. Gene 2006, 380, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Savva, S.C.; Chadjigeorgiou, C.; Hatzis, C.; Kyriakakis, M.; Tsimbinos, G.; Tornaritis, M.; Kafatos, A. Association of adipose tissue arachidonic acid content with BMI and overweight status in children from Cyprus and Crete. Br. J. Nutr. 2004, 91, 643–649. [Google Scholar] [CrossRef]
- Torloni, M.R.; Betrán, A.P.; Daher, S.; Widmer, M.; Dolan, S.M.; Menon, R.; Bergel, E.; Allen, T.; Merialdi, M. Maternal BMI and preterm birth: A systematic review of the literature with meta-analysis. J. Matern. Neonatal Med. 2009, 22, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.G.; Ouyang, F.; Pearson, C.; Gillman, M.W.; Belfort, M.B.; Hong, X.; Wang, G.; Heffner, L.; Zuckerman, B.; Wang, X. Prepregnancy body mass index and risk of preterm birth: Association heterogeneity by preterm subgroups. BMC Pregnancy Childbirth 2014, 14, 153. [Google Scholar] [CrossRef]
- Hendler, I.; Goldenberg, R.L.; Mercer, B.M.; Iams, J.D.; Meis, P.J.; Moawad, A.H.; MacPherson, C.A.; Caritis, S.N.; Miodovnik, M.; Menard, K.M.; et al. The Preterm Prediction study: Association between maternal body mass index and spontaneous and indicated preterm birth. Am. J. Obstet. Gynecol. 2005, 192, 882–886. [Google Scholar] [CrossRef]
- Sharashova, E.E.; Anda, E.E.; Grjibovski, A.M. Early pregnancy body mass index and spontaneous preterm birth in Northwest Russia: A registry-based study. BMC Pregnancy Childbirth 2014, 14, 303. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls Age Matched within 5 Years (15 Weeks) | SPTB (15 Weeks) | Controls Age (within 5 Years) and BMI Matched within (3 Points) (20 Weeks) | Controls Age Matched (within 5 Years) (20 Weeks) | SPTB (20 Weeks) | |
|---|---|---|---|---|---|
| Number of samples | 56 | 50 | 49 | 55 | 49 |
| Age of patients (average) | 30.33 | 30.18 | 30.21 | 30.35 | 30.16 |
| BMI of patients (average) | 24.54 | 25.13 | 24.53 | 24.75 | 24.78 |
| Gestational Age at Delivery (Weeks) | >41 | 22–28 (4 cases) 28–31 (4 cases) 32 ≤ 37 (42 cases) | >41 | >41 | 22–28 (3 cases) 28–31 (4 cases) 32 ≤ 37 (42 cases) |
| Pathway to Delivery | Term | PTL (23) PPROM (27) | Term | Term | PTL (23) PPROM (26) |
| Rank | Feature ID | Biological Grouping | Ranked by Fold Change | Direction of Dysregulation in Cases | VIP Score | VIP Ranking | % Missing Values in Cases | %Missing Values in Controls | Features in Panel A |
|---|---|---|---|---|---|---|---|---|---|
| 1 | neg_1 | VITD/GLU/FA | 1.761188 | DOWN | 4.2515 | 6th | 4 | 4 | |
| 2 | neg_2 | DIAG | 1.696499 | DOWN | 4.0385 | 7th | 55 | 42 | |
| 3 | neg_3 | BA | 1.672851 | UP | 6.6187 | 1st | 33 | 40 | |
| 4 | neg_4 | BA | 1.669295 | UP | 6.5837 | 2nd | 10 | 5 | * |
| 5 | neg_5 | BA | 1.64909 | UP | 6.3849 | 3rd | 33 | 42 | |
| 6 | neg_6 | VITD3 | 1.62416 | DOWN | 3.7802 | 11th | 35 | 29 | |
| 7 | neg_7 | AK/ALD | 1.560826 | DOWN | 3.5345 | 14th | 0 | 0 | |
| 8 | neg_8 | GLP/HFA | 1.541056 | DOWN | 3.4536 | 15th | 55 | 56 | |
| 9 | neg_9 | AG/AK/AA/IND | 1.539377 | DOWN | 3.4467 | 16th | 0 | 0 | |
| 10 | neg_10 | FA | 1.500618 | UP | 4.9245 | 4th | 8 | 7 | * |
| 11 | neg_11 | AG/AK/AA | 1.485738 | DOWN | 3.216 | 19th | 0 | 0 | |
| 12 | neg_12 | VITD | 1.481957 | DOWN | 3.1991 | 20th | 0 | 0 | |
| 13 | neg_13 | STG | 1.458342 | UP | 4.5086 | 5th | 4 | 5 | |
| 14 | neg_14 | VITD | 1.406144 | UP | 3.9951 | 8th | 20 | 9 | * |
| 15 | neg_15 | PG | 1.400314 | UP | 3.9378 | 9th | 0 | 2 | * |
| 16 | neg_16 | AK/AG/AA/HFA | 1.392332 | UP | 3.8593 | 10th | 29 | 22 | |
| 17 | neg_17 | OCT | 1.383006 | UP | 3.7675 | 12th | 31 | 40 |
| Panel | Dataset (GA in Weeks) | Feature Selection Method | % Missing Values | # Features in Final Panel | pAUC | % Specificity (95%CI) | % Sensitivity (95%CI) | p-Value upon 10 Fold cv |
|---|---|---|---|---|---|---|---|---|
| Panel A | 20 vs All Controls | FC and PLSDA +VIP | 7 | 4 | 12.2 (8.4–15.9) | 57.7 (49.0–67.3) | 87.8 (77.6–95.9) | 0.0013 |
| Panel B | 20 vs All controls | MWU | 1 | 5 | 10.3 (7.4–14.0) | 57.7 (48.1–67.3) | 77.6 (65.3–89.8) | 0.09158 |
| Panel C | 15 | FC and PLSDA+VIP | 4 | 4 | 10.4 (6.1–15.2) | 73.2 (60.7–83.9) | 75.5 (63.3–87.8) | 0.24057 |
| Panel D | 15 | MWU | 3 | 4 | 9.7 (5.8–14.9) | 62.5 (50.0–75.0) | 77.6 (65.3–87.8) | 0.59576 |
| Panel A Features | Biological Group | Fold Change | VIP Score | MWU- p Value | % Missing Values | Top Performing Feature in PanelomiX |
|---|---|---|---|---|---|---|
| neg_4 | BA | 1.669295 | 6.5837 | 0.0171 | 6 | **** |
| neg_10 | FA | 1.500618 | 4.9245 | 0.0064 | 7 | - |
| neg_14 | VITD | 1.406144 | 3.9951 | 0.2599 | 12 | - |
| neg_15 | PG | 1.400314 | 3.9378 | 0.0062 | 3 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Considine, E.C.; Khashan, A.S.; Kenny, L.C. Screening for Preterm Birth: Potential for a Metabolomics Biomarker Panel. Metabolites 2019, 9, 90. https://doi.org/10.3390/metabo9050090
Considine EC, Khashan AS, Kenny LC. Screening for Preterm Birth: Potential for a Metabolomics Biomarker Panel. Metabolites. 2019; 9(5):90. https://doi.org/10.3390/metabo9050090
Chicago/Turabian StyleConsidine, Elizabeth C., Ali S. Khashan, and Louise C. Kenny. 2019. "Screening for Preterm Birth: Potential for a Metabolomics Biomarker Panel" Metabolites 9, no. 5: 90. https://doi.org/10.3390/metabo9050090
APA StyleConsidine, E. C., Khashan, A. S., & Kenny, L. C. (2019). Screening for Preterm Birth: Potential for a Metabolomics Biomarker Panel. Metabolites, 9(5), 90. https://doi.org/10.3390/metabo9050090