ADGRL4/ELTD1 Silencing in Endothelial Cells Induces ACLY and SLC25A1 and Alters the Cellular Metabolic Profile

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
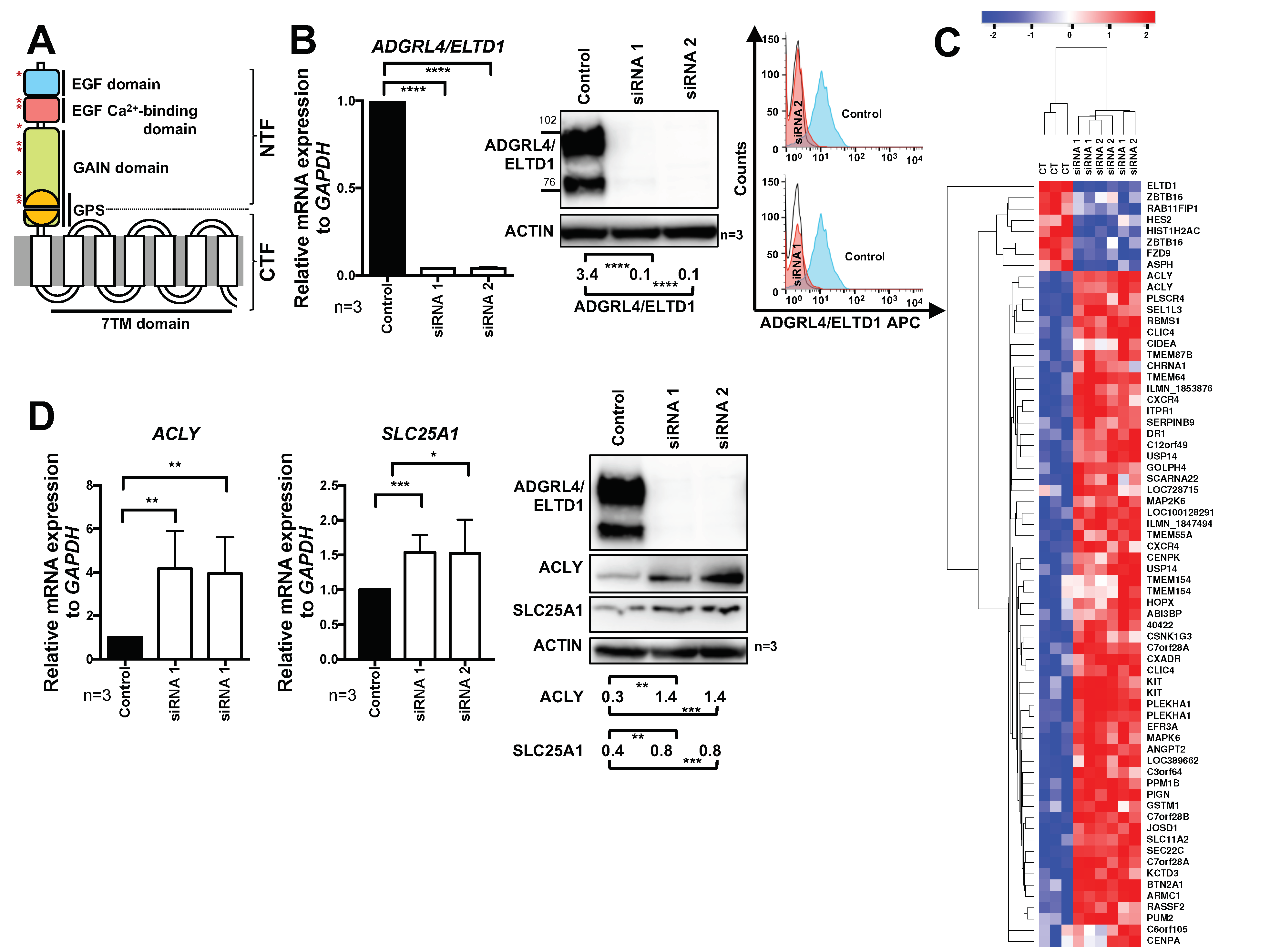
2.1. ADGRL4/ELTD1 Silencing Induces ACLY and SLC25A1 Expression and Affects Expression of KIT and Notch Pathway Genes
2.2. ADGRL4/ELTD1 Silencing Does Not Affect Acetylation Reactions or Lipid Droplet Formation
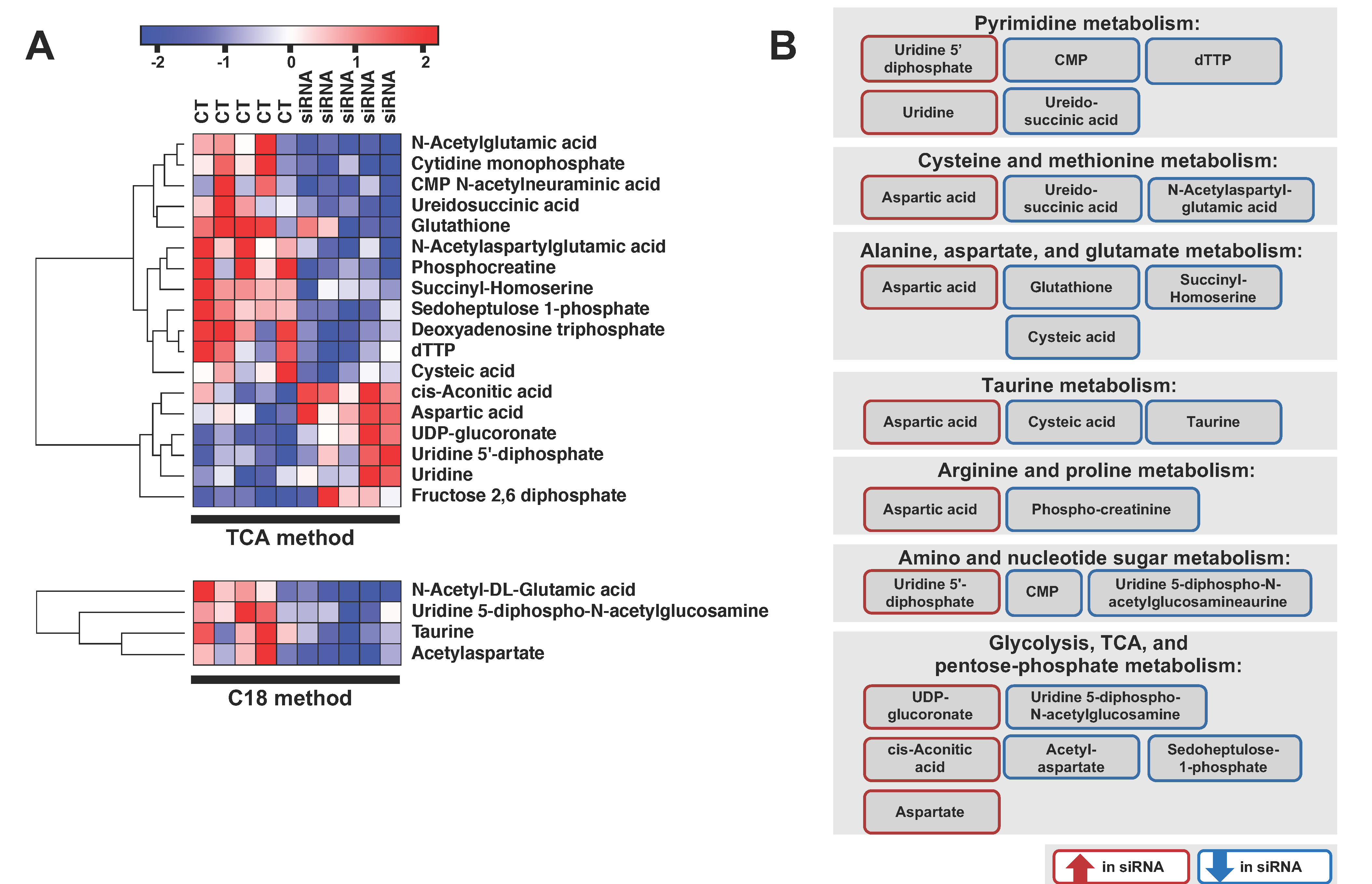
2.3. ADGRL4/ELTD1 Silencing Affects Various Metabolic Pathways
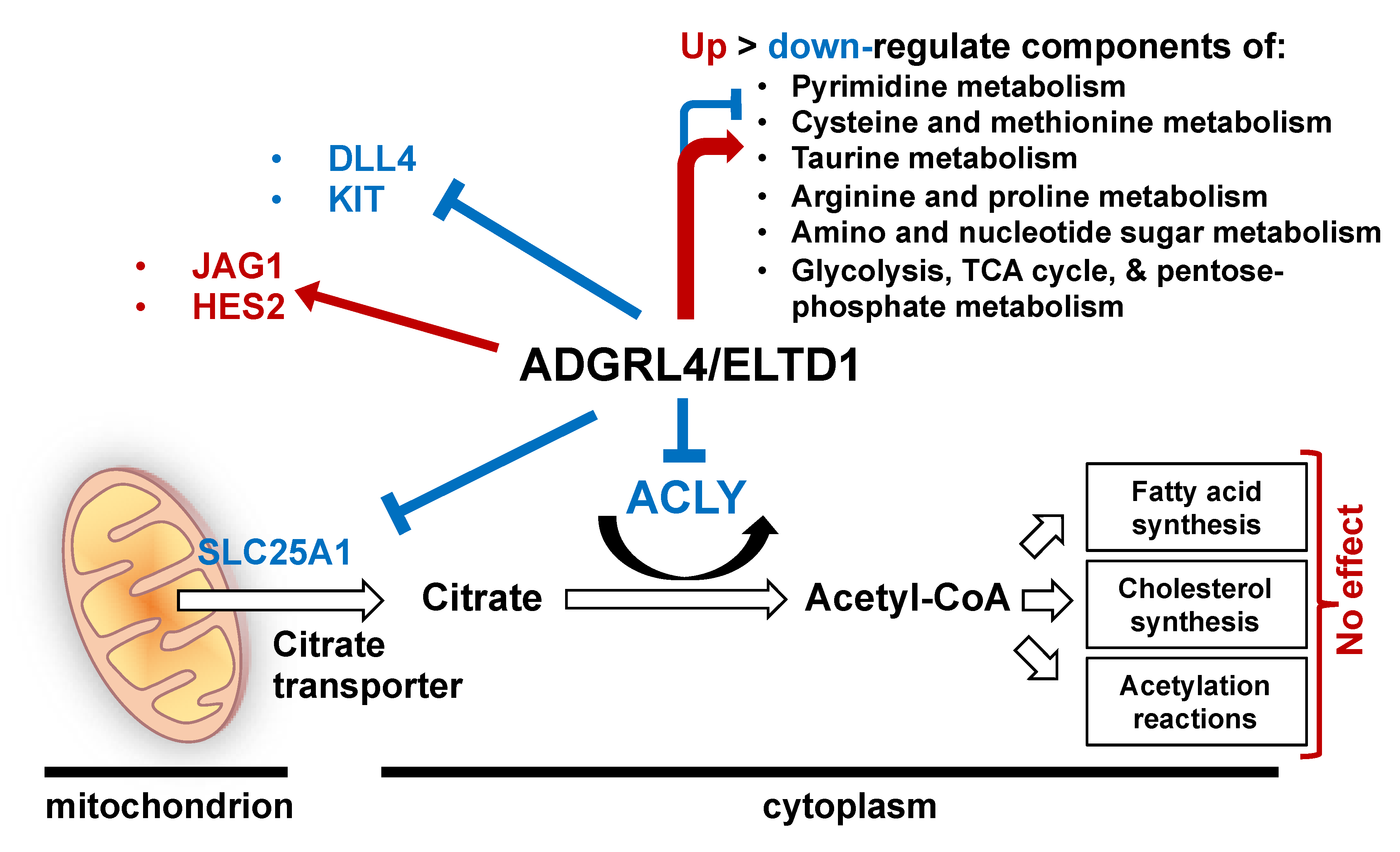
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Maintenance
4.2. Western Blotting
4.3. RNA Analysis and qPCR
4.4. Flow Cytometry
4.5. RNA Interference
4.6. Overexpresssion
4.7. Lipid Droplet Assays
4.8. Acetylation
4.9. Metabolomics
4.10. Microarray Analysis
4.11. Statistical Analyses Using Prism
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 7TM | 7-transmembrane domain |
| AAA | α-Aminoadipic acid |
| aGPCR | adhesion GPCR |
| CTF | C terminal fragment |
| CT | control |
| EMT | endothelial to mesenchymal transition |
| EPC | endothelial progenitor cell |
| FACS | fluorescence-activated cell sorting |
| GAIN | GPCR autoproteolysis inducing domain |
| GPCR | G protein-coupled receptor |
| GPS | GPCR proteolysis site |
| HPC | haematopoietic progenitor cell |
| ICD | intracellular domain |
| LC-MS | liquid chromatography-mass spectrometry |
| NTF | N terminal fragment |
| TCA | trichloroacetic acid/acetone |
| WT | wild type |
References
- Masiero, M.; Simoes, F.C.; Han, H.D.; Snell, C.; Peterkin, T.; Bridges, E.; Mangala, L.S.; Wu, S.Y.; Pradeep, S.; Li, D.; et al. A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 2013, 24, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Bjarnadottir, T.K.; Fredriksson, R.; Hoglund, P.J.; Gloriam, D.E.; Lagerstrom, M.C.; Schioth, H.B. The human and mouse repertoire of the adhesion family of G-protein-coupled receptors. Genomics 2004, 84, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Langenhan, T.; Aust, G.; Hamann, J. Sticky signaling—Adhesion class G protein-coupled receptors take the stage. Sci. Signal. 2013, 6, re3. [Google Scholar] [CrossRef] [PubMed]
- Hamann, J.; Aust, G.; Arac, D.; Engel, F.B.; Formstone, C.; Fredriksson, R.; Hall, R.A.; Harty, B.L.; Kirchhoff, C.; Knapp, B.; et al. International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol. Rev. 2015, 67, 338–367. [Google Scholar] [CrossRef] [PubMed]
- Favara, D.M.; Banham, A.H.; Harris, A.L. ADGRL4/ELTD1 is a highly conserved angiogenesis-associated orphan adhesion GPCR that emerged with the first vertebrates and comprises 3 evolutionary variants. BMC Evol. Biol. 2019, 19, 143. [Google Scholar] [CrossRef]
- Lin, H.H.; Chang, G.W.; Davies, J.Q.; Stacey, M.; Harris, J.; Gordon, S. Autocatalytic Cleavage of the EMR2 Receptor Occurs at a Conserved G Protein-coupled Receptor Proteolytic Site Motif. J. Biol. Chem. 2004, 279, 31823–31832. [Google Scholar] [CrossRef]
- Xiao, J.; Jiang, H.; Zhang, R.; Fan, G.; Zhang, Y.; Jiang, D.; Li, H. Augmented cardiac hypertrophy in response to pressure overload in mice lacking ELTD1. PLoS ONE 2012, 7, e35779. [Google Scholar] [CrossRef]
- Kan, A.; Le, Y.; Zhang, Y.F.; Duan, F.T.; Zhong, X.P.; Lu, L.H.; Ling, Y.H.; Guo, R.P. ELTD1 Function in Hepatocellular Carcinoma is Carcinoma-Associated Fibroblast-Dependent. J. Cancer 2018, 9, 2415–2427. [Google Scholar] [CrossRef]
- Favara, D.M.; Banham, A.H.; Harris, A.L. A review of ELTD1, a pro-angiogenic adhesion GPCR. Biochem. Soc. Trans. 2014, 42, 1658–1664. [Google Scholar] [CrossRef]
- Towner, R.A.; Jensen, R.L.; Colman, H.; Vaillant, B.; Smith, N.; Casteel, R.; Saunders, D.; Gillespie, D.L.; Silasi-Mansat, R.; Lupu, F.; et al. ELTD1, a potential new biomarker for gliomas. Neurosurgery 2013, 72, 77–90. [Google Scholar] [CrossRef]
- Serban, F.; Daianu, O.; Tataranu, L.G.; Artene, S.A.; Emami, G.; Georgescu, A.M.; Alexandru, O.; Purcaru, S.O.; Tache, D.E.; Danciulescu, M.M.; et al. Silencing of epidermal growth factor, latrophilin and seven transmembrane domain-containing protein 1 (ELTD1) via siRNA-induced cell death in glioblastoma. J. Immunoass. Immunochem. 2017, 38, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, J.; Pody, R.; Coutinho de Souza, P.; Evans, B.; Saunders, D.; Smith, N.; Mallory, S.; Njoku, C.; Dong, Y.; Chen, H.; et al. ELTD1, an effective anti-angiogenic target for gliomas: Preclinical assessment in mouse GL261 and human G55 xenograft glioma models. Neuro Oncol. 2017, 19, 175–185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jiang, L.; Boufersaoui, A.; Yang, C.; Ko, B.; Rakheja, D.; Guevara, G.; Hu, Z.; DeBerardinis, R.J. Quantitative metabolic flux analysis reveals an unconventional pathway of fatty acid synthesis in cancer cells deficient for the mitochondrial citrate transport protein. Metab. Eng. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef]
- Verschueren, K.H.G.; Blanchet, C.; Felix, J.; Dansercoer, A.; De Vos, D.; Bloch, Y.; Van Beeumen, J.; Svergun, D.; Gutsche, I.; Savvides, S.N.; et al. Structure of ATP citrate lyase and the origin of citrate synthase in the Krebs cycle. Nature 2019, 568, 571–575. [Google Scholar] [CrossRef]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI BioSystems database. Nucleic Acids Res. 2010, 38, D492–D496. [Google Scholar] [CrossRef]
- Sun, J.; Aluvila, S.; Kotaria, R.; Mayor, J.A.; Walters, D.E.; Kaplan, R.S. Mitochondrial and Plasma Membrane Citrate Transporters: Discovery of Selective Inhibitors and Application to Structure/Function Analysis. Mol. Cell. Pharmacol. 2010, 2, 101–110. [Google Scholar]
- Chypre, M.; Zaidi, N.; Smans, K. ATP-citrate lyase: A mini-review. Biochem. Biophys. Res. Commun. 2012, 422, 1–4. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Abu-Sbeih, H.; Samdani, R.; Nogueras Gonzalez, G.; Raju, G.S.; Richards, D.M.; Gao, J.; Subudhi, S.; Stroehlein, J.; Wang, Y. Can Immune Checkpoint Inhibitors Induce Microscopic Colitis or a Brand New Entity? Inflamm. Bowel Dis. 2019, 25, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Kawagishi, H.; Yan, Y.; Liu, J.; Wells, Q.S.; Edmunds, L.R.; Fergusson, M.M.; Yu, Z.X.; Rovira, I.I.; Brittain, E.L.; et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol. Cell 2018, 69, 689–698. [Google Scholar] [CrossRef]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Depre, C.; Veitch, K.; Hue, L. Role of fructose 2, 6-bisphosphate in the control of glycolysis. Stimulation of glycogen synthesis by lactate in the isolated working rat heart. Acta Cardiol. 1993, 48, 147–164. [Google Scholar] [PubMed]
- Lennartsson, J.; Ronnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef]
- Azzoni, E.; Frontera, V.; McGrath, K.E.; Harman, J.; Carrelha, J.; Nerlov, C.; Palis, J.; Jacobsen, S.E.W.; de Bruijn, M.F. Kit ligand has a critical role in mouse yolk sac and aorta-gonad-mesonephros hematopoiesis. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Solaimani Kartalaei, P.; Yamada-Inagawa, T.; Vink, C.S.; de Pater, E.; van der Linden, R.; Marks-Bluth, J.; van der Sloot, A.; van den Hout, M.; Yokomizo, T.; van Schaick-Solerno, M.L.; et al. Whole-transcriptome analysis of endothelial to hematopoietic stem cell transition reveals a requirement for Gpr56 in HSC generation. J. Exp. Med. 2015, 212, 93–106. [Google Scholar] [CrossRef]
- Harjes, U.; Bridges, E.; McIntyre, A.; Fielding, B.A.; Harris, A.L. Fatty acid-binding protein 4, a point of convergence for angiogenic and metabolic signaling pathways in endothelial cells. J. Biol. Chem. 2014, 289, 23168–23176. [Google Scholar] [CrossRef]
- Huang, Z.; Ruan, H.B.; Xian, L.; Chen, W.; Jiang, S.; Song, A.; Wang, Q.; Shi, P.; Gu, X.; Gao, X. The stem cell factor/Kit signalling pathway regulates mitochondrial function and energy expenditure. Nat. Commun. 2014, 5, 4282. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 2017, 47, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Salazar, C.; Armenta, J.M.; Cortes, D.F.; Shulaev, V. Combination of an AccQ.Tag-ultra performance liquid chromatographic method with tandem mass spectrometry for the analysis of amino acids. Amino Acid Anal. 2012, 828, 13–28. [Google Scholar] [CrossRef]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics (Oxford, England) 2018, 34, 4313–4314. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef]
- Kutmon, M.; Riutta, A.; Nunes, N.; Hanspers, K.; Willighagen, E.L.; Bohler, A.; Melius, J.; Waagmeester, A.; Sinha, S.R.; Miller, R.; et al. WikiPathways: Capturing the full diversity of pathway knowledge. Nucleic Acids Res. 2016, 44, D488–D494. [Google Scholar] [CrossRef]
- Nishimura, D. BioCarta. Biotech Softw. Internet Rep. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer Publishing Company, Incorporated: New York, NY, USA, 2009; p. 216. [Google Scholar]
- Brazma, A.; Hingamp, P.; Quackenbush, J.; Sherlock, G.; Spellman, P.; Stoeckert, C.; Aach, J.; Ansorge, W.; Ball, C.A.; Causton, H.C.; et al. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat. Genet. 2001, 29, 365–371. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Favara, D.M.; Zois, C.E.; Haider, S.; Pires, E.; Sheldon, H.; McCullagh, J.; Banham, A.H.; Harris, A.L. ADGRL4/ELTD1 Silencing in Endothelial Cells Induces ACLY and SLC25A1 and Alters the Cellular Metabolic Profile. Metabolites 2019, 9, 287. https://doi.org/10.3390/metabo9120287
Favara DM, Zois CE, Haider S, Pires E, Sheldon H, McCullagh J, Banham AH, Harris AL. ADGRL4/ELTD1 Silencing in Endothelial Cells Induces ACLY and SLC25A1 and Alters the Cellular Metabolic Profile. Metabolites. 2019; 9(12):287. https://doi.org/10.3390/metabo9120287
Chicago/Turabian StyleFavara, David M., Christos E. Zois, Syed Haider, Elisabete Pires, Helen Sheldon, James McCullagh, Alison H. Banham, and Adrian L. Harris. 2019. "ADGRL4/ELTD1 Silencing in Endothelial Cells Induces ACLY and SLC25A1 and Alters the Cellular Metabolic Profile" Metabolites 9, no. 12: 287. https://doi.org/10.3390/metabo9120287
APA StyleFavara, D. M., Zois, C. E., Haider, S., Pires, E., Sheldon, H., McCullagh, J., Banham, A. H., & Harris, A. L. (2019). ADGRL4/ELTD1 Silencing in Endothelial Cells Induces ACLY and SLC25A1 and Alters the Cellular Metabolic Profile. Metabolites, 9(12), 287. https://doi.org/10.3390/metabo9120287