HR-MAS NMR Based Quantitative Metabolomics in Breast Cancer

 ,
, 

Abstract
1. Introduction
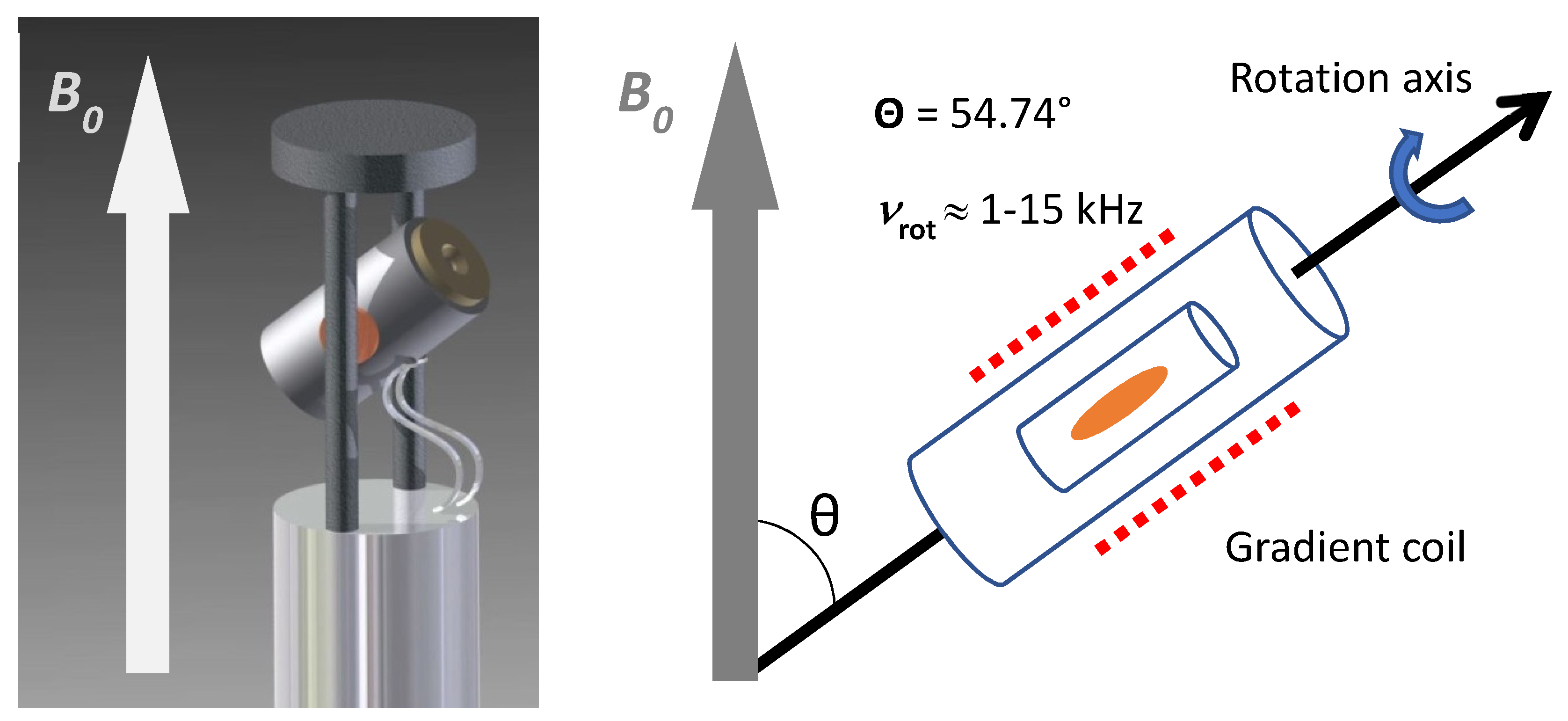
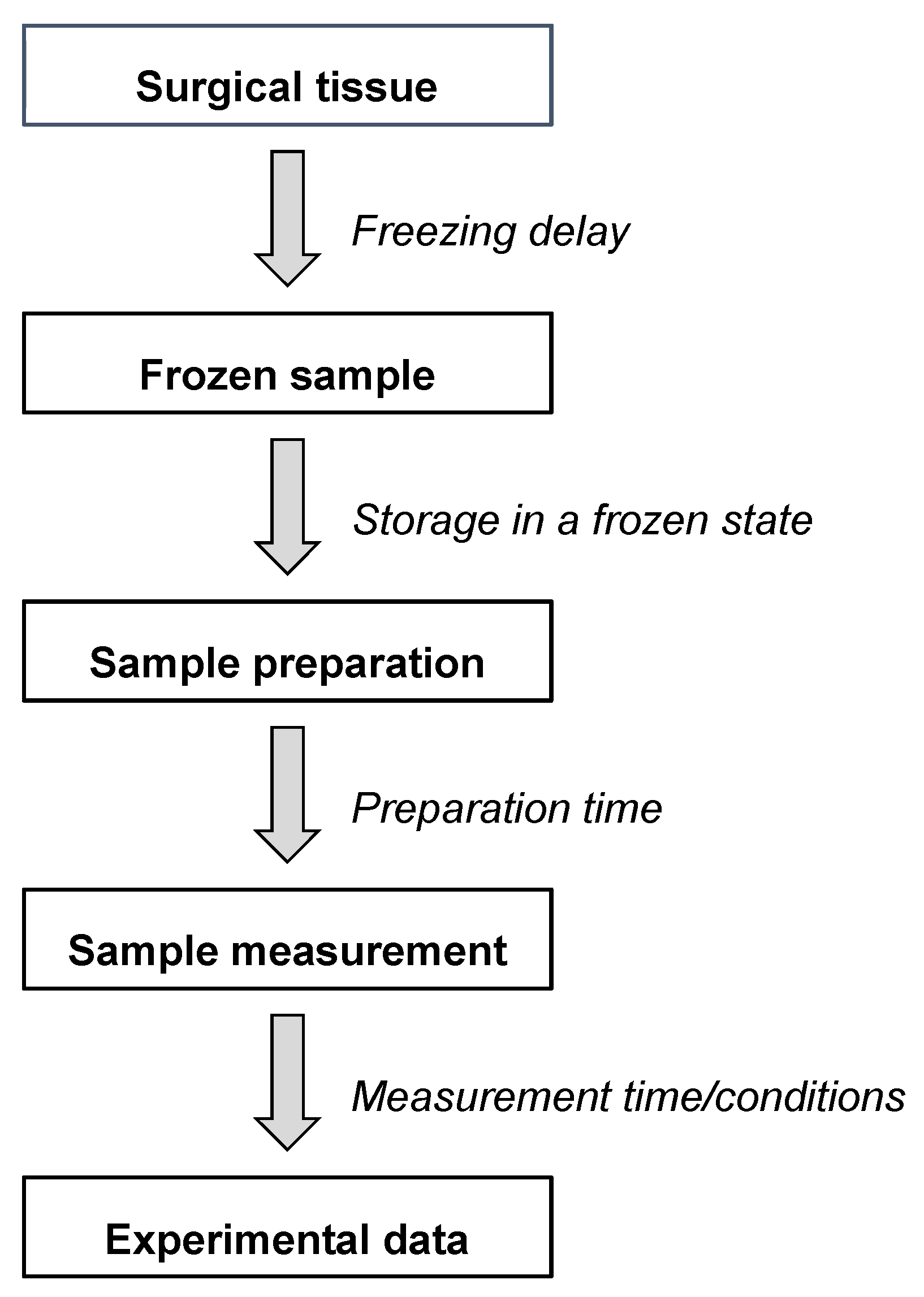
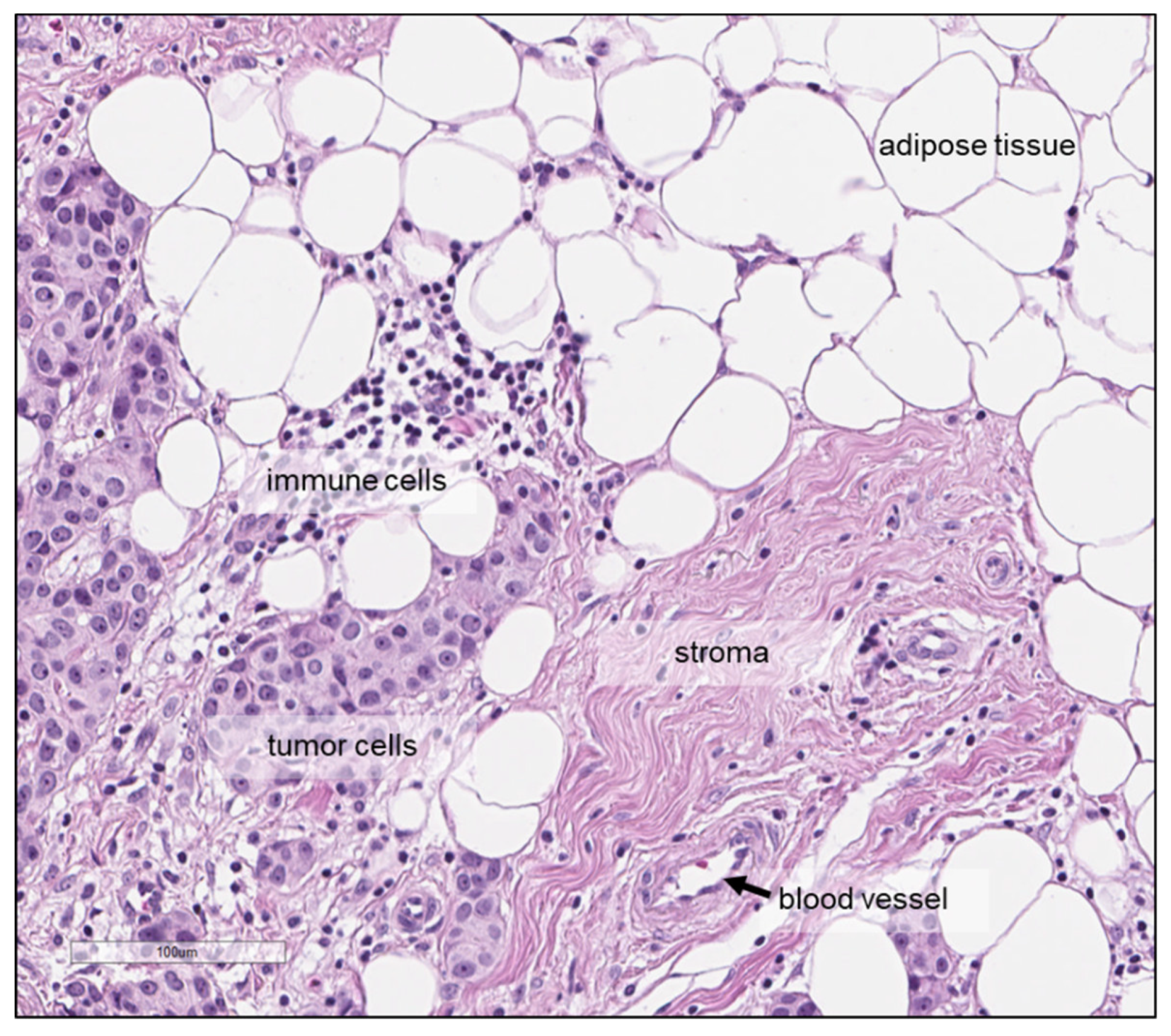
2. Preanalytical Factors and Measurement Conditions
3. NMR Techniques Employed in Tissue Analysis
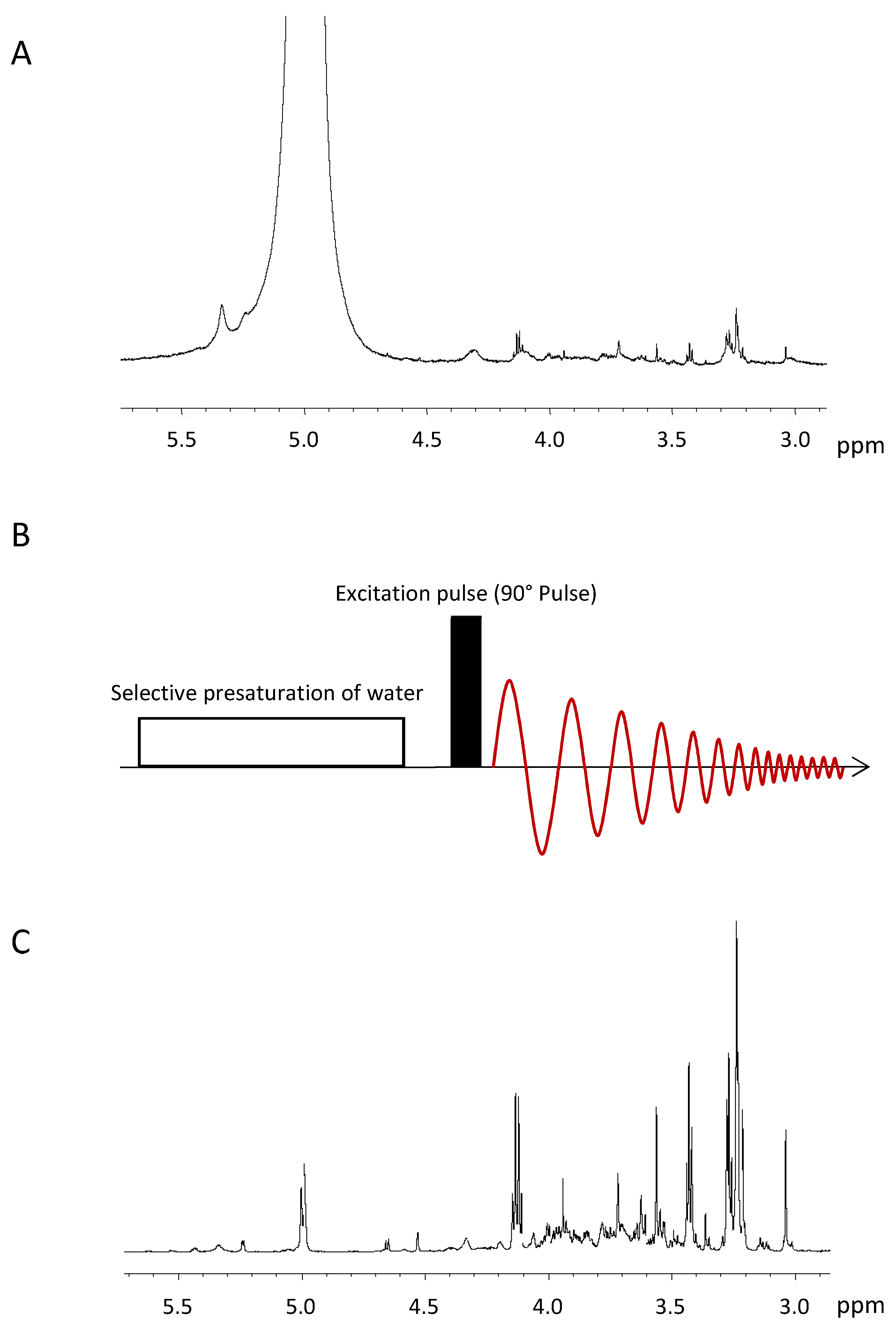
3.1. Water Suppression
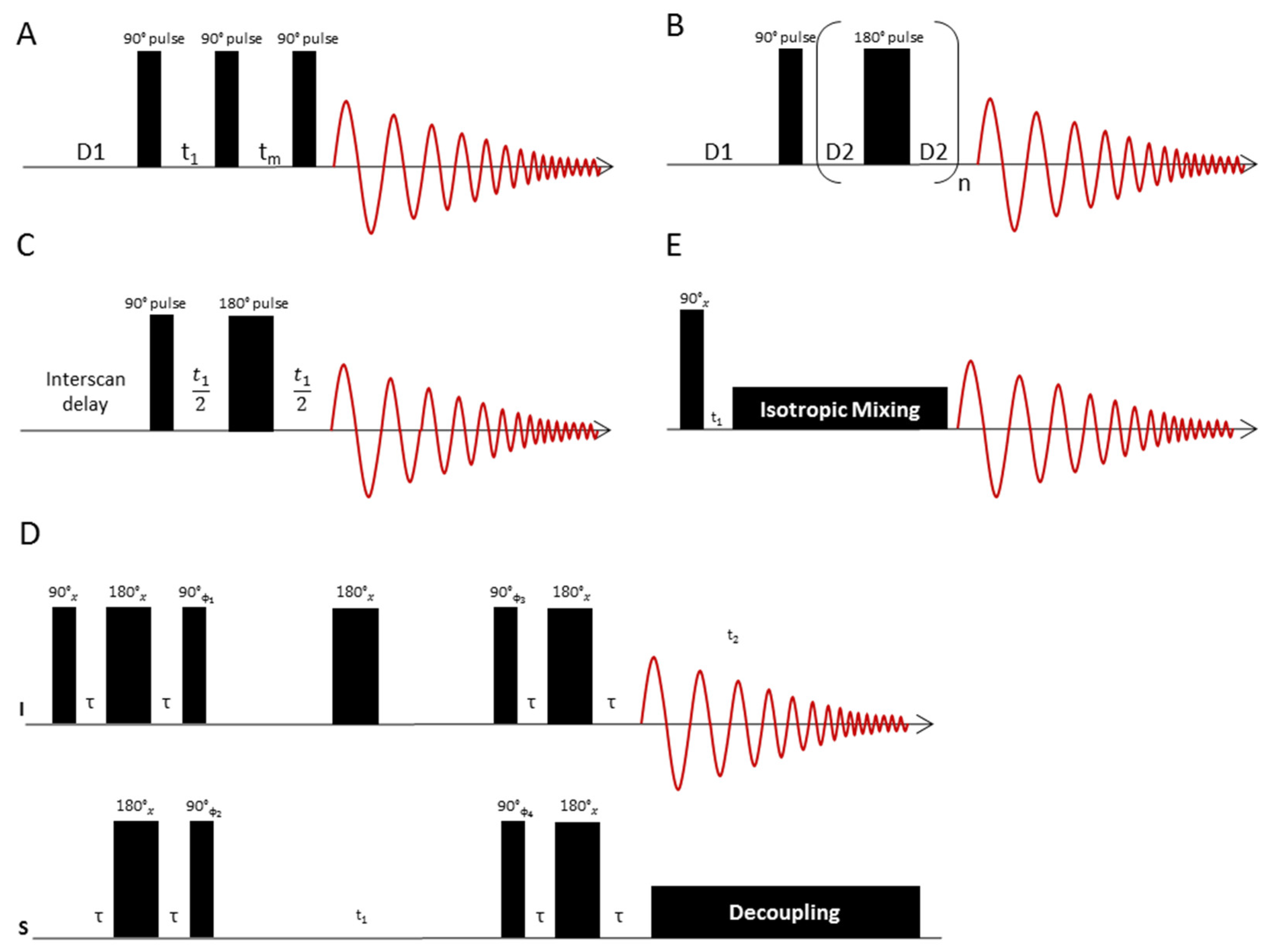
3.2. Pulse Sequences for 1D-NMR
3.3. Pulse Sequences for 2D-NMR
4. Metabolites Identified with HR-MAS NMR in Breast Tumour Tissue
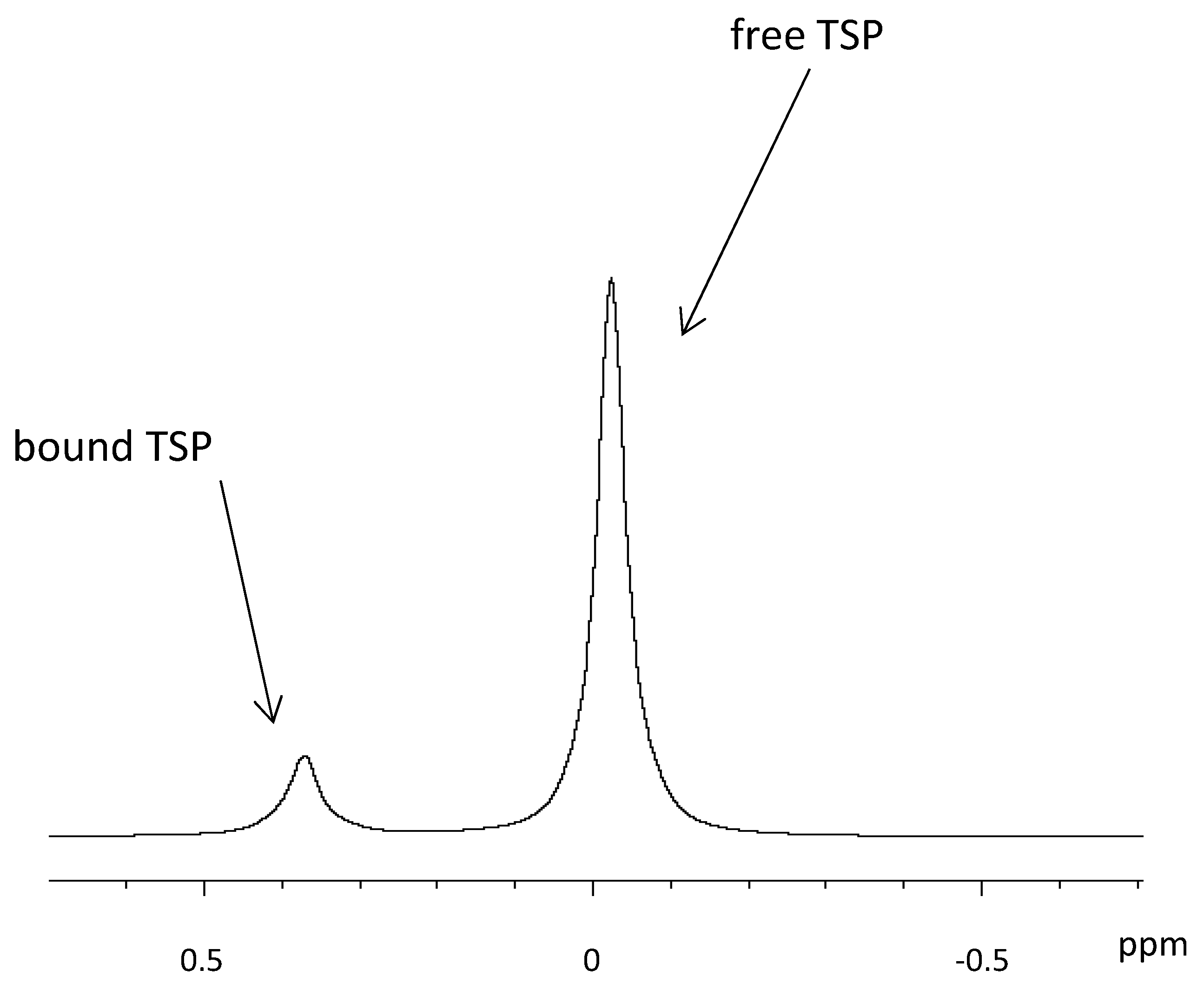
5. Metabolite Quantification with HR-MAS NMR
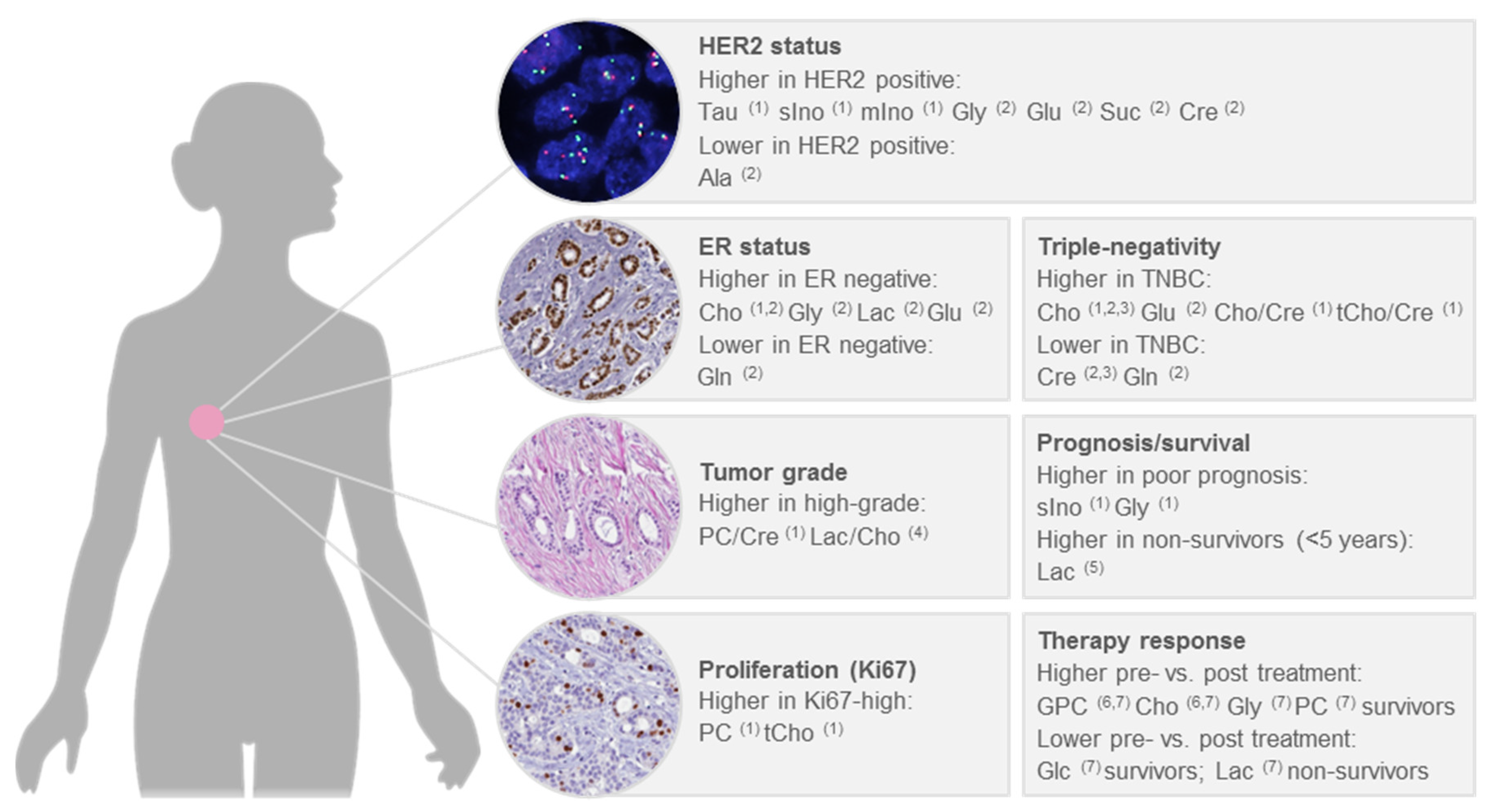
6. Significant Associations with Clinical Factors
7. Summary
Funding
Acknowledgments
Conflicts of Interest
References
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. New Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Filipits, M.; Rudas, M.; Jakesz, R.; Dubsky, P.; Fitzal, F.; Singer, C.F.; Dietze, O.; Greil, R.; Jelen, A.; Sevelda, P.; et al. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin. Cancer Res. An. Off. J. Am. Assoc. Cancer Res. 2011, 17, 6012–6020. [Google Scholar] [CrossRef] [PubMed]
- Sparano, J.A. Prognostic gene expression assays in breast cancer: Are two better than one? NPJ Breast Cancer 2018, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef]
- Nagana Gowda, G.A.; Raftery, D. Can NMR solve some significant challenges in metabolomics? J. Magn. Reson. (San Diego Calif 1997) 2015, 260, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Maniara, G.; Rajamoorthi, K.; Rajan, S.; Stockton, G.W. Method performance and validation for quantitative analysis by (1)h and (31)p NMR spectroscopy. Applications to analytical standards and agricultural chemicals. Anal. Chem. 1998, 70, 4921–4928. [Google Scholar] [CrossRef]
- Mountford, C.; Ramadan, S.; Stanwell, P.; Malycha, P. Proton MRS of the breast in the clinical setting. NMR Biomed. 2009, 22, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Beckonert, O.; Coen, M.; Keun, H.C.; Wang, Y.; Ebbels, T.M.D.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. High-resolution magic-angle-spinning NMR spectroscopy for metabolic profiling of intact tissues. Nat. Protoc. 2010, 5, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.W.; Jachmann, R.C.; Sakellariou, D.; Nielsen, U.G.; Pines, A. High-resolution nuclear magnetic resonance spectroscopy of biological tissues using projected magic angle spinning. Magn. Reson. Med. 2005, 54, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-H.; Enloe, B.M.; Xiao, Y.; Cory, D.G.; Singer, S. Isotropic susceptibility shift under MAS: The origin of the split water resonances in 1H MAS NMR spectra of cell suspensions. Magn. Reson. Med. 2003, 50, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Andrew, E.R.; Bradbury, A.; Eades, R.G. Removal of Dipolar Broadening of Nuclear Magnetic Resonance Spectra of Solids by Specimen Rotation. Nature 1959, 183, 1802–1803. [Google Scholar] [CrossRef]
- Lowe, I.J. Free Induction Decays of Rotating Solids. Phys. Rev. Lett. 1959, 2, 285–287. [Google Scholar] [CrossRef]
- Keifer, P.A.; Baltusis, L.; Rice, D.M.; Tymiak, A.A.; Shoolery, J.N. A Comparison of NMR Spectra Obtained for Solid-Phase-Synthesis Resins Using Conventional High-Resolution, Magic-Angle-Spinning, and High-Resolution Magic-Angle-Spinning Probes. J. Magn. Reson. Ser. A 1996, 119, 65–75. [Google Scholar] [CrossRef]
- Millis, K.K.; Maas, W.E.; Cory, D.G.; Singer, S. Gradient, high-resolution, magic-angle spinning nuclear magnetic resonance spectroscopy of human adipocyte tissue. Magn. Reson. Med. 1997, 38, 399–403. [Google Scholar] [CrossRef]
- Millis, K.; Weybright, P.; Campbell, N.; Fletcher, J.A.; Fletcher, C.D.; Cory, D.G.; Singer, S. Classification of human liposarcoma and lipoma using ex vivo proton NMR spectroscopy. Magn. Reson. Med. 1999, 41, 257–267. [Google Scholar] [CrossRef]
- Cheng, L.L.; Ma, M.J.; Becerra, L.; Ptak, T.; Tracey, I.; Lackner, A.; Gonzalez, R.G. Quantitative neuropathology by high resolution magic angle spinning proton magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. 1997, 94, 6408–6413. [Google Scholar] [CrossRef]
- Cheng, L.L.; Lean, C.L.; Bogdanova, A.; Wright, S.C.; Ackerman, J.L.; Brady, T.J.; Garrido, L. Enhanced resolution of proton NMR spectra of malignant lymph nodes using magic-angle spinning. Magn. Reson. Med. 1996, 36, 653–658. [Google Scholar] [CrossRef]
- Doty, F.D.; Entzminger, G.; Yang, Y.A. Magnetism in high-resolution NMR probe design. I: General methods. Concepts Magn. Reson. 1998, 10, 133–156. [Google Scholar] [CrossRef]
- Doty, F.D.; Entzminger, G.; Yang, Y.A. Magnetism in high-resolution NMR probe design. II: HR MAS. Concepts Magn. Reson. 1998, 10, 239–260. [Google Scholar] [CrossRef]
- Tosi, R.; Tugnoli, V. Nuclear Magnetic Resonance Spectroscopy in the Study of Neoplastic Tissue; Nova Science: New York, NY, USA, 2005. [Google Scholar]
- Sitter, B.; Sonnewald, U.; Spraul, M.; Fjösne, H.E.; Gribbestad, I.S. High-resolution magic angle spinning MRS of breast cancer tissue. NMR Biomed. 2002, 15, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Esteve, V.; Martínez-Granados, B.; Martínez-Bisbal, M.C. Pitfalls to be considered on the metabolomic analysis of biological samples by HR-MAS. Front. Chem. 2014, 2, 33. [Google Scholar] [CrossRef]
- Righi, V.; Schenetti, L.; Maiorana, A.; Libertini, E.; Bettelli, S.; Bonetti, L.R.; Mucci, A. Assessment of freezing effects and diagnostic potential of BioBank healthy and neoplastic breast tissues through HR-MAS NMR spectroscopy. Metabol. Off. J. Metabol. Soc. 2015, 11, 487–498. [Google Scholar] [CrossRef]
- Haukaas, T.H.; Moestue, S.A.; Vettukattil, R.; Sitter, B.; Lamichhane, S.; Segura, R.; Giskeødegård, G.F.; Bathen, T.F. Impact of Freezing Delay Time on Tissue Samples for Metabolomic Studies. Front. Oncol. 2016, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Opstad, K.S.; Bell, B.A.; Griffiths, J.R.; Howe, F.A. An investigation of human brain tumour lipids by high-resolution magic angle spinning 1H MRS and histological analysis. NMR Biomed. 2008, 21, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Middleton, D.A.; Bradley, D.P.; Connor, S.C.; Mullins, P.G.; Reid, D.G. The effect of sample freezing on proton magic-angle spinning NMR spectra of biological tissue. Magn. Reson. Med. 1998, 40, 166–169. [Google Scholar] [CrossRef]
- Waters, N.J.; Garrod, S.; Farrant, R.D.; Haselden, J.N.; Connor, S.C.; Connelly, J.; Lindon, J.C.; Holmes, E.; Nicholson, J.K. High-resolution magic angle spinning (1)H NMR spectroscopy of intact liver and kidney: Optimization of sample preparation procedures and biochemical stability of tissue during spectral acquisition. Anal. Biochem. 2000, 282, 16–23. [Google Scholar] [CrossRef]
- Shabihkhani, M.; Lucey, G.M.; Wei, B.; Mareninov, S.; Lou, J.J.; Vinters, H.V.; Singer, E.J.; Cloughesy, T.F.; Yong, W.H. The procurement, storage, and quality assurance of frozen blood and tissue biospecimens in pathology, biorepository, and biobank settings. Clin. Biochem. 2014, 47, 258–266. [Google Scholar] [CrossRef]
- Jordan, K.W.; He, W.; Halpern, E.F.; Wu, C.-L.; Cheng, L.L. Evaluation of Tissue Metabolites with High Resolution Magic Angle Spinning MR Spectroscopy Human Prostate Samples after Three-Year Storage at −80 °C. BiomarkInsights 2017, 2, 117727190700200. [Google Scholar] [CrossRef]
- Giskeødegård, G.F.; Cao, M.D.; Bathen, T.F. High-resolution magic-angle-spinning NMR spectroscopy of intact tissue. Methods Mol. Biol. (Clifton, NJ) 2015, 1277, 37–50. [Google Scholar] [CrossRef]
- Bertilsson, H.; Angelsen, A.; Viset, T.; Skogseth, H.; Tessem, M.-B.; Halgunset, J. A new method to provide a fresh frozen prostate slice suitable for gene expression study and MR spectroscopy. Prostate 2011, 71, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Gogiashvili, M.; Horsch, S.; Marchan, R.; Gianmoena, K.; Cadenas, C.; Tanner, B.; Naumann, S.; Ersova, D.; Lippek, F.; Rahnenführer, J.; et al. Impact of intratumoral heterogeneity of breast cancer tissue on quantitative metabolomics using high-resolution magic angle spinning 1 H NMR spectroscopy. NMR Biomed. 2018, 31. [Google Scholar] [CrossRef] [PubMed]
- Gogiashvili, M.; Edlund, K.; Gianmoena, K.; Marchan, R.; Brik, A.; Andersson, J.T.; Lambert, J.; Madjar, K.; Hellwig, B.; Rahnenführer, J.; et al. Metabolic profiling of ob/ob mouse fatty liver using HR-MAS 1H-NMR combined with gene expression analysis reveals alterations in betaine metabolism and the transsulfuration pathway. Anal. Bioanal. Chem. 2017, 409, 1591–1606. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.D.; Giskeødegård, G.F.; Bathen, T.F.; Sitter, B.; Bofin, A.; Lønning, P.E.; Lundgren, S.; Gribbestad, I.S. Prognostic value of metabolic response in breast cancer patients receiving neoadjuvant chemotherapy. BMC Cancer 2012, 12, 39. [Google Scholar] [CrossRef]
- Cao, M.D.; Sitter, B.; Bathen, T.F.; Bofin, A.; Lønning, P.E.; Lundgren, S.; Gribbestad, I.S. Predicting long-term survival and treatment response in breast cancer patients receiving neoadjuvant chemotherapy by MR metabolic profiling. NMR Biomed. 2012, 25, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Sitter, B.; Bathen, T.F.; Singstad, T.E.; Fjøsne, H.E.; Lundgren, S.; Halgunset, J.; Gribbestad, I.S. Quantification of metabolites in breast cancer patients with different clinical prognosis using HR MAS MR spectroscopy. NMR Biomed. 2010, 23, 424–431. [Google Scholar] [CrossRef]
- Moestue, S.A.; Borgan, E.; Huuse, E.M.; Lindholm, E.M.; Sitter, B.; Børresen-Dale, A.-L.; Engebraaten, O.; Maelandsmo, G.M.; Gribbestad, I.S. Distinct choline metabolic profiles are associated with differences in gene expression for basal-like and luminal-like breast cancer xenograft models. BMC Cancer 2010, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Sitter, B.; Lundgren, S.; Bathen, T.F.; Halgunset, J.; Fjosne, H.E.; Gribbestad, I.S. Comparison of HR MAS MR spectroscopic profiles of breast cancer tissue with clinical parameters. NMR Biomed. 2006, 19, 30–40. [Google Scholar] [CrossRef]
- Grinde, M.T.; Skrbo, N.; Moestue, S.A.; Rødland, E.A.; Borgan, E.; Kristian, A.; Sitter, B.; Bathen, T.F.; Børresen-Dale, A.-L.; Mælandsmo, G.M.; et al. Interplay of choline metabolites and genes in patient-derived breast cancer xenografts. Breast Cancer Res. BCR 2014, 16, R5. [Google Scholar] [CrossRef] [PubMed]
- Euceda, L.R.; Haukaas, T.H.; Giskeødegård, G.F.; Vettukattil, R.; Engel, J.; Silwal-Pandit, L.; Lundgren, S.; Borgen, E.; Garred, Ø.; Postma, G.; et al. Evaluation of metabolomic changes during neoadjuvant chemotherapy combined with bevacizumab in breast cancer using MR spectroscopy. Metabol. Off. J. Metabol. Soc. 2017, 13, 80. [Google Scholar] [CrossRef]
- Euceda, L.R.; Hill, D.K.; Stokke, E.; Hatem, R.; El Botty, R.; Bièche, I.; Marangoni, E.; Bathen, T.F.; Moestue, S.A. Metabolic Response to Everolimus in Patient-Derived Triple-Negative Breast Cancer Xenografts. J. Proteome Res. 2017, 16, 1868–1879. [Google Scholar] [CrossRef] [PubMed]
- Haukaas, T.H.; Euceda, L.R.; Giskeødegård, G.F.; Lamichhane, S.; Krohn, M.; Jernström, S.; Aure, M.R.; Lingjærde, O.C.; Schlichting, E.; Garred, Ø.; et al. Metabolic clusters of breast cancer in relation to gene- and protein expression subtypes. Cancer Metabol. 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Bathen, T.F.; Jensen, L.R.; Sitter, B.; Fjösne, H.E.; Halgunset, J.; Axelson, D.E.; Gribbestad, I.S.; Lundgren, S. MR-determined metabolic phenotype of breast cancer in prediction of lymphatic spread, grade, and hormone status. Breast Cancer Res. Treat. 2007, 104, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Baek, H.-M.; Kim, S.; Kim, M.J.; Youk, J.H.; Moon, H.J.; Kim, E.-K.; Han, K.H.; Kim, D.-H.; Kim, S.I.; et al. HR-MAS MR spectroscopy of breast cancer tissue obtained with core needle biopsy: Correlation with prognostic factors. PLoS ONE 2012, 7, e51712. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Baek, H.-M.; Kim, S.; Kim, M.J.; Youk, J.H.; Moon, H.J.; Kim, E.-K.; Nam, Y.K. Magnetic resonance metabolic profiling of breast cancer tissue obtained with core needle biopsy for predicting pathologic response to neoadjuvant chemotherapy. PLoS ONE 2013, 8, e83866. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.L.; Chang, I.W.; Smith, B.L.; Gonzalez, R.G. Evaluating human breast ductal carcinomas with high-resolution magic-angle spinning proton magnetic resonance spectroscopy. J. Magn. Reson. (San Diego Calif 1997) 1998, 135, 194–202. [Google Scholar] [CrossRef]
- Park, V.Y.; Yoon, D.; Koo, J.S.; Kim, E.-K.; Kim, S.I.; Choi, J.S.; Park, S.; Park, H.S.; Kim, S.; Kim, M.J. Intratumoral Agreement of High-Resolution Magic Angle Spinning Magnetic Resonance Spectroscopic Profiles in the Metabolic Characterization of Breast Cancer. Medicine 2016, 95, e3398. [Google Scholar] [CrossRef]
- Li, M.; Song, Y.; Cho, N.; Chang, J.M.; Koo, H.R.; Yi, A.; Kim, H.; Park, S.; Moon, W.K. An HR-MAS MR metabolomics study on breast tissues obtained with core needle biopsy. PLoS ONE 2011, 6, e25563. [Google Scholar] [CrossRef]
- Taylor, J.L.; Wu, C.-L.; Cory, D.; Gonzalez, R.G.; Bielecki, A.; Cheng, L.L. High-resolution magic angle spinning proton NMR analysis of human prostate tissue with slow spinning rates. Magn. Reson. Med. 2003, 50, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Weybright, P.; Millis, K.; Campbell, N.; Cory, D.G.; Singer, S. Gradient, high-resolution, magic angle spinning1H nuclear magnetic resonance spectroscopy of intact cells. Magn. Reson. Med. 1998, 39, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Aime, S.; Bruno, E.; Cabella, C.; Colombatto, S.; Digilio, G.; Mainero, V. HR-MAS of cells: A “cellular water shift” due to water-protein interactions? Magn. Reson. Med. 2005, 54, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- André, M.; Dumez, J.-N.; Rezig, L.; Shintu, L.; Piotto, M.; Caldarelli, S. Complete protocol for slow-spinning high-resolution magic-angle spinning NMR analysis of fragile tissues. Anal. Chem. 2014, 86, 10749–10754. [Google Scholar] [CrossRef]
- Chae, E.Y.; Shin, H.J.; Kim, S.; Baek, H.-M.; Yoon, D.; Kim, S.; Shim, Y.E.; Kim, H.H.; Cha, J.H.; Choi, W.J.; et al. The Role of High-Resolution Magic Angle Spinning 1H Nuclear Magnetic Resonance Spectroscopy for Predicting the Invasive Component in Patients with Ductal Carcinoma In Situ Diagnosed on Preoperative Biopsy. PLoS ONE 2016, 11, e0161038. [Google Scholar] [CrossRef] [PubMed]
- Renault, M.; Shintu, L.; Piotto, M.; Caldarelli, S. Slow-spinning low-sideband HR-MAS NMR spectroscopy: Delicate analysis of biological samples. Sci. Rep. 2013, 3, 3349. [Google Scholar] [CrossRef] [PubMed]
- Hoult, D.I. Solvent peak saturation with single phase and quadrature fourier transformation. J. Magn. Reson. (1969) 1976, 21, 337–347. [Google Scholar] [CrossRef]
- Tzika, A.A.; Cheng, L.L.; Goumnerova, L.; Madsen, J.R.; Zurakowski, D.; Astrakas, L.G.; Zarifi, M.K.; Scott, R.M.; Anthony, D.C.; Gonzalez, R.G.; et al. Biochemical characterization of pediatric brain tumors by using in vivo and ex vivo magnetic resonance spectroscopy. J. Neurosurg. 2002, 96, 1023–1031. [Google Scholar] [CrossRef]
- Cheng, L.L.; Chang, I.W.; Louis, D.N.; Gonzalez, R.G. Correlation of high-resolution magic angle spinning proton magnetic resonance spectroscopy with histopathology of intact human brain tumor specimens. Cancer Res. 1998, 58, 1825–1832. [Google Scholar]
- Barton, S.J.; Howe, F.A.; Tomlins, A.M.; Cudlip, S.A.; Nicholson, J.K.; Anthony Bell, B.; Griffiths, J.R. Comparison of in vivo1H MRS of human brain tumours with1H HR-MAS spectroscopy of intact biopsy samples in vitro. MAGMA 1999, 8, 121–128. [Google Scholar] [CrossRef]
- Ludwig, C.; Viant, M.R. Two-dimensional J-resolved NMR spectroscopy: Review of a key methodology in the metabolomics toolbox. Phytochem. Anal. PCA 2010, 21, 22–32. [Google Scholar] [CrossRef]
- Palmer, A.G.; Cavanagh, J.; Wright, P.E.; Rance, M. Sensitivity improvement in proton-detected two-dimensional heteronuclear correlation NMR spectroscopy. J. Magn. Reson. (1969) 1991, 93, 151–170. [Google Scholar] [CrossRef]
- Morris, G.A.; Freeman, R. Enhancement of nuclear magnetic resonance signals by polarization transfer. J. Am. Chem. Soc. 1979, 101, 760–762. [Google Scholar] [CrossRef]
- Ravikumar, M.; Bothner-By, A.A. A two-dimensional NMR experiment for the correlation of spin-locked and free-precession frequencies. J. Am. Chem. Soc. 1993, 115, 7537–7538. [Google Scholar] [CrossRef]
- Kupce, E.; Keifer, P.A.; Delepierre, M. Adiabatic TOCSY MAS in liquids. J. Magn. Reson. (San Diego Calif 1997) 2001, 148, 115–120. [Google Scholar] [CrossRef]
- Wieruszeski, J.M.; Montagne, G.; Chessari, G.; Rousselot-Pailley, P.; Lippens, G. Rotor synchronization of radiofrequency and gradient pulses in high-resolution magic angle spinning NMR. J. Magn. Reson. (San Diego Calif 1997) 2001, 152, 95–102. [Google Scholar] [CrossRef]
- Yoon, H.; Yoon, D.; Yun, M.; Choi, J.S.; Park, V.Y.; Kim, E.-K.; Jeong, J.; Koo, J.S.; Yoon, J.H.; Moon, H.J.; et al. Metabolomics of Breast Cancer Using High-Resolution Magic Angle Spinning Magnetic Resonance Spectroscopy: Correlations with 18F-FDG Positron Emission Tomography-Computed Tomography, Dynamic Contrast-Enhanced and Diffusion-Weighted Imaging MRI. PLoS ONE 2016, 11, e0159949. [Google Scholar] [CrossRef]
- Antzutkin, O.N.; Shekar, S.C.; Levitt, M.H. Two-Dimensional Sideband Separation in Magic-Angle-Spinning NMR. J. Magn. Reson. Ser. A 1995, 115, 7–19. [Google Scholar] [CrossRef]
- Hu, J.Z.; Wang, W.; Liu, F.; Solum, M.S.; Alderman, D.W.; Pugmire, R.J.; Grant, D.M. Magic-Angle-Turning Experiments for Measuring Chemical-Shift-Tensor Principal Values in Powdered Solids. J. Magn. Reson. Ser. A 1995, 113, 210–222. [Google Scholar] [CrossRef]
- Aguilar, J.A.; Nilsson, M.; Bodenhausen, G.; Morris, G.A. Spin echo NMR spectra without J modulation. Chem. Commun. (Camb. Engl.) 2012, 48, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, M.; Bathen, T.F.; Engebråten, O.; Mælandsmo, G.M.; Gribbestad, I.S.; Moestue, S.A. Quantitative (31)P HR-MAS MR spectroscopy for detection of response to PI3K/mTOR inhibition in breast cancer xenografts. Magn. Reson. Med. 2014, 71, 1973–1981. [Google Scholar] [CrossRef]
- Swanson, M.G.; Zektzer, A.S.; Tabatabai, Z.L.; Simko, J.; Jarso, S.; Keshari, K.R.; Schmitt, L.; Carroll, P.R.; Shinohara, K.; Vigneron, D.B.; et al. Quantitative analysis of prostate metabolites using 1H HR-MAS spectroscopy. Magn. Reson. Med. 2006, 55, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Bisbal, M.C.; Monleon, D.; Assemat, O.; Piotto, M.; Piquer, J.; Llácer, J.L.; Celda, B. Determination of metabolite concentrations in human brain tumour biopsy samples using HR-MAS and ERETIC measurements. NMR Biomed. 2009, 22, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Kriat, M.; Confort-Gouny, S.; Vion-Dury, J.; Sciaky, M.; Viout, P.; Cozzone, P.J. Quantitation of metabolites in human blood serum by proton magnetic resonance spectroscopy. A comparative study of the use of formate and TSP as concentration standards. NMR Biomed. 1992, 5, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Albers, M.J.; Butler, T.N.; Rahwa, I.; Bao, N.; Keshari, K.R.; Swanson, M.G.; Kurhanewicz, J. Evaluation of the ERETIC method as an improved quantitative reference for 1H HR-MAS spectroscopy of prostate tissue. Magn. Reson. Med. 2009, 61, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Kostidis, S.; Addie, R.D.; Morreau, H.; Mayboroda, O.A.; Giera, M. Quantitative NMR analysis of intra- and extracellular metabolism of mammalian cells: A tutorial. Anal. Chim. Acta 2017, 980, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Nowick, J.S.; Khakshoor, O.; Hashemzadeh, M.; Brower, J.O. DSA: A new internal standard for NMR studies in aqueous solution. Org. Lett. 2003, 5, 3511–3513. [Google Scholar] [CrossRef] [PubMed]
- Alum, M.F.; Shaw, P.A.; Sweatman, B.C.; Ubhi, B.K.; Haselden, J.N.; Connor, S.C. 4,4-Dimethyl-4-silapentane-1-ammonium trifluoroacetate (DSA), a promising universal internal standard for NMR-based metabolic profiling studies of biofluids, including blood plasma and serum. Metabol. Off. J. Metabol. Soc. 2008, 4, 122–127. [Google Scholar] [CrossRef]
- Barker, P.B.; Soher, B.J.; Blackband, S.J.; Chatham, J.C.; Mathews, V.P.; Bryan, R.N. Quantitation of proton NMR spectra of the human brain using tissue water as an internal concentration reference. NMR Biomed. 1993, 6, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Barantin, L.; Le Pape, A.; Akoka, S. A new method for absolute quantitation MRS metabolites. Magn. Reson. Med. 1997, 38, 179–182. [Google Scholar] [CrossRef]
- Akoka, S.; Barantin, L.; Trierweiler, M. Concentration Measurement by Proton NMR Using the ERETIC Method. Anal. Chem. 1999, 71, 2554–2557. [Google Scholar] [CrossRef] [PubMed]
- Wider, G.; Dreier, L. Measuring protein concentrations by NMR spectroscopy. J. Am. Chem. Soc. 2006, 128, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Borgan, E.; Sitter, B.; Lingjærde, O.C.; Johnsen, H.; Lundgren, S.; Bathen, T.F.; Sørlie, T.; Børresen-Dale, A.-L.; Gribbestad, I.S. Merging transcriptomics and metabolomics—Advances in breast cancer profiling. BMC Cancer 2010, 10, 628. [Google Scholar] [CrossRef] [PubMed]
- Giskeødegård, G.F.; Lundgren, S.; Sitter, B.; Fjøsne, H.E.; Postma, G.; Buydens, L.M.C.; Gribbestad, I.S.; Bathen, T.F. Lactate and glycine-potential MR biomarkers of prognosis in estrogen receptor-positive breast cancers. NMR Biomed. 2012, 25, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- He, Q.H.; Shungu, D.C.; Vanzijl, P.C.M.; Bhujwalla, Z.M.; Glickson, J.D. Single-Scan in Vivo Lactate Editing with Complete Lipid and Water Suppression by Selective Multiple-Quantum-Coherence Transfer (Sel-MQC) with Application to Tumors. J. Magn. Reson. Ser. B 1995, 106, 203–211. [Google Scholar] [CrossRef]
- Holbach, M.; Lambert, J.; Suter, D. Optimized multiple-quantum filter for robust selective excitation of metabolite signals. J. Magn. Reson. (San Diego Calif 1997) 2014, 243, 8–16. [Google Scholar] [CrossRef]
- Holbach, M.; Lambert, J.; Johst, S.; Ladd, M.E.; Suter, D. Optimized selective lactate excitation with a refocused multiple-quantum filter. J. Magn. Reson. (San Diego Calif 1997) 2015, 255, 34–38. [Google Scholar] [CrossRef]
- Maximov, I.I.; Tosner, Z.; Nielsen, N.C. Optimal control design of NMR and dynamic nuclear polarization experiments using monotonically convergent algorithms. J. Chem. Phys. 2008, 128, 184505. [Google Scholar] [CrossRef]
- Ye, T.; Zheng, C.; Zhang, S.; Gowda, G.A.N.; Vitek, O.; Raftery, D. “Add to subtract”: A simple method to remove complex background signals from the 1H nuclear magnetic resonance spectra of mixtures. Anal. Chem. 2012, 84, 994–1002. [Google Scholar] [CrossRef]
- Provencher, S.W. Estimation of metabolite concentrations from localizedin vivo proton NMR spectra. Magn. Reson. Med. 1993, 30, 672–679. [Google Scholar] [CrossRef]
- Provencher, S.W. Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed. 2001, 14, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Bathen, T.F.; Geurts, B.; Sitter, B.; Fjøsne, H.E.; Lundgren, S.; Buydens, L.M.; Gribbestad, I.S.; Postma, G.; Giskeødegård, G.F. Feasibility of MR metabolomics for immediate analysis of resection margins during breast cancer surgery. PLoS ONE 2013, 8, e61578. [Google Scholar] [CrossRef] [PubMed]
- Martelotto, L.G.; Ng, C.K.Y.; Piscuoglio, S.; Weigelt, B.; Reis-Filho, J.S. Breast cancer intra-tumor heterogeneity. Breast Cancer Res. BCR 2014, 16, 210. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.K.Y.; Pemberton, H.N.; Reis-Filho, J.S. Breast cancer intratumor genetic heterogeneity: Causes and implications. Expert Rev. Anticancer Ther. 2012, 12, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.D.; Lamichhane, S.; Lundgren, S.; Bofin, A.; Fjøsne, H.; Giskeødegård, G.F.; Bathen, T.F. Metabolic characterization of triple negative breast cancer. BMC Cancer 2014, 14, 941. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Burstein, H.J.; P Winer, E.; Gnant, M.; Dubsky, P.; Loibl, S.; Colleoni, M.; Regan, M.M.; Piccart-Gebhart, M.; Senn, H.-J.; et al. De-escalating and escalating treatments for early-stage breast cancer: The St. Gallen International Expert Consensus Conference on the Primary Therapy of Early Breast Cancer 2017. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Giskeødegård, G.F.; Grinde, M.T.; Sitter, B.; Axelson, D.E.; Lundgren, S.; Fjøsne, H.E.; Dahl, S.; Gribbestad, I.S.; Bathen, T.F. Multivariate modeling and prediction of breast cancer prognostic factors using MR metabolomics. J. Proteome Res. 2010, 9, 972–979. [Google Scholar] [CrossRef]
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological complete response and long-term clinical benefit in breast cancer: The CTNeoBC pooled analysis. Lancet 2014, 384, 164–172. [Google Scholar] [CrossRef]
- Bingol, K. Recent Advances in Targeted and Untargeted Metabolomics by NMR and MS/NMR Methods. High.-Throughput 2018, 7. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gogiashvili et al. 2018 # (A) | Tayyari et al. 2018 # | Euceda et al. 2017a # | Euceda et al. 2017b $ | Park et al. 2016 # (B) | Yoon 2016 # (B) | Haukaas 2016a $ | Haukaas 2016b # | Chae 2016 # (B) | Cao et al. 2014 # | Grinde et al. 2014 $ (C) | Choi et al. 2013 # (B) | Borgan et al. 2013 $ (A) | Bathen et al. 2013 # | Giskeødegård et al. 2012 # | Cao et al. 2012b # | Cao et al. 2012a # (A) | Choi et al. 2012 # (B) | Li et al. 2011 # | Sitter et al. 2010 # (A) | Moestue et al. 2010 # $ (A) | Borgan et al. 2010 # | Giskeødegård et al. 2010 # | Bathen et al. 2007 # | Sitter et al. 2006 # (B) | Sitter et al. 2002 # | Cheng et al. 1998 # | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3-Hydroxybutyrate | Q | ||||||||||||||||||||||||||
| Acetate | Q | Q | Q | R | I | R | I | ||||||||||||||||||||
| Adipate | Q | ||||||||||||||||||||||||||
| Alanine | Q | R | R | R | Q | Q | R | R | Q | R | Q | R | I | I | Q | I | I | I | I | I | |||||||
| Arginine | R | Q | Q | I | |||||||||||||||||||||||
| Ascorbate | Q | R | R | R | R | R | I | I | |||||||||||||||||||
| Asparagine | Q | Q | I | I | |||||||||||||||||||||||
| Aspartate | Q | Q | Q | I | I | I | I | ||||||||||||||||||||
| ATP | R | ||||||||||||||||||||||||||
| Betaine | Q | Q | Q | I | |||||||||||||||||||||||
| Choline | Q | R | R | R | Q | Q | R | R | Q | R | Q | Q | Q | R | I | R | Q | Q | I | Q | Q | I | I | I | Q | I | I |
| Creatine | Q | R | R | R | Q | Q | R | R | Q | R | I | Q | Q | R | I | I | I | Q | I | Q | Q | I | I | I | Q | I | I |
| Ethanol | Q | ||||||||||||||||||||||||||
| Ethanolamine | Q | Q | |||||||||||||||||||||||||
| Formate | I | ||||||||||||||||||||||||||
| Fumarate | Q | Q | |||||||||||||||||||||||||
| Glucose | Q | R | R | R | Q | Q | R | R | I | I | R | I | R | I | Q | I | I | Q | I | ||||||||
| Glutamate | Q | R | R | R | Q | Q | R | R | I | R | I | I | R | I | I | I | I | I | |||||||||
| Glutamine | Q | R | R | R | Q | Q | R | R | I | R | I | R | I | I | I | ||||||||||||
| Glutathione | Q | R | R | R | R | R | |||||||||||||||||||||
| Glycerol | Q | Q | I | ||||||||||||||||||||||||
| Glycine | Q | R | R | R | Q | Q | R | R | Q | R | I | Q | Q | R | R | R | Q | Q | I | Q | Q | I | I | I | Q | I | I |
| GPC | Q | R | R | Q | Q | R | R | Q | I | Q | Q | Q | R | I | R | Q | Q | Q | Q | I | I | I | Q | I | |||
| Histidine | Q | Q | I | ||||||||||||||||||||||||
| Inosine | Q | I | |||||||||||||||||||||||||
| Isoleucine | Q | Q | Q | I | I | I | I | ||||||||||||||||||||
| Lactate | Q | R | R | R | Q | Q | R | R | I | R | I | I | Q | R | R | R | I | I | I | Q | I | I | I | I | I | I | |
| Leucine | Q | R | Q | Q | I | I | I | I | |||||||||||||||||||
| Lysine | Q | R | Q | Q | I | I | I | I | |||||||||||||||||||
| Methionine | Q | R | Q | Q | |||||||||||||||||||||||
| myo-Inositol | Q | R | R | R | Q | Q | R | R | Q | I | I | Q | R | Q | I | Q | I | I | Q | I | |||||||
| O-Phosphocholine | Q | R | R | R | Q | Q | R | R | Q | R | Q | Q | Q | R | I | R | Q | Q | I | Q | Q | I | I | I | Q | I | I |
| O-Phosphoethanolamine | Q | Q | Q | I | I | I | I | ||||||||||||||||||||
| Phenylalanine | Q | R | Q | Q | I | I | |||||||||||||||||||||
| Proline | Q | Q | Q | ||||||||||||||||||||||||
| scyllo-Inositol | Q | R | R | I | R | Q | I | I | I | ||||||||||||||||||
| Serine | Q | Q | Q | I | |||||||||||||||||||||||
| Succinate | Q | R | R | R | R | Q | R | I | Q | I | |||||||||||||||||
| Taurine | Q | R | R | R | Q | Q | R | R | Q | I | I | Q | Q | R | I | R | Q | Q | I | Q | Q | I | I | I | Q | I | I |
| Threonine | Q | R | Q | Q | |||||||||||||||||||||||
| Tyrosine | Q | R | R | R | Q | Q | R | R | I | I | |||||||||||||||||
| Uracil | Q | Q | I | ||||||||||||||||||||||||
| Uridine | R | ||||||||||||||||||||||||||
| Valine | Q | R | Q | Q | I | I | I | I | I |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gogiashvili, M.; Nowacki, J.; Hergenröder, R.; Hengstler, J.G.; Lambert, J.; Edlund, K. HR-MAS NMR Based Quantitative Metabolomics in Breast Cancer. Metabolites 2019, 9, 19. https://doi.org/10.3390/metabo9020019
Gogiashvili M, Nowacki J, Hergenröder R, Hengstler JG, Lambert J, Edlund K. HR-MAS NMR Based Quantitative Metabolomics in Breast Cancer. Metabolites. 2019; 9(2):19. https://doi.org/10.3390/metabo9020019
Chicago/Turabian StyleGogiashvili, Mikheil, Jessica Nowacki, Roland Hergenröder, Jan G. Hengstler, Jörg Lambert, and Karolina Edlund. 2019. "HR-MAS NMR Based Quantitative Metabolomics in Breast Cancer" Metabolites 9, no. 2: 19. https://doi.org/10.3390/metabo9020019
APA StyleGogiashvili, M., Nowacki, J., Hergenröder, R., Hengstler, J. G., Lambert, J., & Edlund, K. (2019). HR-MAS NMR Based Quantitative Metabolomics in Breast Cancer. Metabolites, 9(2), 19. https://doi.org/10.3390/metabo9020019