Palmitate and Stearate are Increased in the Plasma in a 6-OHDA Model of Parkinson’s Disease

 and
and
Abstract
1. Introduction
2. Results
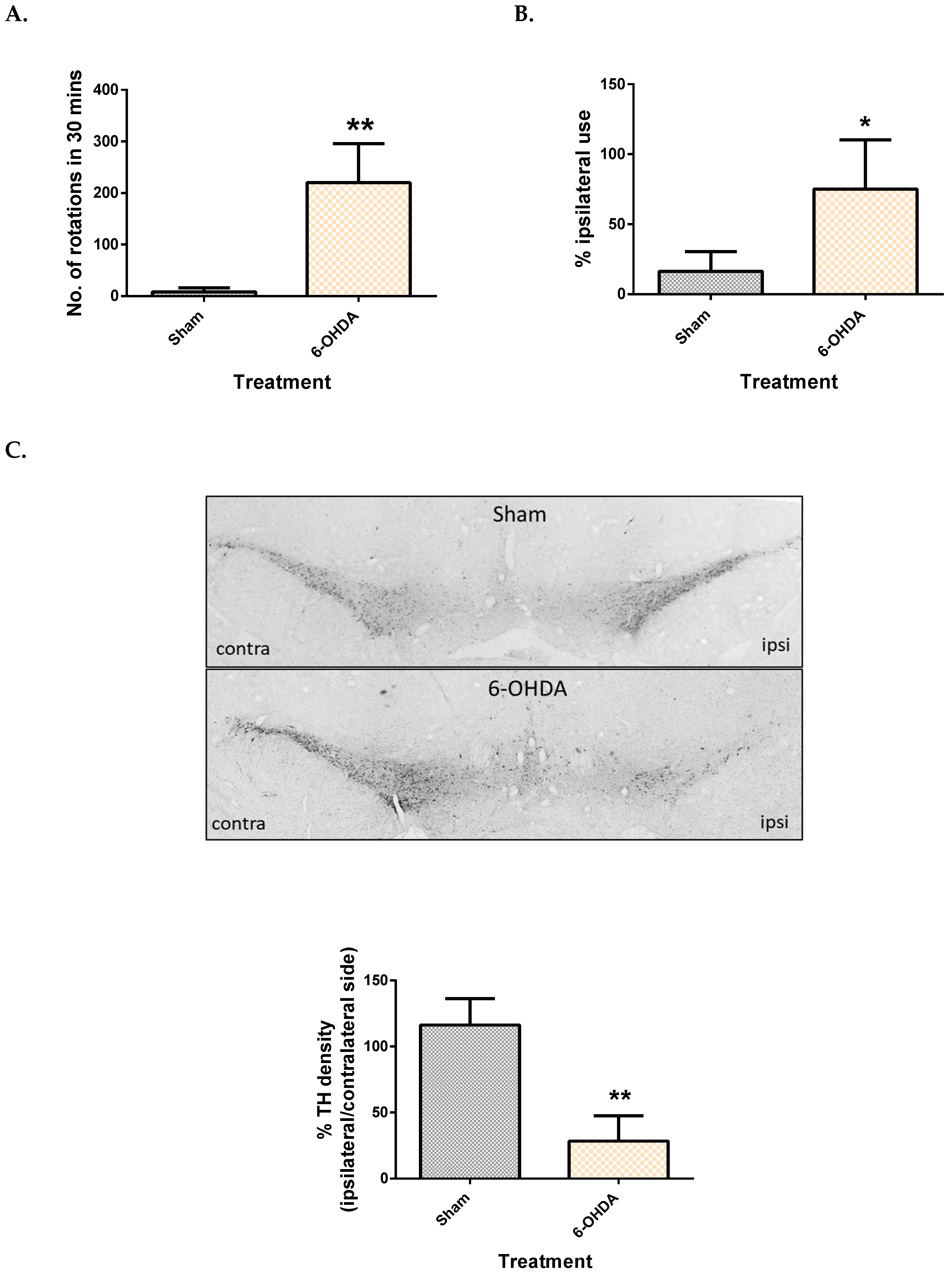
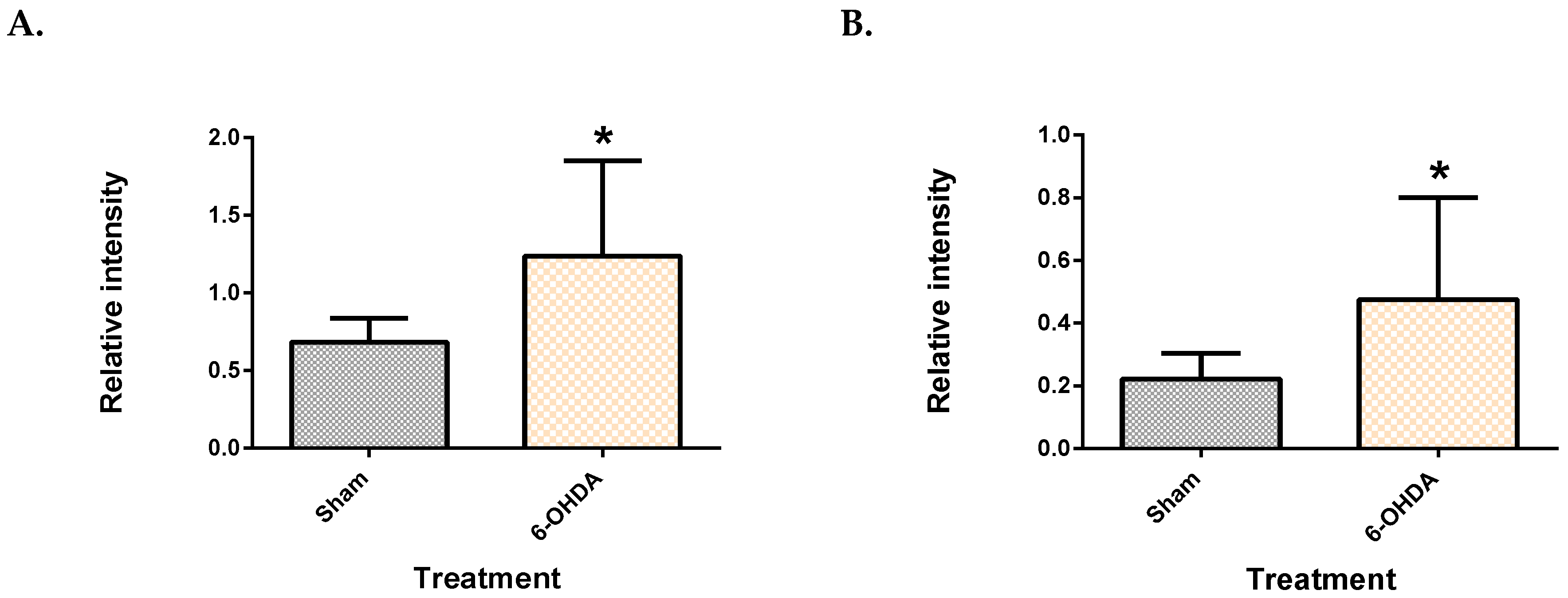
2.1. Validation of the 6-OHDA Model
2.2. Metabolomic Method Validation and Feature Selection
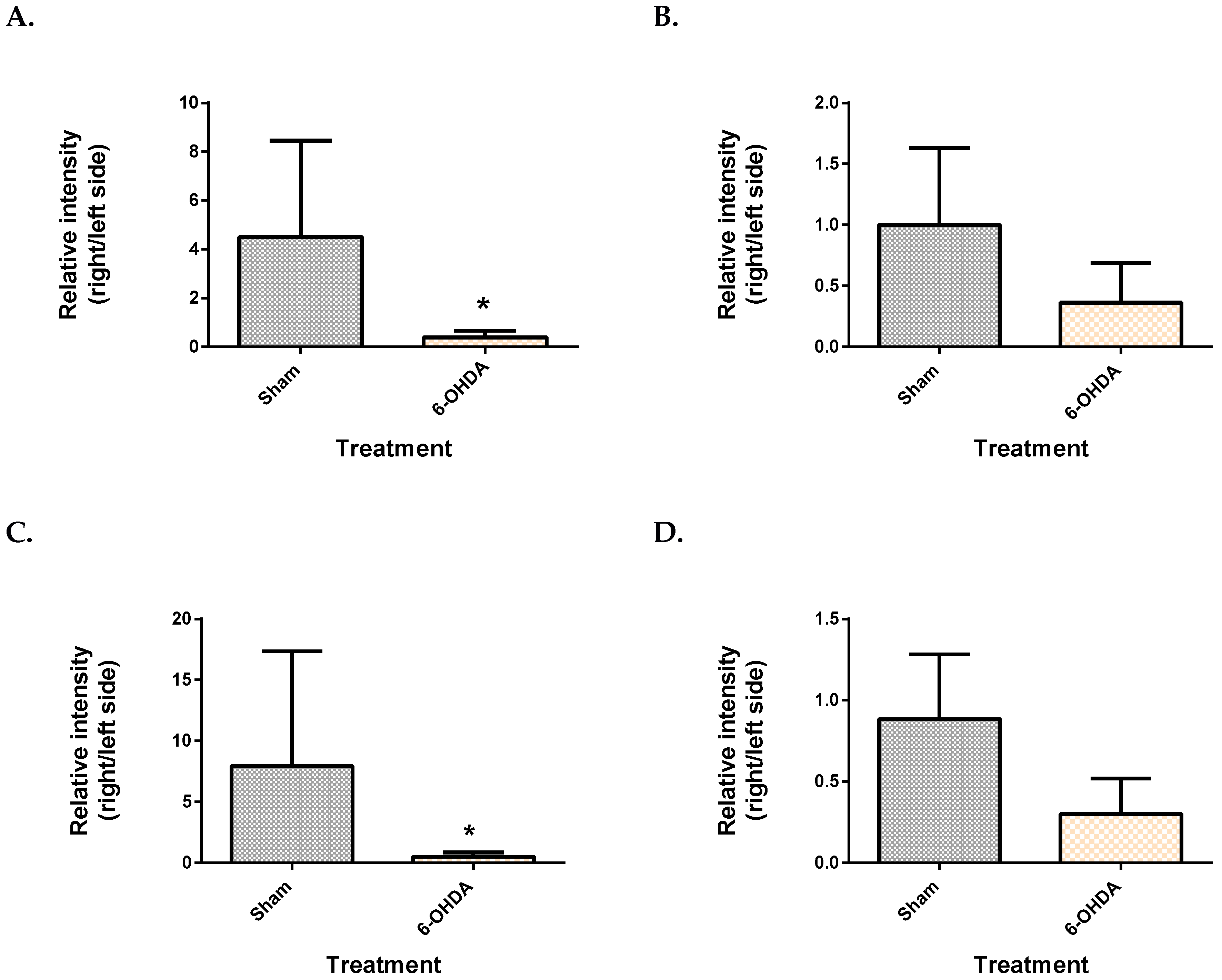
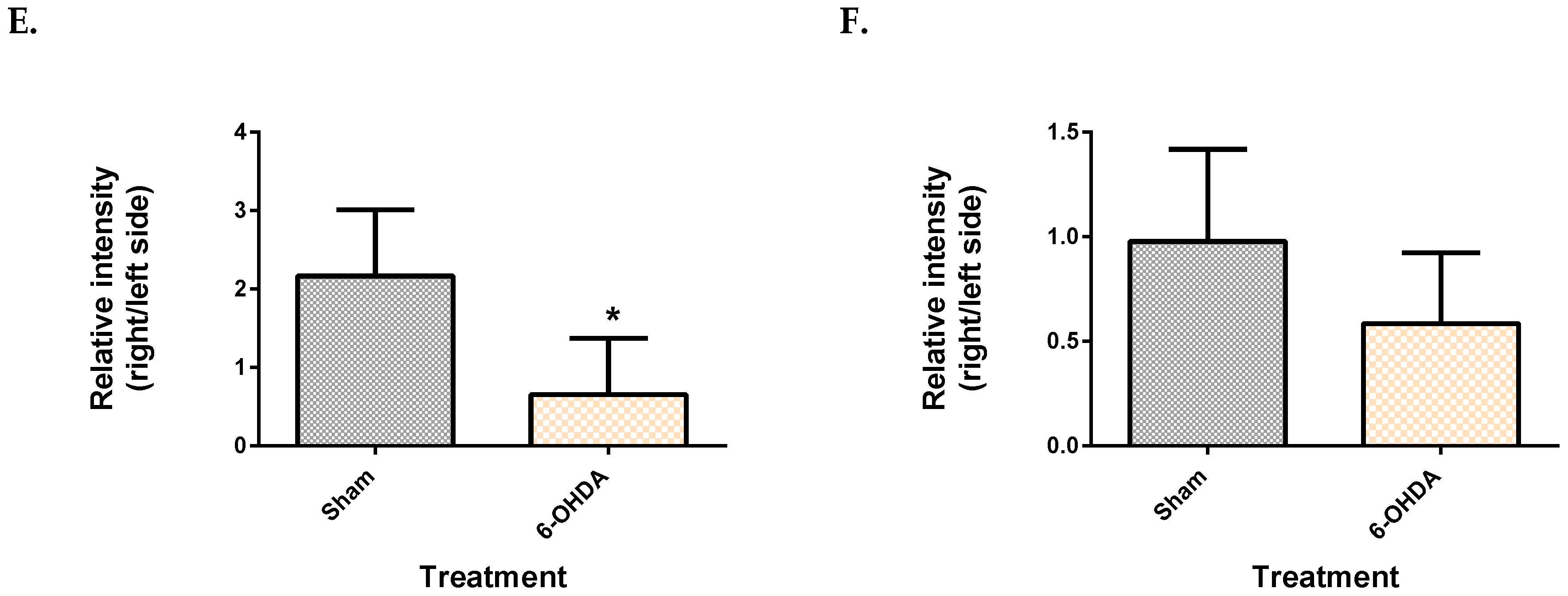
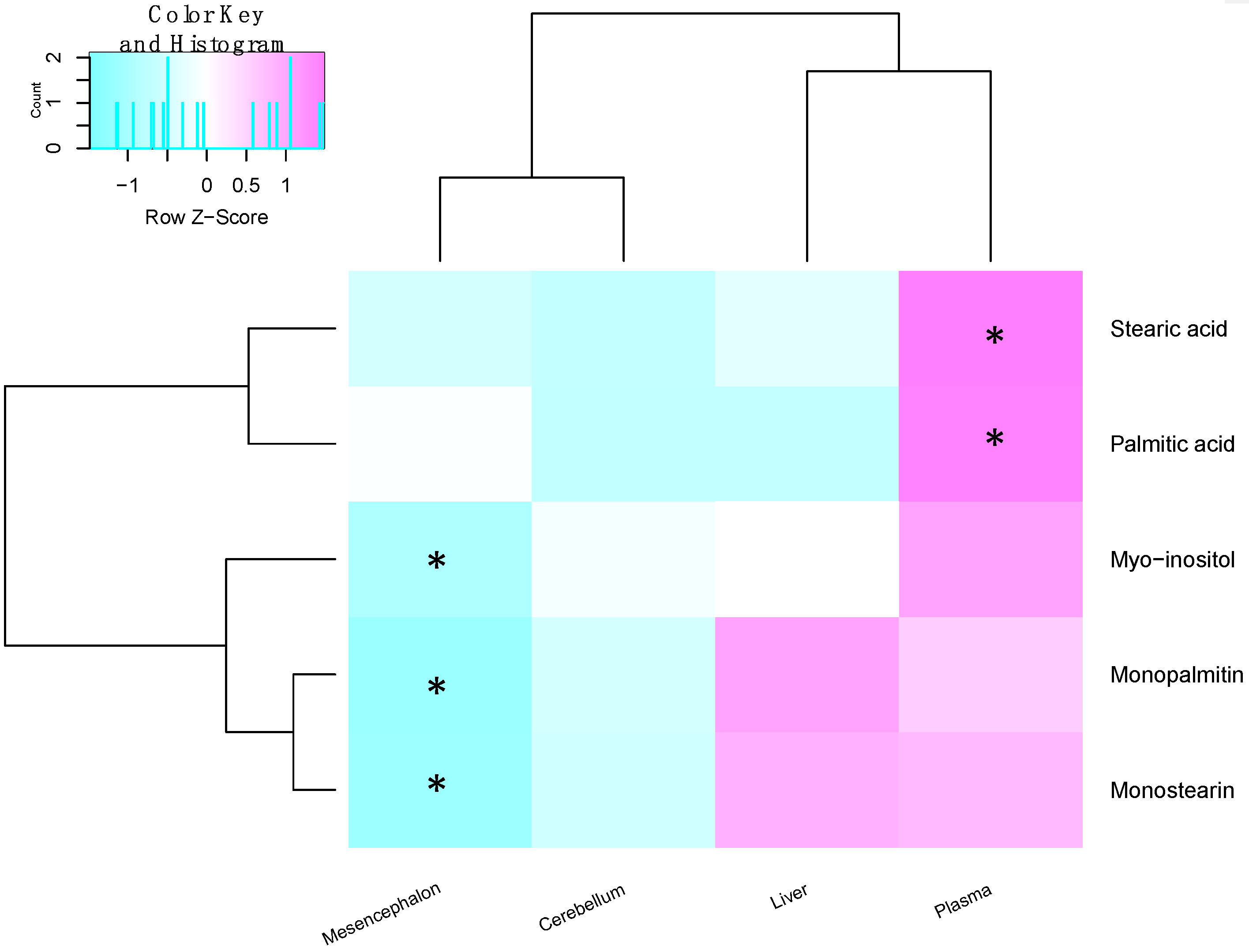
2.3. Metabolite Levels in the Plasma, Brain and Liver
2.4. Correlation of Plasma and Midbrain Features with Motor Dysfunction
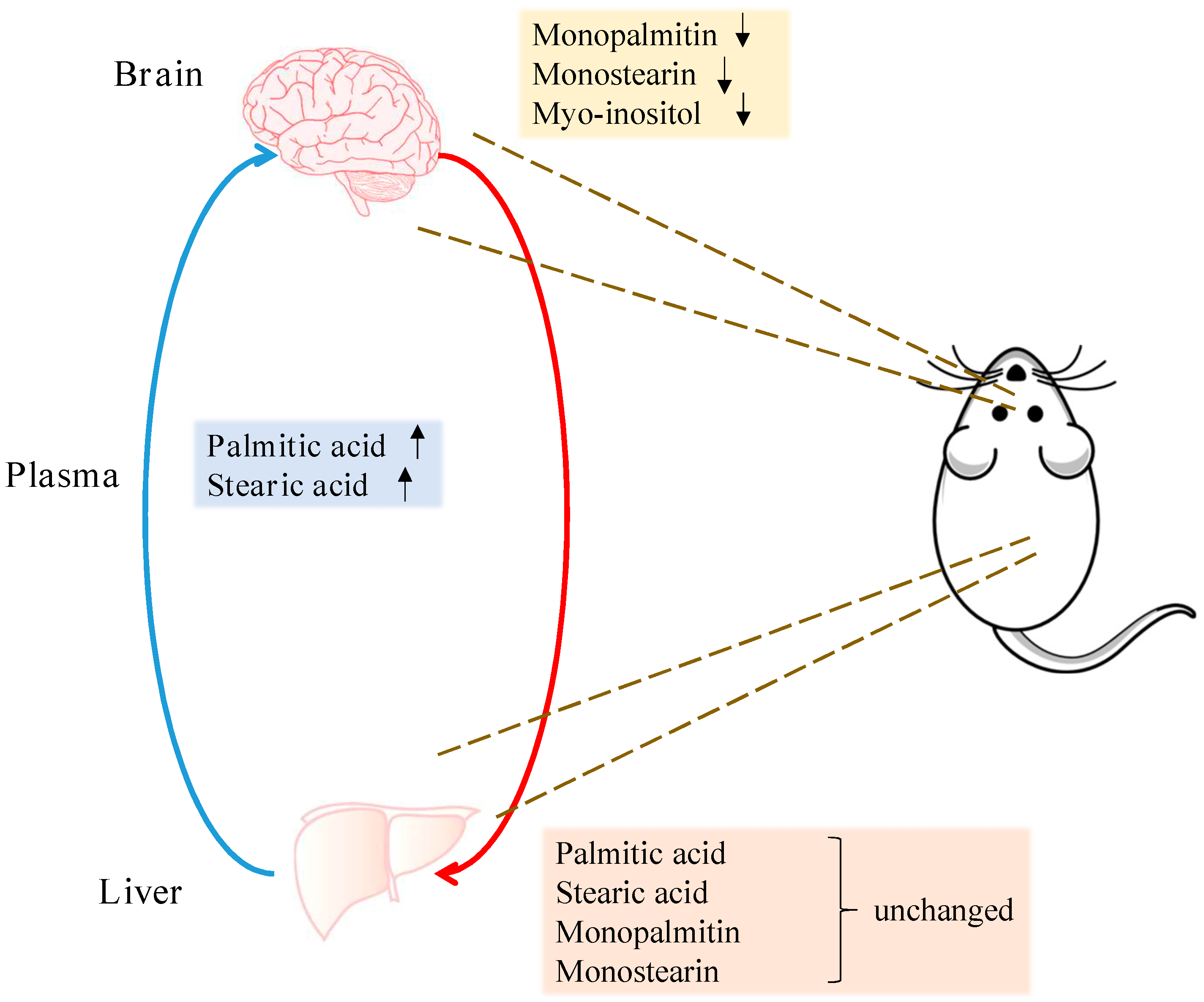
3. Discussion
4. Materials and Methods
4.1. Stereotactic Injection of 6-OHDA
4.2. Behavioural Assessment
4.3. Immunohistochemistry
4.4. Tissue Harvest
4.5. Sample Extraction for Metabolomics
4.6. Derivatization of Tissues for GC-MS Analysis
4.7. GC-MS Analysis
4.8. Data Processing and Metabolite Identification
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dorsey, E.R.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; Siderowf, A.; et al. Projected Number of People with Parkinson Disease in the Most Populous Nations, 2005 through 2030. Neurology 2007, 68, 384–386. [Google Scholar] [CrossRef] [PubMed]
- McCrone, P.; Allcock, L.M.; Burn, D.J. Predicting the Cost of Parkinson’s Disease. Mov. Disord. 2007, 22, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Kowal, S.L.; Dall, T.M.; Chakrabarti, R.; Storm, M.V.; Jain, A. The Current and Projected Economic Burden of Parkinson’s Disease in the United States. Mov. Disord. 2013, 28, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Lill, C.M. Genetics of Parkinson’s Disease. Mol. Cell. Probes 2016, 30, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, M.; Matson, W.R.; Wang, L.; Matson, T.; Saunders-Pullman, R.; Bressman, S.S.; Beal, M.F. Metabolomic Profiling to Develop Blood Biomarkers for Parkinson’s Disease. Brain 2008, 131, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Parkinson’s Disease: Clinical Features and Diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Houghton, D.; Lowes, H.; Pyle, A.; Nesbitt, S.; Yarnall, A.; Yu-Wai-Man, P.; Burn, D.J.; Santibanez-Koref, M.; Hudson, G. Metabolic Profiling of Parkinson’s Disease and Mild Cognitive Impairment. Mov. Disord. 2017, 32, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Lewitt, P.A.; Lu, M.; Auinger, P. Metabolomic biomarkers as strong correlates of Parkinson disease progression. Neurology 2017, 88, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Hatano, T.; Saiki, S.; Okuzumi, A.; Mohney, R.P.; Hattori, N. Identification of Novel Biomarkers for Parkinson’s Disease by Metabolomic Technologies. J. Neurol. Neurosurg. Psychiatry 2016, 87, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Luan, H.; Liu, L.F.; Tang, Z.; Zhang, M.; Chua, K.K.; Song, J.X.; Mok, V.C.T.; Li, M.; Cai, Z. Comprehensive Urinary Metabolomic Profiling and Identification of Potential Noninvasive Marker for Idiopathic Parkinson s Disease. Sci. Rep. 2015, 1–11. [Google Scholar]
- Öhman, A.; Forsgren, L. NMR Metabonomics of Cerebrospinal Fluid Distinguishes between Parkinson’s Disease and Controls. Neurosci. Lett. 2015, 594, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Trupp, M.; Jonsson, P.; Ohrfelt, A.; Zetterberg, H.; Obudulu, O.; Malm, L.; Wuolikainen, A.; Linder, J.; Moritz, T.; Blennow, K.; et al. Metabolite and Peptide Levels in Plasma and CSF Differentiating Healthy Controls from Patients with Newly Diagnosed Parkinson’s Disease. J. Parkinsons. Dis. 2014, 4, 549–560. [Google Scholar] [PubMed]
- Lewitt, P.A.; Li, J.; Lu, M.; Beach, T.G.; Adler, C.H.; Guo, L. 3-Hydroxykynurenine and Other Parkinson’s Disease Biomarkers Discovered by Metabolomic Analysis. Mov. Disord. 2013, 28, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Poliquin, P.O.; Chen, J.; Cloutier, M.; Trudeau, L.É.; Jolicoeur, M. Metabolomics and In-Silico Analysis Reveal Critical Energy Deregulations in Animal Models of Parkinson’s Disease. PLoS ONE 2013, 8, e69146. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Zhang, S.N.; Lu, F.; Liu, C.F.; Wang, Y.; Bai, Y.; Wang, N.; Liu, S.M. Cerebral Metabonomics Study on Parkinson’s Disease Mice Treated with Extract of Acanthopanax Senticosus Harms. Phytomedicine 2013, 20, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Farmer, K.; Smith, C.A.; Hayley, S.; Smith, J. Major Alterations of Phosphatidylcholine and Lysophosphotidylcholine Lipids in the Substantia Nigra Using an Early Stage Model of Parkinson’s Disease. Int. J. Mol. Sci. 2015, 16, 18865–18877. [Google Scholar] [CrossRef]
- Lu, Z.; Wang, J.; Li, M.; Liu, Q.; Wei, D.; Yang, M.; Kong, L. 1H NMR-Based Metabolomics Study on a Goldfish Model of Parkinson’s Disease Induced by 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP). Chem. Biol. Interact. 2014, 223, 18–26. [Google Scholar] [CrossRef]
- Tyurina, Y.Y.; Polimova, A.M.; Maciel, E.; Tyurin, V.A.; Kapralova, V.I.; Winnica, D.E.; Vikulina, A.S.; Rosario, M.; Mccoy, J.; Sanders, L.H.; et al. LC/MS analysis of cardiolipins in substantia nigra and plasma of rotenone-treated rats: implication for mitochondrial dysfunction in Parkinson’s disease. Free Radic. Res. 2015, 49, 681–691. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Kamp, F. How Are Free Fatty Acids Transported in Membranes? Diabetes 1999, 48, 2255–2269. [Google Scholar] [CrossRef]
- Spector, R. Fatty Acid Transport through the Blood–Brain Barrier. J. Neurochem. 1988, 50, 639–643. [Google Scholar] [CrossRef]
- Conner, W.E.; Lin, D.S.; Colvis, C. Differential Mobilization of Fatty Acids from Adipose Tissue. J. Lipid Res. 1996, 37, 290–298. [Google Scholar] [PubMed]
- Hellerstein, M.K.; Christiansen, M.; Kaempfer, S.; Kletke, C.; Wu, K.; Reid, J.S.; Mulligan, K.; Hellerstein, N.S.; Shackleton, C.H.L. Measurement of De Novo Hepatic Lipogenesis in Humans Using Stable Isotopes. J. Clin. Investig. 1991, 87, 1841–1852. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Wanders, R.J.A. A General Introduction to the Biochemistry of Mitochondrial Fatty Acid β-Oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef]
- Gu, M.; Owen, A.D.; Toffa, S.E.K.; Cooper, J.M.; Dexter, D.T.; Jenner, P.; Marsden, C.D.; Schapira, A.H.V. Mitochondrial Function, GSH and Iron in Neurodegeneration and Lewy Body Diseases. J. Neurol. Sci. 1998, 158, 24–29. [Google Scholar] [CrossRef]
- Park, H.-R.; Kim, J.-Y.; Park, K.-Y.; Lee, J.-W. Lipotoxicity of Palmitic Acid on Neural Progenitor Cells and Hippocampal Neurogenesis. Toxicol. Res. 2011, 27, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Claycombe, K.; Schommer, J.; Collins, D.; Ghribi, O. Palmitate-Induced Endoplasmic Reticulum Stress and Subsequent C/EBP α Homologous Protein Activation Attenuates Leptin and Insulin-like Growth Factor 1 Expression in the Brain. Cell. Signal. 2016, 28, 1789–1805. [Google Scholar] [CrossRef]
- Patil, S.; Chan, C. Palmitic and Stearic Fatty Acids Induce Alzheimer-like Hyperphosphorylation of Tau in Primary Rat Cortical Neurons. Neurosci. Lett. 2005, 384, 288–293. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, Y.; Fang, Q.; Zhong, P.; Li, W.; Wang, L.; Fu, W.; Zhang, Y.; Xu, Z.; Li, X.; et al. Saturated Palmitic Acid Induces Myocardial Inflammatory Injuries through Direct Binding to TLR4 Accessory Protein MD2. Nat. Commun. 2017, 8, 13997. [Google Scholar] [CrossRef]
- Grimsgaard, S.; Jacobsen, B.K.; Bjerve, K.S. Plasma saturated and linoleic fatty acids are Independently Associated with Blood Pressure. Hypertension 1999, 34, 478–483. [Google Scholar] [CrossRef]
- Nestel, P.; Clifton, P.; Noakes, M. Effects of Increasing Dietary Palmitoleic Acid Compared with Palmitic and Oleic Acids on Plasma Lipids of Hypercholesterolemic Men. J. Lipid Res. 1994, 35, 656–662. [Google Scholar]
- King, I.B.; Song, X.; Ma, W.; Wu, J.H.Y.; Wang, Q.; Lemaitre, R.N.; Mukamal, K.J.; Djousse, L.; Biggs, M.L.; Delaney, J.A.; et al. Prospective Association of Fatty Acids in the de Novo Lipogenesis Pathway with Risk of Type 2 Diabetes: The Cardiovascular Health Study 1–5. Am. J. Clin. Nutr. 2015, 101, 153–163. [Google Scholar]
- Mu, Y.M.; Yanase, T.; Nishi, Y.; Tanaka, A.; Saito, M.; Jin, C.H.; Mukasa, C.; Okabe, T.; Nomura, M.; Goto, K.; et al. Saturated FFAs, Palmitic Acid and Stearic Acid, Induce Apoptosis in Human Granulosa Cells. Endocrinology 2001, 142, 3590–3597. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; Méndez-Giménez, L.; Fernández-Formoso, J.A.; Fernández, S.; Rodríguez, A. Regulation of Adipocyte Lipolysis. Nutr. Res. Rev. 2014, 27, 63–93. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Jeong, I.H.; Kong, B.S.; Lee, J.E.; Kim, K.H.; Lee, D.Y.; Kim, H.J. Disease Type- and Status-Specific Alteration of CSF Metabolome Coordinated with Clinical Parameters in Inflammatory Demyelinating Diseases of CNS. PLoS ONE 2016, 11, e0166277. [Google Scholar] [CrossRef] [PubMed]
- Prestel, J.; Gempel, K.; Hauser, T.K.; Schweitzer, K.; Prokisch, H.; Ahting, U.; Freudenstein, D.; Bueltmann, E.; Naegele, T.; Berg, D.; et al. Clinical and Molecular Characterisation of a Parkinson Family with a Novel PINK1 Mutation. J. Neurol. 2008, 255, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Badar-Goffer, R.S.; Ben-Yoseph, O.; Bachelard, H.S.; Morris, P.G. Neuronal-Glial Metabolism under Depolarizing Conditions. A 13C-N.M.R. Study. Biochem. J. 1992, 282 (Pt 1), 225–230. [Google Scholar] [CrossRef]
- Berridge, M.J.; Taylor, C.W. Inositol Trisphosphate and Calcium Signaling. Cold Spring Harb. Symp. Quant. Biol. 1988, 53, 927–933. [Google Scholar] [CrossRef]
- Shukla, A.K.; Ratnasekhar, C.; Pragya, P.; Chaouhan, H.S.; Patel, D.K.; Chowdhuri, D.K.; Mudiam, M.K.R. Metabolomic Analysis Provides Insights on Paraquat-Induced Parkinson-Like Symptoms in Drosophila Melanogaster. Mol. Neurobiol. 2016, 53, 254–269. [Google Scholar] [CrossRef]
- Torres, E.M.; Lane, E.L.; Heuer, A.; Smith, G.A.; Murphy, E.; Dunnett, S.B. Increased Efficacy of the 6-Hydroxydopamine Lesion of the Median Forebrain Bundle in Small Rats, by Modification of the Stereotaxic Coordinates. J. Neurosci. Methods 2011, 200, 29–35. [Google Scholar] [CrossRef]
- Schallert, T.; Fleming, S.M.; Leasure, J.L.; Tillerson, J.L.; Bland, S.T. CNS Plasticity and Assessment of Forelimb Sensorimotor Outcome in Unilateral Rat Models of Stroke, Cortical Ablation, Parkinsonism and Spinal Cord Injury. Neuropharmacology 2000, 39, 777–787. [Google Scholar] [CrossRef]
- Chiu, K.; Lau, W.M.; Lau, H.T.; So, K.-F.; Chang, R.C.-C. Micro-Dissection of Rat Brain for RNA or Protein Extraction from Specific Brain Region. J. Vis. Exp. 2007, 269. [Google Scholar] [CrossRef] [PubMed]
- Whiley, L.; Godzien, J.; Ruperez, F.J.; Legido-Quigley, C.; Barbas, C. In-Vial Dual Extraction for Direct LC-MS Analysis of Plasma for Comprehensive and Highly Reproducible Metabolic Fingerprinting. Anal. Chem. 2012, 84, 5992–5999. [Google Scholar] [CrossRef] [PubMed]
- Ebshiana, A.A.; Snowden, S.G.; Thambisetty, M.; Parsons, R.; Hye, A.; Legido-Quigley, C. Metabolomic Method: UPLC-q-ToF Polar and Non-Polar Metabolites in the Healthy Rat Cerebellum Using an in-Vial Dual Extraction. PLoS ONE 2015, 10, e0122883. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Site | Spearman’s Correlation Coefficient | q-Value |
|---|---|---|---|
| Palmitic acid | Plasma | 0.674 | 0.035 |
| Stearic acid | Plasma | 0.649 | 0.027 |
| Monopalmitin | Midbrain | −0.578 | 0.07 |
| Monostearin | Midbrain | −0.439 | 0.205 |
| Myo-inositol | Midbrain | −0.205 | 0.438 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, A.; Han, P.; Wong, M.-Y.; Chang, R.C.-C.; Legido-Quigley, C. Palmitate and Stearate are Increased in the Plasma in a 6-OHDA Model of Parkinson’s Disease. Metabolites 2019, 9, 31. https://doi.org/10.3390/metabo9020031
Shah A, Han P, Wong M-Y, Chang RC-C, Legido-Quigley C. Palmitate and Stearate are Increased in the Plasma in a 6-OHDA Model of Parkinson’s Disease. Metabolites. 2019; 9(2):31. https://doi.org/10.3390/metabo9020031
Chicago/Turabian StyleShah, Anuri, Pei Han, Mung-Yee Wong, Raymond Chuen-Chung Chang, and Cristina Legido-Quigley. 2019. "Palmitate and Stearate are Increased in the Plasma in a 6-OHDA Model of Parkinson’s Disease" Metabolites 9, no. 2: 31. https://doi.org/10.3390/metabo9020031
APA StyleShah, A., Han, P., Wong, M.-Y., Chang, R. C.-C., & Legido-Quigley, C. (2019). Palmitate and Stearate are Increased in the Plasma in a 6-OHDA Model of Parkinson’s Disease. Metabolites, 9(2), 31. https://doi.org/10.3390/metabo9020031