Mass Spectrometry Data Repository Enhances Novel Metabolite Discoveries with Advances in Computational Metabolomics

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Overview of Data Analysis Workflow
2.2. Mass Spectrometry Data
2.3. Software Programs
2.4. In-Silico MS/MS Spectral Libraries
3. Results and Discussion
3.1. Novel Lipid Characterizations in Algae with Enriched In-Silico Spectral Libraries
3.2. Strategy to Link Untargeted- and Targeted Analyses for Increasing Lipid Coverage
3.3. Showcase of Newly Resolved Lipid Profiles by MS/MS-Centric Data Analysis
3.4. Comparison of Untargeted- and Targeted Analysis Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Haug, K.; Salek, R.M.; Steinbeck, C. Global Open Data Management in Metabolomics. Curr. Opin. Chem. Biol. 2017, 36, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An International Repository for Metabolomics Data and Metadata, Metabolite Standards, Protocols, Tutorials and Training, and Analysis Tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef] [PubMed]
- Kale, N.S.; Haug, K.; Conesa, P.; Jayseelan, K.; Moreno, P.; Rocca-Serra, P.; Nainala, V.C.; Spicer, R.A.; Williams, M.; Li, X.; et al. MetaboLights: An Open-Access Database Repository for Metabolomics Data. Curr. Protoc. Bioinform. 2016, 53, 14.13.1–14.13.18. [Google Scholar]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Spicer, R.A.; Steinbeck, C. A Lost Opportunity for Science: Journals Promote Data Sharing in Metabolomics but Do Not Enforce It. Metabolomics 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-Independent MS/MS Deconvolution for Comprehensive Metabolome Analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Ikeda, K.; Tanaka, W.; Senoo, Y.; Arita, M.; Arita, M. Comprehensive Identification of Sphingolipid Species by in Silico Retention Time and Tandem Mass Spectral Library. J. Cheminform. 2017, 9, 19. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Arita, M. The importance of bioinformatics for connecting data-driven lipidomics and biological insights. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 762–765. [Google Scholar] [CrossRef]
- Tsugawa, H.; Nakabayashi, R.; Mori, T.; Yamada, Y.; Takahashi, M.; Rai, A.; Sugiyama, R.; Yamamoto, H.; Nakaya, T.; Yamazaki, M.; et al. A Cheminformatics Approach to Characterize Metabolomes in Stable-Isotope-Labeled Organisms. Nat. Methods 2019, 16, 295–298. [Google Scholar] [CrossRef]
- Tsugawa, H.; Arita, M.; Kanazawa, M.; Ogiwara, A.; Bamba, T.; Fukusaki, E. MRMPROBS: A Data Assessment and Metabolite Identification Tool for Large-Scale Multiple Reaction Monitoring Based Widely Targeted Metabolomics. Anal. Chem. 2013, 85, 5191–5199. [Google Scholar] [CrossRef]
- Tsugawa, H.; Kanazawa, M.; Ogiwara, A.; Arita, M. MRMPROBS Suite for Metabolomics Using Large-Scale MRM Assays. Bioinformatics 2014, 30, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Tsugawa, H.; Wohlgemuth, G.; Mehta, S.; Mueller, M.; Zheng, Y.; Ogiwara, A.; Meissen, J.; Showalter, M.; Takeuchi, K.; et al. Identifying Metabolites by Integrating Metabolome Databases with Mass Spectrometry Cheminformatics. Nat. Methods 2017, 15, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Mahieu, N.G.; Genenbacher, J.L.; Patti, G.J. A Roadmap for the XCMS Family of Software Solutions in Metabolomics. Curr. Opin. Chem. Biol. 2016, 30, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular Framework for Processing, Visualizing, and Analyzing Mass Spectrometry-Based Molecular Profile Data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cai, Y.; Guo, Y.; Chen, F.; Zhu, Z.J. MetDIA: Targeted Metabolite Extraction of Multiplexed MS/MS Spectra Generated by Data-Independent Acquisition. Anal. Chem. 2016, 88, 8757–8764. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, M.; da Veiga Leprevost, F.; Squizzato, S.; Park, Y.M.; Haug, K.; Carroll, A.J.; Spalding, D.; Paschall, J.; Wang, M.; et al. Discovering and Linking Public Omics Data Sets Using the Omics Discovery Index. Nat. Biotechnol. 2017, 35, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Koellensperger, G.; Guijas, C.; Benton, H.P.; Huan, T.; Wolan, D.W.; Warth, B.; Aisporna, A.E.; Hermann, G.; Domingo-Almenara, X.; Spilker, M.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar]
- Kind, T.; Liu, K.-H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in Silico Tandem Mass Spectrometry Database for Lipid Identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef]
- Eichenberger, W.; Gribi, C. Lipids of Pavlova Lutheri: Cellular Site and Metabolic Role of DGCC. Phytochemistry 1997, 45, 1561–1567. [Google Scholar] [CrossRef]
- Schleyer, G.; Shahaf, N.; Ziv, C.; Dong, Y.; Meoded, R.A.; Helfrich, E.J.N.; Schatz, D.; Rosenwasser, S.; Rogachev, I.; Aharoni, A.; et al. In Plaque-Mass Spectrometry Imaging of a Bloom-Forming Alga during Viral Infection Reveals a Metabolic Shift towards Odd-Chain Fatty Acid Lipids. Nat. Microbiol. 2019, 4, 527–538. [Google Scholar] [CrossRef]
- Aoyagi, R.; Ikeda, K.; Isobe, Y.; Arita, M. Comprehensive Analyses of Oxidized Phospholipids Using a Measured MS/MS Spectra Library. J. Lipid Res. 2017, 58, 2229–2237. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, Y.; Otsuki, H.; Narisawa, T.; Kobayashi, M.; Sawai, S.; Kamide, Y.; Kusano, M.; Aoki, T.; Hirai, M.Y.; Saito, K. A New Class of Plant Lipid Is Essential for Protection against Phosphorus Depletion. Nat. Commun. 2013, 4, 1510. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lou, Y.; Mu, T.; Xu, J.; Zhou, C.; Yan, X. Simultaneous Structural Identification of Diacylglyceryl-N-Trimethylhomoserine (DGTS) and Diacylglycerylhydroxymethyl-N, N, N-Trimethyl-β-Alanine (DGTA) in Microalgae Using Dual Li+/H+ Adduct Ion Mode by Ultra-Performance Liquid Chromatography/Quadrupole Time-Of-Flight Mass Spectrometry. Rapid Commun. Mass Spectrom. 2017, 31, 824. [Google Scholar] [PubMed]
- Okazaki, Y.; Nishizawa, T.; Takano, K.; Ohnishi, M.; Mimura, T.; Saito, K. Induced accumulation of glucuronosyldiacylglycerol in tomato and soybean under phosphorus deprivation. Physiol. Plant. 2015, 155, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Roughan, P.G.; Slack, C.R.; Holland, R. Generation of phospholipid artefacts during extraction of developing soybean seeds with methanolic solvents. Lipids 1978, 13, 497–503. [Google Scholar] [CrossRef]
- Reis, A.; Spickett, C.M. Chemistry of Phospholipid Oxidation. Biochim. Biophys. Acta-Biomembr. 2012, 1818, 2374–2387. [Google Scholar] [CrossRef]
- Bonnera, R.; Hopfgartner, G. SWATH data independent acquisition mass spectrometry for metabolomics. TrAc Trends Anal. Chem. 2018. [Google Scholar] [CrossRef]
- Ohlrogge, J.; Browse, J. Lipid Biosynthesis. Plant Cell 2007, 7, 957–970. [Google Scholar] [CrossRef]
- Matsuda, F.; Tsugawa, H.; Fukusaki, E. Method for Assessing the Statistical Significance of Mass Spectral Similarities Using Basic Local Alignment Search Tool Statistics. Anal. Chem. 2013, 85, 8291–8297. [Google Scholar] [CrossRef]
- Palmer, A.; Phapale, P.; Chernyavsky, I.; Lavigne, R.; Fay, D.; Tarasov, A.; Kovalev, V.; Fuchser, J.; Nikolenko, S.; Pineau, C.; et al. FDR-Controlled Metabolite Annotation for High-Resolution Imaging Mass Spectrometry. Nat. Methods 2016, 14, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Scheubert, K.; Hufsky, F.; Petras, D.; Wang, M.; Nothias, L.-F.; Dührkop, K.; Bandeira, N.; Dorrestein, P.C.; Böcker, S. Significance Estimation for Large Scale Metabolomics Annotations by Spectral Matching. Nat. Commun. 2017, 8, 1494. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.; Zaeem, M.; Fillier, T.A.; Nadeem, M.; Vidal, N.P.; Manful, C.; Cheema, S.; Cheema, M.; Thomas, R.H. Targeting Modified Lipids during Routine Lipidomics Analysis Using HILIC and C30 Reverse Phase Liquid Chromatography Coupled to Mass Spectrometry. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Lísa, M.; Holčapek, M. High-Throughput and Comprehensive Lipidomic Analysis Using Ultrahigh-Performance Supercritical Fluid Chromatography-Mass Spectrometry. Anal. Chem. 2015, 87, 7187–7195. [Google Scholar] [CrossRef] [PubMed]
- Blaženović, I.; Shen, T.; Mehta, S.S.; Kind, T.; Ji, J.; Piparo, M.; Cacciola, F.; Mondello, L.; Fiehn, O. Increasing Compound Identification Rates in Untargeted Lipidomics Research with Liquid Chromatography Drift Time-Ion Mobility Mass Spectrometry. Anal. Chem. 2018, 90, 10758–10764. [Google Scholar] [CrossRef] [PubMed]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A Public Repository for Sharing Mass Spectral Data for Life Sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass Spectral and Retention Index Libraries for Metabolomics Based on Quadrupole and Time-of-Flight Gas Chromatography/Mass Spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef]
- Tsugawa, H. Advances in Computational Metabolomics and Databases Deepen the Understanding of Metabolisms. Curr. Opin. Biotechnol. 2018, 54, 10–17. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
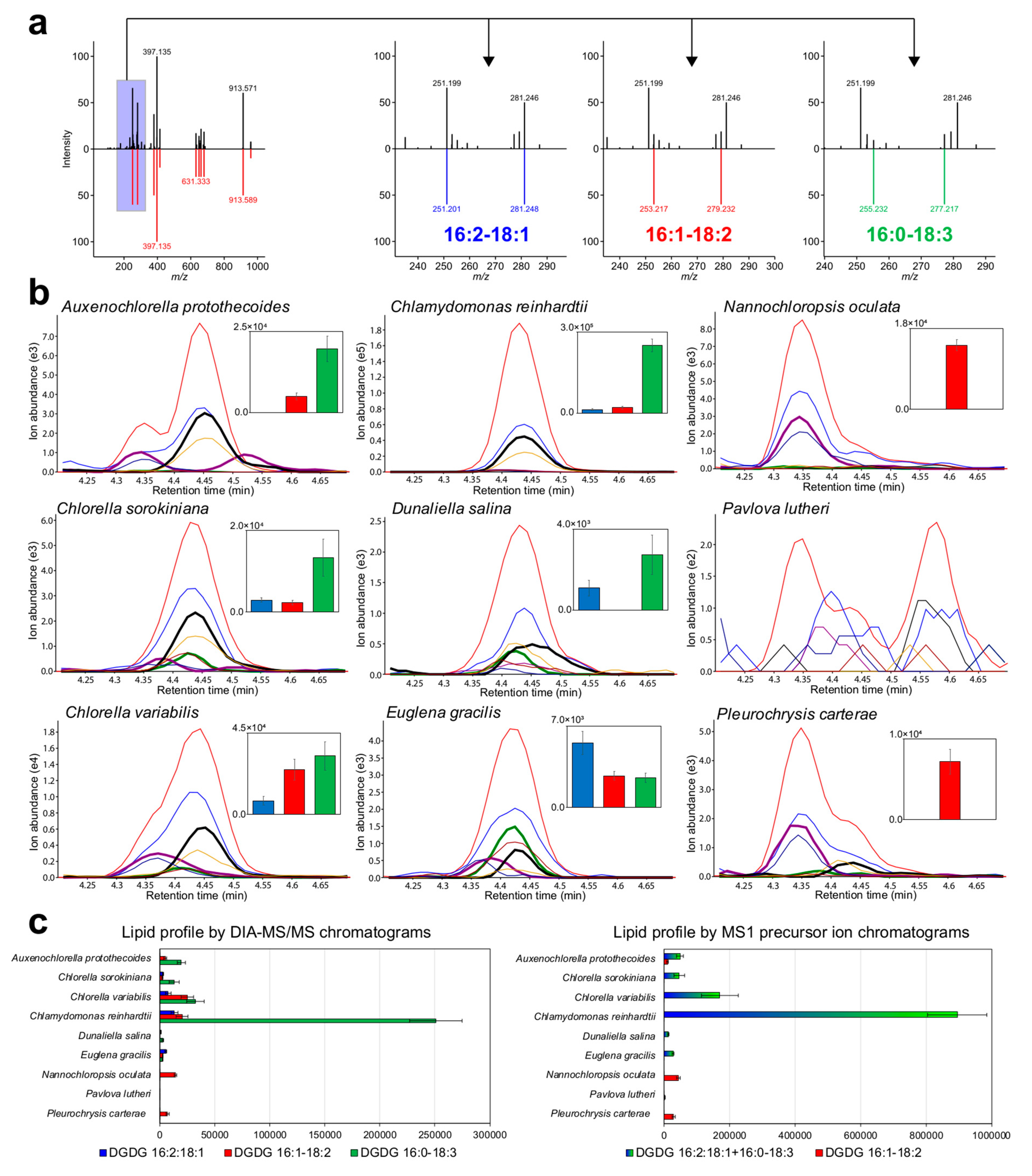{kind=link}
{kind=link}
| Super Class | Class | Auxenochlorella protothecoides | Chlorella sorokiniana | Chlorella variabilis | Chlamydomonas reinhardtii | Dunaliella salina | Euglena gracilis | Nannochloropsis oculata | Pavlova lutheri | Pleurochrysis carterae | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fatty acids | FA | 5 | 5 | 5 | 5 | 4 | 9 | 6 | 6 | 9 | 11 |
| Glycerolipids | DAG | 13 | 18 | 21 | 16 | 2 | 61 | 27 | 5 | 13 | 100 |
| Glycerolipids | TAG | 91 | 80 | 144 | 97 | 152 | 481 | 231 | 126 | 121 | 622 |
| Phospholipids | PC | 25 | 21 | 26 | 0 | 10 | 33 | 42 | 1 | 0 | 75 |
| Phospholipids | PE | 12 | 18 | 19 | 5 | 5 | 14 | 16 | 4 | 1 | 46 |
| Phospholipids | PG | 12 | 16 | 13 | 9 | 9 | 9 | 13 | 8 | 1 | 28 |
| Phospholipids | PS | 3 | 2 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 6 |
| Phospholipids | PI | 8 | 8 | 9 | 8 | 6 | 7 | 14 | 8 | 1 | 19 |
| Phospholipids | PMeOH | 0 | 0 | 0 | 0 | 0 | 29 | 0 | 0 | 0 | 29 |
| Phospholipids | LPC | 4 | 8 | 6 | 0 | 0 | 1 | 6 | 0 | 0 | 10 |
| Phospholipids | LPE | 2 | 6 | 2 | 1 | 0 | 0 | 3 | 0 | 0 | 8 |
| Phospholipids | LPG | 0 | 2 | 1 | 2 | 0 | 1 | 0 | 0 | 0 | 2 |
| Phospholipids | LPS | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Oxidized lipids | OxPC | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
| Oxidized lipids | OxPE | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
| Oxidized lipids | OxPG | 0 | 2 | 6 | 0 | 1 | 0 | 0 | 0 | 0 | 7 |
| Oxidized lipids | OxPI | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
| Algal lipids | LDGTS/LDGTA | 0 | 0 | 0 | 14 | 4 | 9 | 21 | 1 | 3 | 34 |
| Algal lipids | DGTS/DGTA | 0 | 0 | 0 | 68 | 12 | 12 | 68 | 14 | 4 | 134 |
| Algal lipids | LDGCC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 14 | 16 |
| Algal lipids | DGCC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 35 | 38 | 47 |
| Plant lipids | MGDG | 25 | 40 | 35 | 26 | 16 | 42 | 18 | 24 | 27 | 82 |
| Plant lipids | DGDG | 19 | 22 | 28 | 23 | 15 | 39 | 26 | 12 | 28 | 64 |
| Plant lipids | SQDG | 16 | 15 | 10 | 20 | 8 | 25 | 22 | 16 | 24 | 41 |
| Plant lipids | GlcADG | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 1 | 4 | 5 |
| Ceramides | Cer-AP | 8 | 8 | 8 | 7 | 8 | 8 | 8 | 8 | 8 | 10 |
| Ceramides | Cer-NP | 8 | 7 | 8 | 7 | 6 | 6 | 10 | 7 | 6 | 10 |
| Ceramides | Cer-NS | 0 | 0 | 0 | 0 | 0 | 1 | 4 | 0 | 2 | 5 |
| Ceramides | Cer-NDS | 1 | 2 | 4 | 1 | 2 | 3 | 4 | 2 | 2 | 6 |
| Ceramides | Cer-AS | 1 | 1 | 1 | 1 | 1 | 3 | 1 | 2 | 1 | 4 |
| Ceramides | Cer-ADS | 0 | 0 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 4 |
| Ceramides | HexCer-AP | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 2 |
| Total | 253 | 282 | 362 | 310 | 261 | 798 | 540 | 289 | 307 | 1437 | |
| Lipid Class | Ion Mode | Adduct Type | Example | Diagnostic Ions (Lipid Class) | Diagnostic Ions (Acyl Chains) |
|---|---|---|---|---|---|
| LDGCC | Positive | [M+H]+ | LDGCC 18:0 | m/z 104.107 C5H14NO+, m/z 132.102 C6H14NO2+ | - |
| DGCC | Positive | [M+H]+ | DGCC 18:0-20:4 | m/z 104.107 C5H14NO+, m/z 132.102 C6H14NO2+ | NL of SN1 (m/z 538.374 C30H52NO7+), NL of SN1+H2O (m/z 520.363 C30H50NO6+), NL of SN2 (m/z 518.405 C28H56NO7+), NL of SN2+H2O (m/z 500.395 C28H54NO6+) |
| OxPC | Negative | [M+HCOO]− | OxPC 18:0-20:4+2O | NL of HCOO+CH3 (m/z 826.56 C45H81NO10P−) | SN1 (m/z 283.264 C18H35O2−), SN2 (m/z 335.223, C20H31O4−), SN2−H2O (m/z 317.212 C20H29O3−) *SN2−2H2O (m/z 299.202 C20H27O2−) |
| OxPE | Negative | [M−H]− | OxPE 18:0-20:4+2O | m/z 196.038 C5H11NO5P− | SN1 (m/z 283.264 C18H35O2−), SN2 (m/z 335.223, C20H31O4−), SN2−H2O (m/z 317.212 C20H29O3−) *SN2−2H2O (m/z 299.202 C20H27O2−) |
| OxPG | Negative | [M−H]− | OxPG 18:0-20:4+2O | m/z 152.995 C3H6O5P− | SN1 (m/z 283.264 C18H35O2−), SN2 (m/z 335.223, C20H31O4−), SN2−H2O (m/z 317.212 C20H29O3−) *SN2−2H2O (m/z 299.202 C20H27O2−) |
| OxPI | Negative | [M−H]− | OxPI 18:0-20:4+2O | m/z 297.038 C9H14O9P−, m/z 241.012 C6H10O8P− | SN1 (m/z 283.264 C18H35O2−), SN2 (m/z 335.223, C20H31O4−), SN2−H2O (m/z 317.212 C20H29O3−) *SN2−2H2O (m/z 299.202 C20H27O2−) |
| PMeOH | Negative | [M−H]− | PMeOH 18:0-20:4 | m/z 167.012 C4H8O5P−, m/z 110.985 CH4O4P− | SN1 (m/z 283.264 C18H35O2−), SN2 (m/z 303.233 C20H31O2−) |
| GlcADG | Negative | [M−H]− | GlcADG 18:0-20:4 | m/z 249.062 C9H13O7 | SN1 (m/z 283.264 C18H35O2−), SN2 (m/z 303.233 C20H31O2−) |
| LPG | Negative | [M−H]− | LPG 18:0 | m/z 152.995 C3H6O5P− | SN1 || SN2 (m/z 283.264 C18H35O2−) |
| LPS | Negative | [M−H]− | LPS 18:0 | NL of C3H6NO2 (m/z 437.267 C21H42O7P−) | SN1 || SN2 (m/z 283.264 C18H35O2−) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsugawa, H.; Satoh, A.; Uchino, H.; Cajka, T.; Arita, M.; Arita, M. Mass Spectrometry Data Repository Enhances Novel Metabolite Discoveries with Advances in Computational Metabolomics. Metabolites 2019, 9, 119. https://doi.org/10.3390/metabo9060119
Tsugawa H, Satoh A, Uchino H, Cajka T, Arita M, Arita M. Mass Spectrometry Data Repository Enhances Novel Metabolite Discoveries with Advances in Computational Metabolomics. Metabolites. 2019; 9(6):119. https://doi.org/10.3390/metabo9060119
Chicago/Turabian StyleTsugawa, Hiroshi, Aya Satoh, Haruki Uchino, Tomas Cajka, Makoto Arita, and Masanori Arita. 2019. "Mass Spectrometry Data Repository Enhances Novel Metabolite Discoveries with Advances in Computational Metabolomics" Metabolites 9, no. 6: 119. https://doi.org/10.3390/metabo9060119
APA StyleTsugawa, H., Satoh, A., Uchino, H., Cajka, T., Arita, M., & Arita, M. (2019). Mass Spectrometry Data Repository Enhances Novel Metabolite Discoveries with Advances in Computational Metabolomics. Metabolites, 9(6), 119. https://doi.org/10.3390/metabo9060119