CFM-ID 3.0: Significantly Improved ESI-MS/MS Prediction and Compound Identification

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
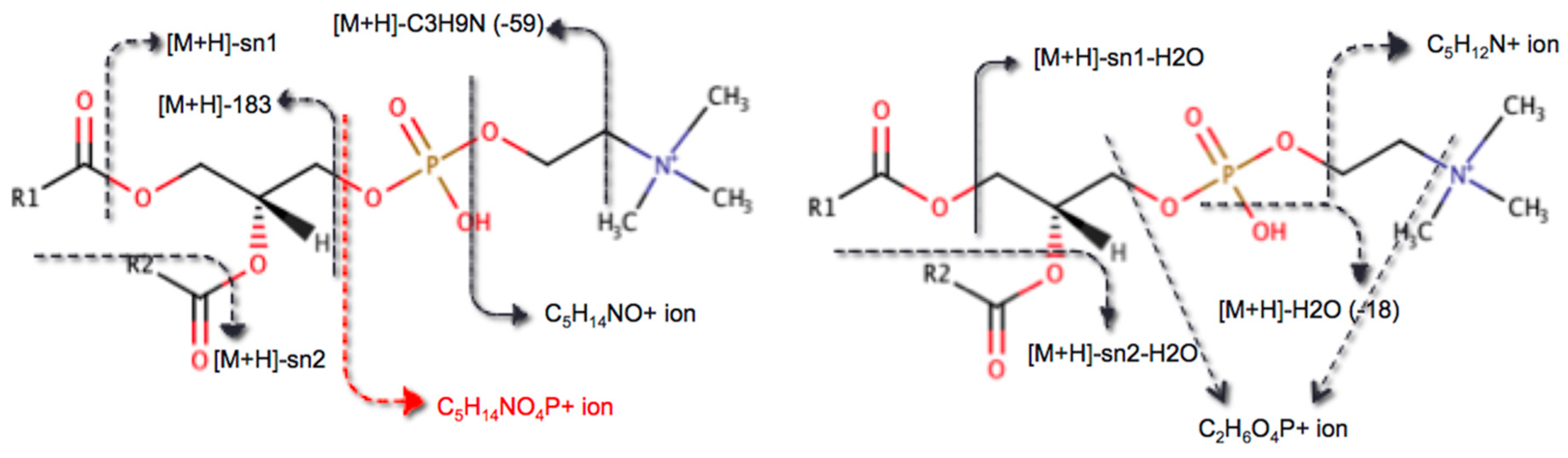
2.1. Encoding Lipid Fragmentation Rules
2.2. The New CFM-ID 3.0 Spectral Library
2.3. Performance Testing
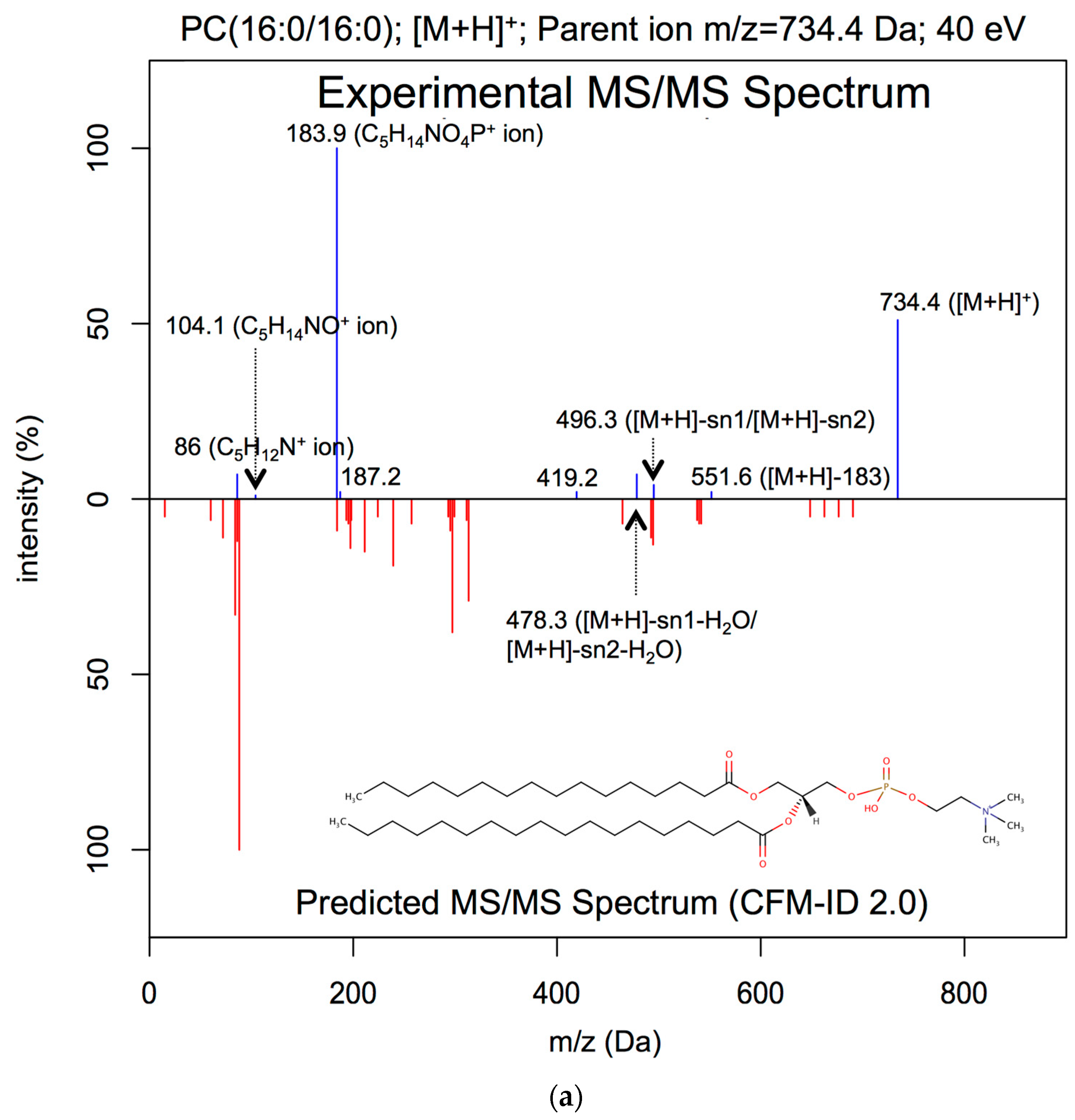
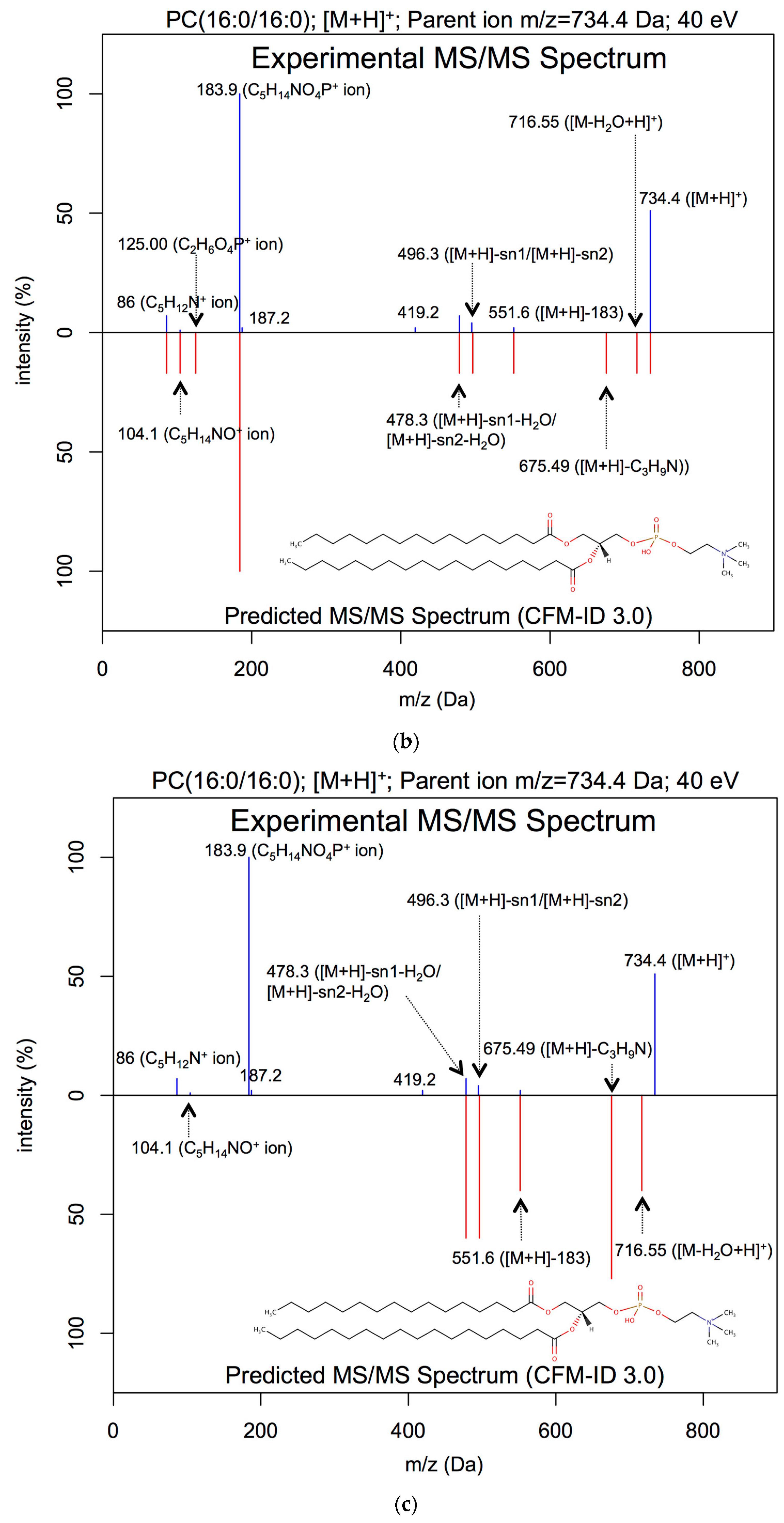
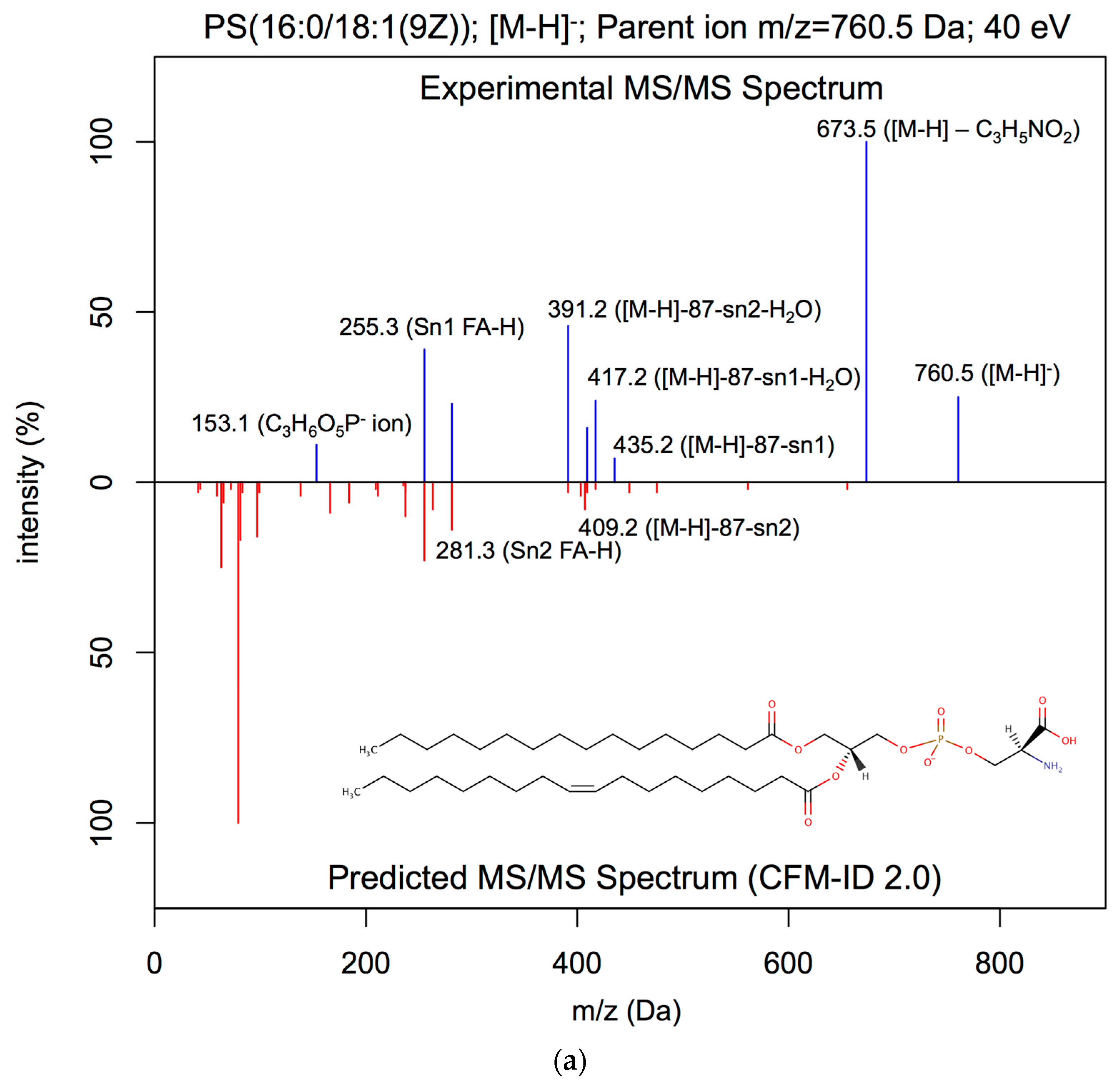
2.3.1. Lipid ESI-MS/MS Spectral Prediction
2.3.2. Compound Identification Using the New Scoring Functions
2.3.3. Compound Chemical Classification
3. Discussion
3.1. ESI-MS/MS Lipid Spectra Prediction
3.2. Compound Identification and Chemical Class Prediction
4. Materials and Methods
4.1. Encoding Lipid Fragmentation Rules
4.1.1. Acquisition of Reference Lipid MS/MS Spectra
4.1.2. Annotation of Reference Lipid MS/MS Spectra
4.1.3. Implementation of Lipid Fragmentation Rules
4.2. Integration of Chemical Classification
4.3. Collection of Compound Citations
4.4. Redesigning and Expanding of CFM-ID’s Spectral Library
4.4.1. Collection of Experimental MS/MS Spectra from External Sources
4.4.2. Compilation of Predicted ESI-MS/MS Spectra
4.5. Modifying CFM-ID’s Scoring Function and Ranking Schema
4.6. Performance Testing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lynn, K.S.; Cheng, M.L.; Chen, Y.R.; Hsu, C.; Chen, A.; Lih, T.M.; Chang, H.Y.; Huang, C.J.; Shiao, M.S.; Pan, W.H.; et al. Metabolite identification for mass spectrometry-based metabolomics using multiple types of correlated ion information. Anal. Chem. 2015, 87, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Allard, P.M.; Péresse, T.; Bisson, J.; Gindro, K.; Marcourt, L.; Pham, V.C.; Roussi, F.; Litaudon, M.; Wolfender, J.L. Integration of Molecular Networking and In-Silico MS/MS Fragmentation for Natural Products Dereplication. Anal. Chem. 2016, 88, 3317–3323. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.A.; Jones, O.A.H.; Beale, D.J.; Boughton, B.A.; Benheim, D.; Kouremenos, K.A.; Wolfender, J.L.; Wishart, D.S. Current and future perspectives on the structural identification of small molecules in biological systems. Metabolites 2016, 6, 46. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Singer, H.P.; Longrée, P.; Loos, M.; Ruff, M.; Stravs, M.A.; Ripollés Vidal, C.; Hollender, J. Strategies to characterize polar organic contamination in wastewater: Exploring the capability of high resolution mass spectrometry. Environ. Sci. Technol. 2014, 48, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.R.; Dorrestein, P.C.; Quinn, R.A. Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. USA 2015, 112, 12549–12550. [Google Scholar] [CrossRef]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [PubMed]
- Ridder, L.; Van Der Hooft, J.J.J.; Verhoeven, S.; De Vos, R.C.H.; Van Schaik, R.; Vervoort, J. Substructure-based annotation of high-resolution multistage MSn spectral trees. Rapid Commun. Mass Spectrom. 2012, 26, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminform. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, W94–W99. [Google Scholar] [CrossRef]
- Allen, F.; Greiner, R.; Wishart, D. Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics 2014, 11, 98–110. [Google Scholar] [CrossRef]
- Allen, F.; Pon, A.; Greiner, R.; Wishart, D. Computational Prediction of Electron Ionization Mass Spectra to Assist in GC/MS Compound Identification. Anal. Chem. 2016, 88, 7689–7697. [Google Scholar] [CrossRef]
- Hufsky, F.; Böcker, S. Mining molecular structure databases: Identification of small molecules based on fragmentation mass spectrometry data. Mass Spectrom. Rev. 2017, 36, 624–633. [Google Scholar] [CrossRef]
- Heinonen, M.; Rantanen, A.; Mielikäinen, T.; Kokkonen, J.; Kiuru, J.; Ketola, R.A.; Rousu, J. FiD: A software for ab initio structural identification of product ions from tandem mass spectrometric data. Rapid Commun. Mass Spectrom. 2008, 22, 3043–3052. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Kind, T.; Liu, K.H.; Lee, D.Y.; Defelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Ikeda, K.; Tanaka, W.; Senoo, Y.; Arita, M.; Arita, M. Comprehensive identification of sphingolipid species by in silico retention time and tandem mass spectral library. J. Cheminform. 2017, 9. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Ruttkies, C.; Krauss, M.; Brouard, C.; Kind, T.; Dührkop, K.; Allen, F.; Vaniya, A.; Verdegem, D.; Böcker, S.; et al. Critical Assessment of Small Molecule Identification 2016: Automated methods. J. Cheminform. 2017, 9. [Google Scholar] [CrossRef]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 1–20. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Phytohub v1.4. Available online: http://phytohub.eu/ (accessed on 10 February 2018).
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- MassBank of North America (MoNA), Fiehn Lab, UC Davis, CA 95618. Available online: http://mona.fiehnlab.ucdavis.edu/ (accessed on 26 February 2019).
- Nikolic, D.; Jones, M.; Sumner, L.; Dunn, W. CASMI 2014: Challenges, solutions and results. Curr. Metab. 2017, 5, 5–17. [Google Scholar] [CrossRef]
- Tsugawa, H.; Kind, T.; Nakabayashi, R.; Yukihira, D.; Tanaka, W.; Cajka, T.; Saito, K.; Fiehn, O.; Arita, M. Hydrogen rearrangement rules: Computational MS/MS fragmentation and structure elucidation using MS-FINDER software. Anal. Chem. 2016, 88, 7946. [Google Scholar] [CrossRef] [PubMed]
- Witting, M.; Ruttkies, C.; Neumann, S.; Schmitt-Kopplin, P. LipidFrag: Improving reliability of in silico fragmentation of lipids and application to the Caenorhabditis elegans lipidome. PLoS ONE 2017, 12, e0172311. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.H.; Shimizu, T.; Spener, F.; Van Meer, G.; Wakelam, M.J.O.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef]
- Pi, J.; Wu, X.; Feng, Y. Fragmentation patterns of five types of phospholipids by ultra-high-performance liquid chromatography electrospray ionization quadrupole time-of-flight tandem mass spectrometry. Anal. Methods 2016, 8, 1319–1332. [Google Scholar] [CrossRef]
- Murphy, R.C. Tandem Mass Spectrometry of Lipids: Molecular Analysis of Complex Lipids; Royal Society of Chemistry: London, UK, 2014. [Google Scholar] [CrossRef]
- Blaženović, I.; Kind, T.; Torbašinović, H.; Obrenović, S.; Mehta, S.S.; Tsugawa, H.; Wermuth, T.; Schauer, N.; Jahn, M.; Biedendieck, R. Comprehensive comparison of in silico MS/MS fragmentation tools of the CASMI contest: Database boosting is needed to achieve 93% accuracy. J. Cheminform. 2017, 9, 32. [Google Scholar] [CrossRef]
- SMARTS Theory Manual, Daylight Chemical Information Systems, Inc., Laguna Niguel, CA 92677. Available online: http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html (accessed on 20 December 2018).
- SMIRKS Theory Manual, Daylight Chemical Information Systems, Inc., Laguna Niguel, CA 92677. Available online: http://www.daylight.com/dayhtml/doc/theory/theory.smirks.html (accessed on 29 October 2018).
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- mzCloud—Advanced Mass Spectral Database, HighChem, Bratislava, Slovakia. Available online: https://www.mzcloud.org/ (accessed on 18 December 2018).
- Han, X. Lipidomics: Comprehensive Mass Spectrometry of Lipids; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Jeliazkova, N.; Kochev, N. AMBIT-SMARTS: Efficient searching of chemical structures and fragments. Mol. Inform. 2011, 30, 707–720. [Google Scholar] [CrossRef]
- RDKit: Open-Source Cheminformatics Software. Available online: https://www.rdkit.org/ (accessed on 6 September 2018).
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Pence, H.E.; Williams, A. Chemspider: An online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016, 44, D1214–D1219. [Google Scholar] [CrossRef] [PubMed]
- McEachran, A.D.; Sobus, J.R.; Williams, A.J. Identifying known unknowns using the US EPA’s CompTox Chemistry Dashboard. Anal. Bioanal. Chem. 2017, 409, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.; Arndt, D.; Pon, A.; Sajed, T.; Guo, A.C.; Djoumbou, Y.; Knox, C.; Wilson, M.; Liang, Y.; Grant, J.; et al. T3DB: The toxic exposome database. Nucleic Acids Res. 2015, 43, D928–D934. [Google Scholar] [CrossRef]
- ContaminantDB v1.0, TMIC, University of Alberta, Canada. Available online: http://contaminantdb.ca/ (accessed on 28 March 2019).
- FooDB v1.0, TMIC, University of Alberta, Canada. Available online: http://foodb.ca/ (accessed on 28 March 2019).
- Sajed, T.; Marcu, A.; Ramirez, M.; Pon, A.; Guo, A.C.; Knox, C.; Wilson, M.; Grant, J.R.; Djoumbou, Y.; Wishart, D.S. ECMDB 2.0: A richer resource for understanding the biochemistry of E. coli. Nucleic Acids Res. 2016, 44, D495–D501. [Google Scholar] [CrossRef]
- Ramirez-Gaona, M.; Marcu, A.; Pon, A.; Guo, A.C.; Sajed, T.; Wishart, N.A.; Karu, N.; Feunang, Y.D.; Arndt, D.; Wishart, D.S. YMDB 2.0: A significantly expanded version of the yeast metabolome database. Nucleic Acids Res. 2017, 45, D440–D445. [Google Scholar] [CrossRef]
- Sawada, Y.; Nakabayashi, R.; Yamada, Y.; Suzuki, M.; Sato, M.; Sakata, A.; Akiyama, K.; Sakurai, T.; Matsuda, F.; Aoki, T.; et al. RIKEN tandem mass spectral database (ReSpect) for phytochemicals: A plant-specific MS/MS-based data resource and database. Phytochemistry 2012, 82, 38–45. [Google Scholar] [CrossRef]
- Dodder, N.G. Organic/Biological Mass Spectrometry Data Analysis. Available online: https://cran.r-project.org/web/packages/OrgMassSpecR/index.html (accessed on 28 March 2019).
- Djoumbou-Feunang, Y.; Fiamoncini, J.; Gil-de-la-Fuente, A.; Greiner, R.; Manach, C.; Wishart, D.S. BioTransformer: A comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J. Cheminform. 2019, 11. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
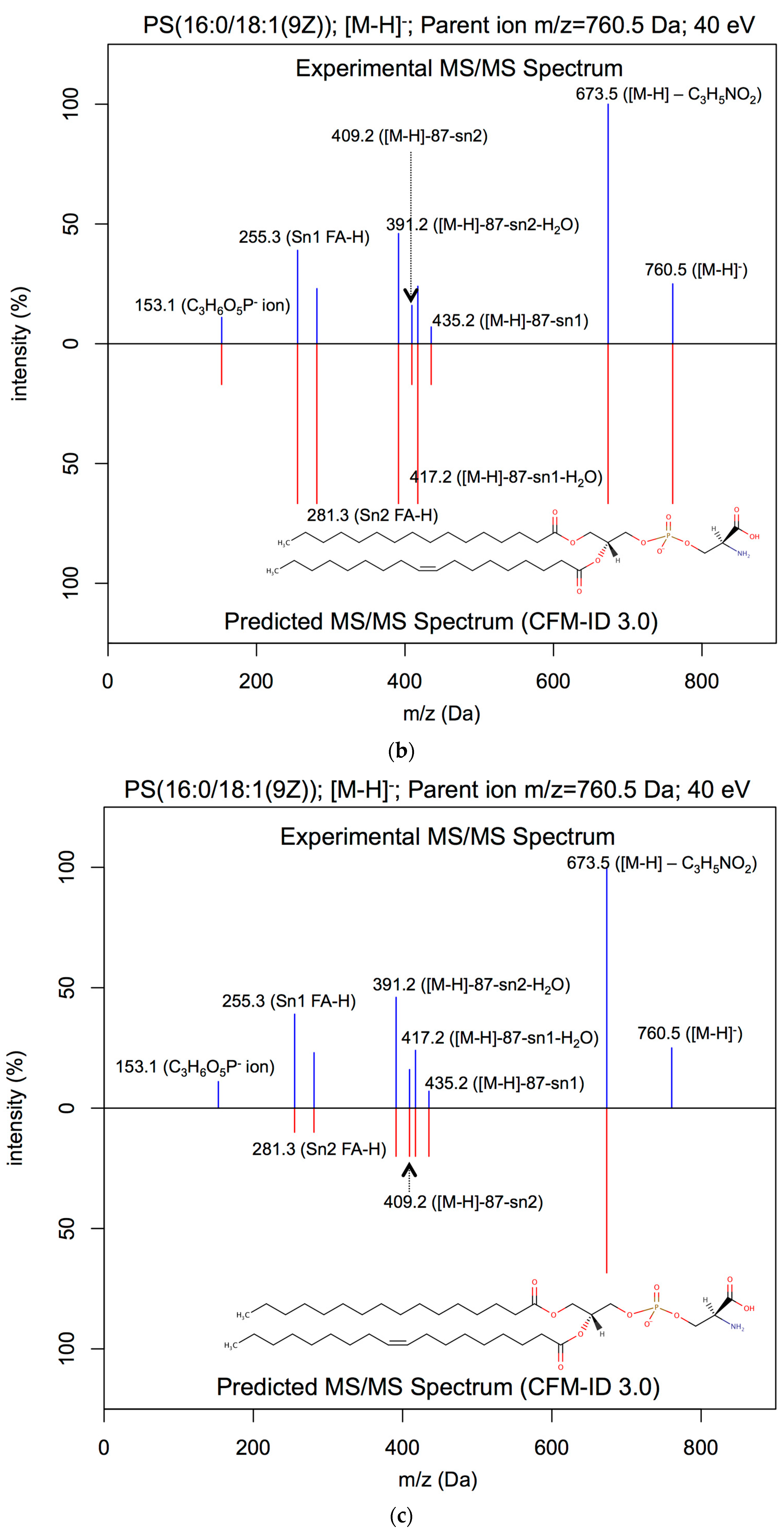{kind=link}
| Lipid Class | Number of Covered Rules | Number of Covered Adduct Types |
|---|---|---|
| 1-Monoacylglycerols | 8 | [M+Li]+; [M+NH4]+ |
| 2-Monoacylglycerols | 11 | [M+H]+; [M+NH4]+; [M+Na]+ |
| 1,2-Diacylglycerols | 10 | [M+NH4]+; [M+Na]+ |
| Triacylglycerols | 19 | [M+Na]+; [M+NH4]+; [M+Li]+ |
| Phosphatidic acids | 22 | [M+H]+; [M+Na]+; [M−H]− |
| Phosphatidylcholines | 41 | [M+H]+; [M+Na]+; [M+Li]+; [M+Cl]− |
| Phosphatidylethanolamines | 24 | [M+H]+; [M+Na]+; [M−H]− |
| Lysophosphatidylcholines | 29 | [M+H]+; [M+Na]+; [M+Li]+; [M+Cl]− |
| Lysophosphatidic acids | 12 | [M+H]+; [M−H]− |
| Phosphatidylserines | 28 | [M+H]+; [M+Li]+; [M+Na]+; [M−H]− |
| Ceramides | 17 | [M+H]+; [M+Li]+; [M−H]− |
| Sphingomyelins | 13 | [M+H]+; [M+Li]+; [M+Na]+ |
| Cardiolipins | 13 | [M−2H](2H)− |
| Phosphatidylglycerols | 11 | [M−H]− |
| Lysophosphatidylglycerols | 7 | [M−H]− |
| Plasmanyl-PC (1-alkyl,2-acylglycero-3-phosphocholines) | 17 | [M+H]+; [M+Cl]− |
| Plasmenyl-PC (1-(1Z-alkenyl)-glycero-3-phosphocholines) | 17 | [M+H]+; [M+Cl]− |
| 1-Alkanylglycerophosphocholines (Monoalkylglycerophosphocholines) | 15 | [M+H]+; [M+Cl]−; [M+Na]+ |
| 1-Alkenylglycerophosphocholines (1-(1Z-alkenyl)-glycero-3-phosphocholines) | 13 | [M+H]+; [M+Cl]− |
| Phosphatidylinositols | 9 | [M−H]− |
| Lysophosphatidylinositols | 8 | [M−H]− |
| Total | 344 | 50 |
| Feature | Value |
|---|---|
| Total number of unique compounds | 229,084 |
| Total number of unique ESI-MS/MS spectra | 397,679 |
| Total number of experimental ESI-MS/MS spectra | 87,570 |
| Total number of predicted ESI-MS/MS spectra | 310,109 |
| Number of compounds with ≥1 experimental ESI-MS/MS spectra | 13,537 |
| Number of compounds with ≥1 predicted ESI-MS/MS spectra | 108,972 |
| Number of compounds with ≥2 citations | 229,084 |
| Average number of citations per compound | 272 |
| Number of compounds with chemical classification assignments | 229,084 |
| Average number of chemical category assignments/compound | 25 |
| Compound | Adduct | Energy (eV) | CFM-ID 3.0 (Score) | CFM-ID 2.0 (Score) | LipidBlast (Score) |
|---|---|---|---|---|---|
| PA(16:0/18:1(9Z)) | [M−H]− | 10 | 1.00 | 0.36 | 0.00 |
| PS(16:0/18:1(9Z)) | [M−H]− | 10 | 1.00 | 0.31 | 0.00 |
| CL(18:0/18:0/18:0/18:0) | [M−2H](2H) | 10 | 0.98 | N/A | 0.00 |
| DG(18:0/20:4/0:0) | [M+Na]+ | 10 | 0.92 | 0.00 | N/A |
| PA(16:0/18:1(9Z)) | [M−H]− | 20 | 0.55 | 0.02 | 0.00 |
| PS(16:0/18:1(9Z)) | [M−H]− | 20 | 0.98 | 0.03 | 0.00 |
| CL(18:0/18:0/18:0/18:0) | [M−2H](2H) | 20 | 0.97 | N/A | 0.12 |
| DG(18:0/20:4/0:0) | [M+Na]+ | 20 | 0.93 | 0.00 | N/A |
| PA(16:0/18:1(9Z)) | [M−H]− | 40 | 0.96 | 0.03 | 0.90 |
| PS(16:0/18:1(9Z)) | [M−H]− | 40 | 0.92 | 0.10 | 0.91 |
| CL(18:0/18:0/18:0/18:0) | [M−2H](2H) | 40 | 0.91 | N/A | 0.89 |
| DG(18:0/20:4/0:0) | [M+Na]+ | 40 | 0.18 | 0.00 | N/A |
| PC(16:0/16:0) | [M+H]+ | 40 | 0.88 | 0.07 | 0.13 |
| TG(18:1/18:1/18:2) | [M+NH4]+ | 40 | 0.78 | 0.01 | 0.84 |
| Version | # Top 1 | # Top 3 | # Top 10 | Average Rank | Median Rank | # Correct Classifications |
|---|---|---|---|---|---|---|
| CFM-ID 3.0 | 149 | 194 | 204 | 1.8 | 1 | 168 |
| CFM-ID 2.0-2019 | 123 | 171 | 201 | 2.4 | 1 | N/A |
| CFM-ID 2.0-2016 | 120 | 160 | 182 | 13.64 | 1 | N/A |
| MS-FINDER | 146 | 162 | 174 | 6.4 | 1 | N/A |
| CFM-ID 3.0 * | 208 | 208 | 208 | 1 | 1 | N/A |
| CFM-ID 2.0-2019 * | 208 | 208 | 208 | 1 | 1 | N/A |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djoumbou-Feunang, Y.; Pon, A.; Karu, N.; Zheng, J.; Li, C.; Arndt, D.; Gautam, M.; Allen, F.; Wishart, D.S. CFM-ID 3.0: Significantly Improved ESI-MS/MS Prediction and Compound Identification. Metabolites 2019, 9, 72. https://doi.org/10.3390/metabo9040072
Djoumbou-Feunang Y, Pon A, Karu N, Zheng J, Li C, Arndt D, Gautam M, Allen F, Wishart DS. CFM-ID 3.0: Significantly Improved ESI-MS/MS Prediction and Compound Identification. Metabolites. 2019; 9(4):72. https://doi.org/10.3390/metabo9040072
Chicago/Turabian StyleDjoumbou-Feunang, Yannick, Allison Pon, Naama Karu, Jiamin Zheng, Carin Li, David Arndt, Maheswor Gautam, Felicity Allen, and David S. Wishart. 2019. "CFM-ID 3.0: Significantly Improved ESI-MS/MS Prediction and Compound Identification" Metabolites 9, no. 4: 72. https://doi.org/10.3390/metabo9040072
APA StyleDjoumbou-Feunang, Y., Pon, A., Karu, N., Zheng, J., Li, C., Arndt, D., Gautam, M., Allen, F., & Wishart, D. S. (2019). CFM-ID 3.0: Significantly Improved ESI-MS/MS Prediction and Compound Identification. Metabolites, 9(4), 72. https://doi.org/10.3390/metabo9040072