WebSpecmine: A Website for Metabolomics Data Analysis and Mining



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
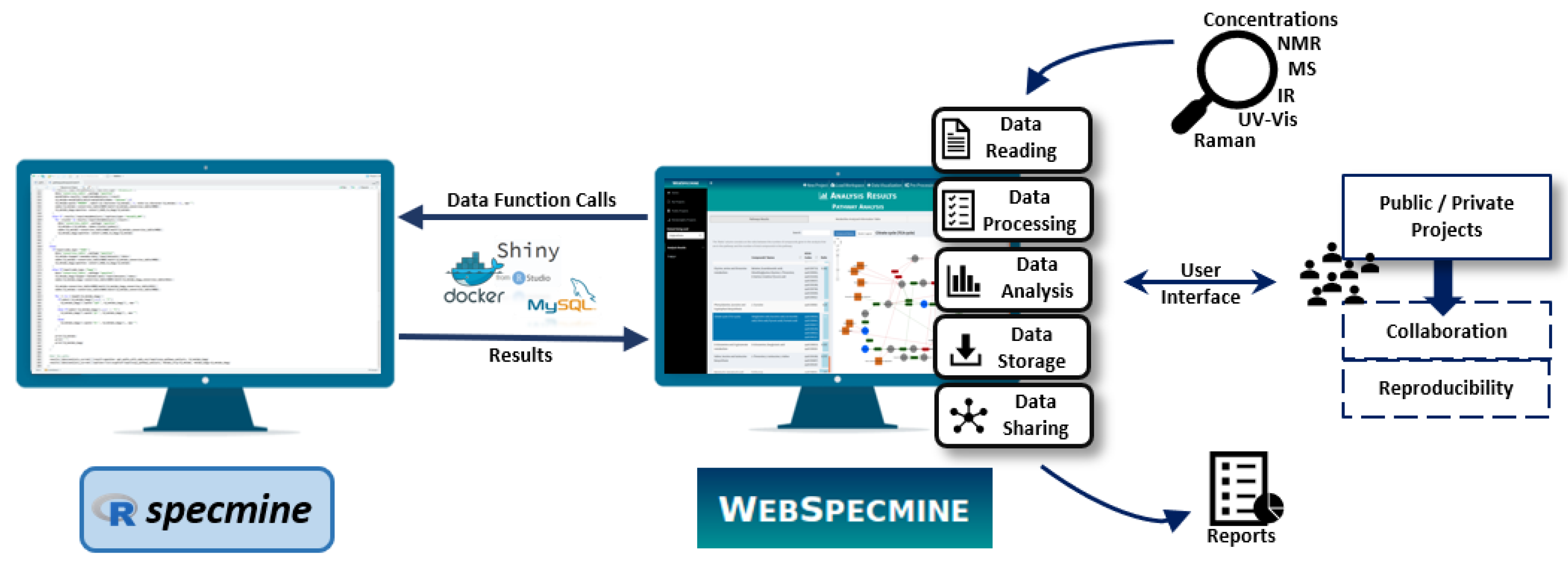
2. Results
2.1. User Accounts
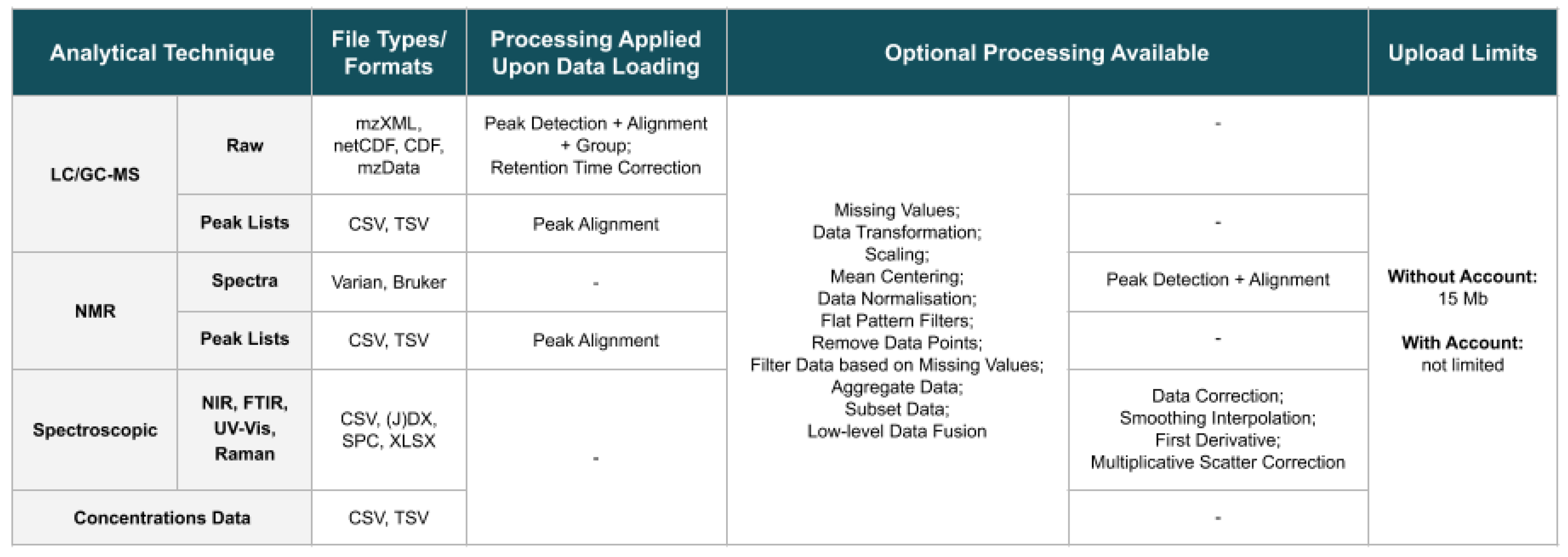
2.2. Loading and Visualising Data
2.3. Pre-Processing
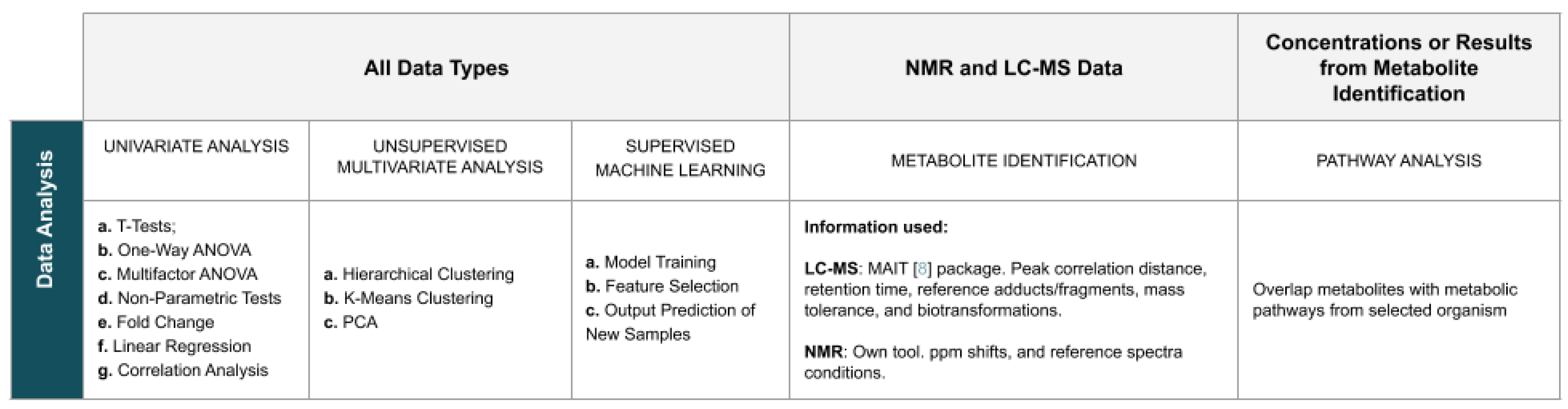
2.4. Data Analysis
2.5. Application of WebSpecmine to a Case Study
3. Discussion
4. Materials and Methods
4.1. Website Implementation
4.2. Desktop Version
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of Variance |
| CSV | Comma Separated Values |
| FID | Free Induction Decay |
| FTIR | Fourier-Transform Infrared |
| GC | Gas Chromatography |
| HMDB | Human Metabolome Database |
| LC | Liquid Chromatography |
| LDA | Linear Discriminant Analysis |
| MS | Mass Spectrometry |
| NIR | Near Infrared |
| NMR | Nuclear Magnetic Resonance |
| NN | Neural Networks |
| PCA | Principal Component Analysis |
| PLS | Partial Least Squares |
| SVMs | Support Vector Machines |
| TSV | Tab Separated Values |
| UV | Ultra-Violet |
References
- Costa, C.; Maraschin, M.; Rocha, M. An R package for the integrated analysis of metabolomics and spectral data. Comput. Methods Progr. Biomed. 2016, 129, 117–124. [Google Scholar] [CrossRef] [PubMed]
- WebSpecmine. Available online: https://webspecmine.bio.di.uminho.pt/ (accessed on 18 October 2019).
- Helmus, J.J.; Jaroniec, C.P. Nmrglue: An open source Python package for the analysis of multidimensional NMR data. J. Biomol. NMR 2013, 55, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Astle, W.; De Iorio, M.; Ebbels, T.M. BATMAN—An R package for the automated quantification of metabolites from nuclear magnetic resonance spectra using a Bayesian model. Bioinformatics 2012, 28, 2088–2090. [Google Scholar] [CrossRef]
- Hanson, B.A. ChemoSpec: Exploratory Chemometrics for Spectroscopy. Available online: https://bryanhanson.github.io/ChemoSpec/ (accessed on 18 October 2019).
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Gowda, H.; Ivanisevic, J.; Johnson, C.H.; Kurczy, M.E.; Benton, H.P.; Rinehart, D.; Nguyen, T.; Ray, J.; Kuehl, J.; Arevalo, B.; et al. Interactive XCMS Online: Simplifying advanced metabolomic data processing and subsequent statistical analyses. Anal. Chem. 2014, 86, 6931–6939. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Salek, R.M.; Conesa, P.; Hastings, J.; de Matos, P.; Rijnbeek, M.; Mahendraker, T.; Williams, M.; Neumann, S.; Rocca-Serra, P.; et al. MetaboLights—An open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2012, 41, D781–D786. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Albert, F.; Llorach, R.; Andrés-Lacueva, C.; Perera, A. An R package to analyse LC/MS metabolomic data: MAIT (Metabolite Automatic Identification Toolkit). Bioinformatics 2014, 30, 1937–1939. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.L.; Weber, R.J.; Liu, H.; Sharma-Oates, A.; Viant, M.R. Galaxy-M: A Galaxy workflow for processing and analyzing direct infusion and liquid chromatography mass spectrometry-based metabolomics data. GigaScience 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Giacomoni, F.; Le Corguillé, G.; Monsoor, M.; Landi, M.; Pericard, P.; Pétéra, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.F.; Jacob, D.; et al. Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics 2014, 31, 1493–1495. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.; Bradbury, J.; Bergmann, S.; Capuccini, M.; Cascante, M.; de Atauri, P.; Ebbels, T.M.; Foguet, C.; Glen, R.; Gonzalez-Beltran, A.; et al. PhenoMeNal: Processing and analysis of metabolomics data in the cloud. GigaScience 2018, 8, giy149. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Cheng, J.; Allaire, J.; Xie, Y.; McPherson, J. Shiny: Web Application Framework for R. Available online: https://cran.r-project.org/web/packages/shiny/index.html (accessed on 18 October 2019).



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso, S.; Afonso, T.; Maraschin, M.; Rocha, M. WebSpecmine: A Website for Metabolomics Data Analysis and Mining. Metabolites 2019, 9, 237. https://doi.org/10.3390/metabo9100237
Cardoso S, Afonso T, Maraschin M, Rocha M. WebSpecmine: A Website for Metabolomics Data Analysis and Mining. Metabolites. 2019; 9(10):237. https://doi.org/10.3390/metabo9100237
Chicago/Turabian StyleCardoso, Sara, Telma Afonso, Marcelo Maraschin, and Miguel Rocha. 2019. "WebSpecmine: A Website for Metabolomics Data Analysis and Mining" Metabolites 9, no. 10: 237. https://doi.org/10.3390/metabo9100237
APA StyleCardoso, S., Afonso, T., Maraschin, M., & Rocha, M. (2019). WebSpecmine: A Website for Metabolomics Data Analysis and Mining. Metabolites, 9(10), 237. https://doi.org/10.3390/metabo9100237