A Simple and Convenient Synthesis of Unlabeled and 13C-Labeled 3-(3-Hydroxyphenyl)-3-Hydroxypropionic Acid and Its Quantification in Human Urine Samples

 ,
, 
Abstract
1. Introduction
2. Results and Discussion
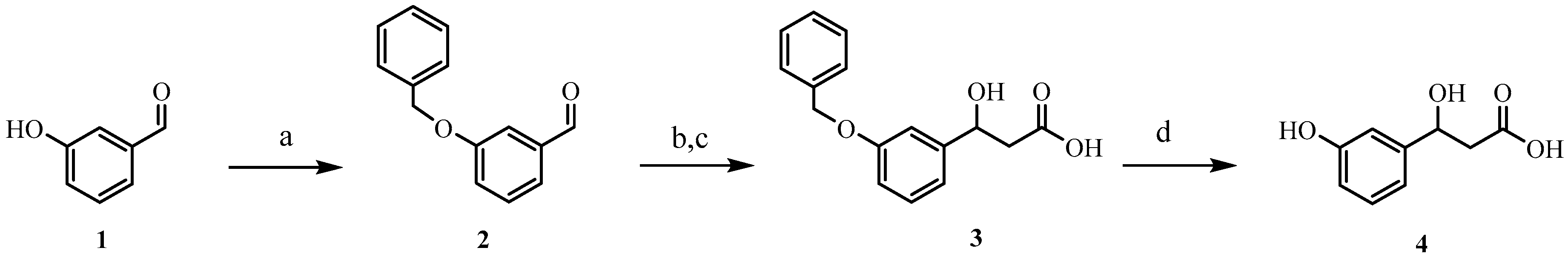
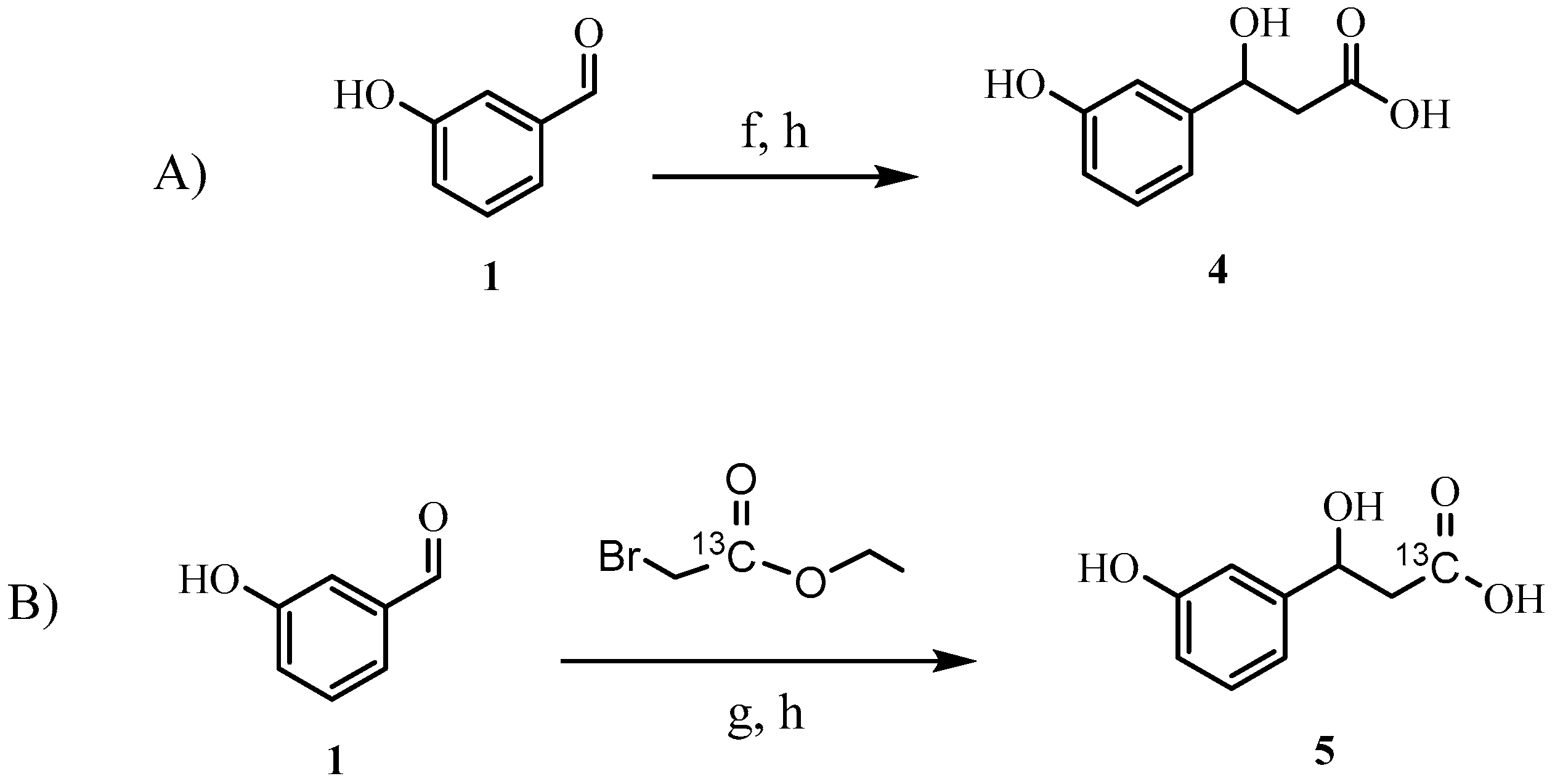
2.1. Synthesis of HPHPA
2.2. Assay Validation-HPHPA
2.3. LC-MS/MS Assay Validation of HPHPA on Random Urine Samples
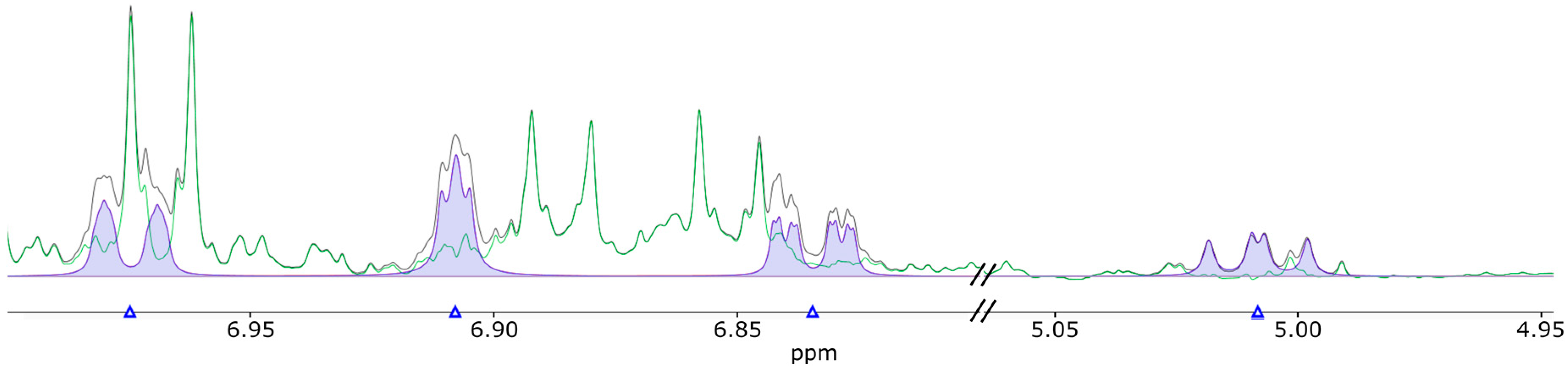
2.4. Cross-Validation with NMR on Human Urine Samples
3. Materials and Methods
3.1. Materials and Methods for HPHPA Synthesis
3.2. Materials and Methods for the LC-MS/MS Assay
3.3. Materials and Methods for NMR Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–D610. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- de Angelis, M.; Francavilla, R.; Piccolo, M.; De Giacomo, A.; Gobbetti, M. Autism spectrum disorders and intestinal microbiota. Gut Microbes 2015, 6, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Keşli, R.; Gökçen, C.; Buluǧ, U.; Terzi, Y. Investigation of the relation between anaerobic bacteria genus clostridium and late-onset autism etiology in children. J. Immunoass. Immunochem. 2014, 35, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Shaw, W. Increased urinary excretion of a 3-(3-hydroxyphenyl)-3-hydroxypropionic acid (HPHPA), an abnormal phenylalanine metabolite of Clostridia spp. in the gastrointestinal tract, in urine samples from patients with autism and schizophrenia. Nutr. Neurosci. 2010, 13, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Liu, D.; Wang, Y.; Zeng, T.; Peng, Y. Urinary 3-(3-Hydroxyphenyl)-3-hydroxypropionic Acid, 3-Hydroxyphenylacetic Acid, and 3-Hydroxyhippuric Acid Are Elevated in Children with Autism Spectrum Disorders. BioMed Res. Int. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pineiro, R.; Dong, Y.W.; Wishart, D.S. Solid phase synthesis of acylglycine human metabolites. Bioorg. Med. Chem. Lett. 2009, 19, 6706–6708. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.D.; Shaw, K. The occurrence hydracrylic acid in human urine. J. Biol. Chem. 1957, 225, 269–278. [Google Scholar] [PubMed]
- Shen, Z.; Zhang, J.; Zou, H.; Yang, M. A Novel One-Pot Reformatsky Type Reaction via Bismuth Salt in Aqueous Media. Tetrahedron Lett. 1997, 38, 2733–2736. [Google Scholar] [CrossRef]
- Gabriel, T.; Wessjohann, L. The Chromium-Reformatsky Reaction: Asymmetric Synthesis of the Aldol Fragment of the Cytotoxic Epothilons from 3-(2-Bromoacyl)-2-oxazolidinones. Tetrahedron Lett. 1997, 38, 1363–1366. [Google Scholar] [CrossRef]
- Schick, H.; Ludwig, R.; Schwarz, K.-H.; Kleiner, K.; Kunath, A. Novel Synthesis of Tetrasubstituted β-Lactones: The Use of Indium in the Electrochemically Supported Reformatsky Reaction. Angew. Chem. Int. Ed. Engl. 1993, 32, 1191–1193. [Google Scholar] [CrossRef]
- Cahiez, G.; Chavant, P.-Y. Organomanganese (II) Reagents XX: Manganese Mediated Barbier and Reformatsky Like Reactions an Efficient Route to Homoallylic Alcohols and β-acetoxyesters. Tetrahedron Lett. 1989, 30, 7373–7376. [Google Scholar] [CrossRef]
- Araki, S.; Butsugan, Y. Enantioseiective Synthesis of β-Hydroxy Esters by indium-induced Reformatsky Reaction. J. Chem. Soc. Perkin Trans. 1 1992, 711–713. [Google Scholar]
- Fürstner, A.; Bogdanović, B. New developments in the chemistry of low-valent titanium. Angew. Chem. Int. Ed. Engl. 1996, 35, 2442–2469. [Google Scholar] [CrossRef]
- Fürstner, A. Recent Advancements in the Reformatsky Reaction. Synthesis (Stuttg). 1989, 1989, 571–590. [Google Scholar] [CrossRef]
- Lee, P.H.; Bang, K.; Lee, K.; Lee, C.-H.; Chang, S. In-mediated synthesis of 2-(2-hydroxyethyl)homoallenylsilanes. Tetrahedron Lett. 2000, 41, 7521–7524. [Google Scholar] [CrossRef]
- Li, C.J. Aqueous Barbier-Grignard type reaction: Scope, mechanism, and synthetic applications. Tetrahedron 1996, 52, 5643–5668. [Google Scholar] [CrossRef]
- Li, C.J. Organic Reactions in Aqueous Media—With a Focus on Carbon-Carbon Bond Formation. Chem. Rev. 1993, 93, 2023–2035. [Google Scholar] [CrossRef]
- Jun, C.; Chan, T. Organic Syntheses Using Indium-Mediated and Catalyzed Reactions in Aqueous Media. Tetrahedron 1999, 55, 11149–11176. [Google Scholar] [CrossRef]
- Han, B.H.; Boudjouk, P. Organic Sonochemistry. Sonic Acceleration of the Reformatsky Reaction. J. Org. Chem. 1982, 47, 5030–5032. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Urine ID | HPHPA Concentrations Measured by LC-MS (µM) | HPHPA Concentrations Measured by NMR (µM) | Concentration Differences between LC-MS and NMR (%) |
|---|---|---|---|
| F1 | 76.1 | 78.3 | 2.75 |
| F2 | 41.5 | 39.4 | 5.40 |
| F3 | 37.6 | 38.3 | 1.70 |
| F4 | 54.3 | 56.8 | 4.32 |
| M1 | 59.3 | 59.4 | 0.13 |
| M2 | 89.2 | 88.0 | 1.36 |
| M3 | 42.5 | 39.5 | 7.59 |
| M4 | 5.48 | 5.38 | 1.95 |
| M5 | 77.0 | 71.5 | 7.69 |
| M6 | 173 | 172 | 0.73 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khaniani, Y.; Lipfert, M.; Bhattacharyya, D.; Perez Pineiro, R.; Zheng, J.; Wishart, D.S. A Simple and Convenient Synthesis of Unlabeled and 13C-Labeled 3-(3-Hydroxyphenyl)-3-Hydroxypropionic Acid and Its Quantification in Human Urine Samples. Metabolites 2018, 8, 80. https://doi.org/10.3390/metabo8040080
Khaniani Y, Lipfert M, Bhattacharyya D, Perez Pineiro R, Zheng J, Wishart DS. A Simple and Convenient Synthesis of Unlabeled and 13C-Labeled 3-(3-Hydroxyphenyl)-3-Hydroxypropionic Acid and Its Quantification in Human Urine Samples. Metabolites. 2018; 8(4):80. https://doi.org/10.3390/metabo8040080
Chicago/Turabian StyleKhaniani, Yeganeh, Matthias Lipfert, Dipanjan Bhattacharyya, Rolando Perez Pineiro, Jiamin Zheng, and David S. Wishart. 2018. "A Simple and Convenient Synthesis of Unlabeled and 13C-Labeled 3-(3-Hydroxyphenyl)-3-Hydroxypropionic Acid and Its Quantification in Human Urine Samples" Metabolites 8, no. 4: 80. https://doi.org/10.3390/metabo8040080
APA StyleKhaniani, Y., Lipfert, M., Bhattacharyya, D., Perez Pineiro, R., Zheng, J., & Wishart, D. S. (2018). A Simple and Convenient Synthesis of Unlabeled and 13C-Labeled 3-(3-Hydroxyphenyl)-3-Hydroxypropionic Acid and Its Quantification in Human Urine Samples. Metabolites, 8(4), 80. https://doi.org/10.3390/metabo8040080