Computational Approaches for Integrative Analysis of the Metabolome and Microbiome



{kind=link}
Abstract
1. Introduction
2. Metabolomics Data Integration in the Microbiome
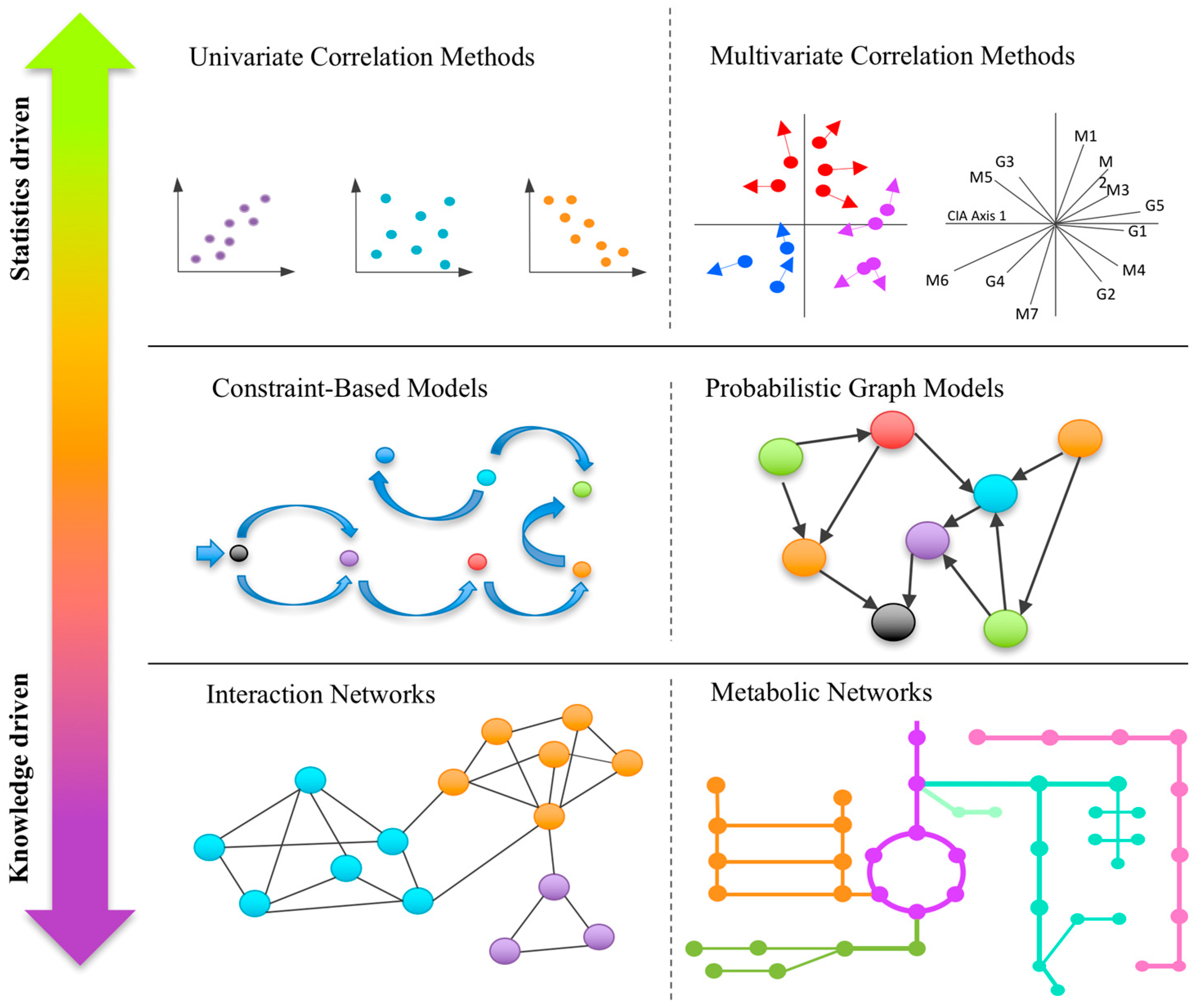
2.1. Univariate Correlation-Based Approaches
2.2. Multivariate Correlation-Based Approaches
3. Knowledge-Based Approaches for Omics Integration
3.1. Correlation/Interaction-Based Community Networks
3.2. Metabolic Networks
3.2.1. Topological Analysis of Metabolic Models
3.2.2. Community Metabolic Models
4. Summary and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Martin, F.-P.J.; Sprenger, N.; Yap, I.K.; Wang, Y.; Bibiloni, R.; Rochat, F.; Rezzi, S.; Cherbut, C.; Kochhar, S.; Lindon, J.C.; et al. Panorganismal gut microbiome—Host metabolic crosstalk. J. Proteome Res. 2009, 8, 2090–2105. [Google Scholar] [CrossRef] [PubMed]
- Candela, M.; Guidotti, M.; Fabbri, A.; Brigidi, P.; Franceschi, C.; Fiorentini, C. Human intestinal microbiota: Cross-talk with the host and its potential role in colorectal cancer. Crit. Rev. Microbiol. 2011, 37, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.R.; Pop, M.; DeBoy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Consortium, H.M.P. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1131. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Morgan, X.C.; Segata, N.; Waldron, L.; Reyes, J.; Earl, A.M.; Giannoukos, G.; Boylan, M.R.; Ciulla, D.; Gevers, D.; et al. Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. USA 2014, 111, E2329–E2338. [Google Scholar] [CrossRef] [PubMed]
- Verberkmoes, N.C.; Russell, A.L.; Shah, M.; Godzik, A.; Rosenquist, M.; Halfvarsson, J.; Lefsrud, M.G.; Apajalahti, J.; Tysk, C.; Hettich, R.L.; et al. Shotgun Metaproteomics of the Human Distal Gut Microbiota; Ernest Orlando Lawrence Berkeley National Laboratory: Berkeley, CA, USA, 2008. [Google Scholar]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. In Functional Genomics; Springer: Berlin/Heidelberg, Germany, 2002; pp. 155–171. [Google Scholar]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. Microbiomeanalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Mandal, R.; Sinelnikov, I.V.; Broadhurst, D.; Wishart, D.S. Metaboanalyst 2.0—A comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012, 40, W127–W133. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. Metaboanalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. Metaboanalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Qiime allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E., Jr.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Li, J.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to clostridium difficile infection. Nat. Commun. 2014, 5, 3114. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.Y.; Huo, W.J.; Zhu, W.Y. Microbiome–metabolome analysis reveals unhealthy alterations in the composition and metabolism of ruminal microbiota with increasing dietary grain in a goat model. Environ. Microbiol. 2016, 18, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Trygg, J.; Wold, S. O2-pls, a two-block (x–y) latent variable regression (LVR) method with an integral OSC filter. J. Chemom. 2003, 17, 53–64. [Google Scholar] [CrossRef]
- Bylesjö, M.; Eriksson, D.; Kusano, M.; Moritz, T.; Trygg, J. Data integration in plant biology: The O2PLS method for combined modeling of transcript and metabolite data. Plant J. 2007, 52, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- El Bouhaddani, S.; Houwing-Duistermaat, J.; Salo, P.; Perola, M.; Jongbloed, G.; Uh, H.-W. Evaluation of o2pls in Omics data integration. In BMC Bioinformatics; BioMed Central Ltd.: London, UK, 2016; p. 11. [Google Scholar]
- El Aidy, S.; Derrien, M.; Merrifield, C.A.; Levenez, F.; Doré, J.; Boekschoten, M.V.; Dekker, J.; Holmes, E.; Zoetendal, E.G.; Van Baarlen, P.; et al. Gut bacteria–host metabolic interplay during conventionalisation of the mouse germfree colon. ISME J. 2013, 7, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Hotelling, H. Relations between two sets of variates. Biometrika 1936, 28, 321–377. [Google Scholar] [CrossRef]
- Dolédec, S.; Chessel, D. Co-inertia analysis: An alternative method for studying species–environment relationships. Freshw. Biol. 1994, 31, 277–294. [Google Scholar] [CrossRef]
- Lin, D.; Zhang, J.; Li, J.; Calhoun, V.D.; Deng, H.-W.; Wang, Y.-P. Group sparse canonical correlation analysis for genomic data integration. BMC Bioinform. 2013, 14, 245. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Vert, J.-P.; Kanehisa, M. Protein network inference from multiple genomic data: A supervised approach. Bioinformatics 2004, 20, i363–i370. [Google Scholar] [CrossRef] [PubMed]
- De Bie, T.; De Moor, B. On the regularization of canonical correlation analysis. Int. Sympos. ICA BSS 2003, 785–790. [Google Scholar]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyötyläinen, T.; Hämäläinen, A.-M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I.; et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Thioulouse, J. Simultaneous analysis of a sequence of paired ecological tables: A comparison of several methods. Ann. Appl. Stat. 2011, 5, 2300–2325. [Google Scholar] [CrossRef]
- Hill, C.J.; Lynch, D.B.; Murphy, K.; Ulaszewska, M.; Jeffery, I.B.; O’Shea, C.A.; Watkins, C.; Dempsey, E.; Mattivi, F.; Tuohy, K.; et al. Evolution of gut microbiota composition from birth to 24 weeks in the infantmet cohort. Microbiome 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Hong, J.; Xu, X.; Feng, Q.; Zhang, D.; Gu, Y.; Shi, J.; Zhao, S.; Liu, W.; Wang, X.; et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 2017, 23, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Gower, J.C. Generalized procrustes analysis. Psychometrika 1975, 40, 33–51. [Google Scholar] [CrossRef]
- McHardy, I.H.; Goudarzi, M.; Tong, M.; Ruegger, P.M.; Schwager, E.; Weger, J.R.; Graeber, T.G.; Sonnenburg, J.L.; Horvath, S.; Huttenhower, C.; et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome 2013, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.A.; Navas-Molina, J.A.; Hyde, E.R.; Song, S.J.; Vázquez-Baeza, Y.; Humphrey, G.; Gaffney, J.; Minich, J.J.; Melnik, A.V.; Herschend, J.; et al. From sample to multi-omics conclusions in under 48 hours. mSystems 2016, 1, e00038-00016. [Google Scholar] [CrossRef] [PubMed]
- Garali, I.; Adanyeguh, I.M.; Ichou, F.; Perlbarg, V.; Seyer, A.; Colsch, B.; Moszer, I.; Guillemot, V.; Durr, A.; Mochel, F.; et al. A strategy for multimodal data integration: Application to biomarkers identification in spinocerebellar ataxia. Brief. Bioinform. 2017, bbx060. [Google Scholar] [CrossRef] [PubMed]
- Agler, M.T.; Ruhe, J.; Kroll, S.; Morhenn, C.; Kim, S.-T.; Weigel, D.; Kemen, E.M. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016, 14, e1002352. [Google Scholar] [CrossRef] [PubMed]
- Greenblum, S.; Turnbaugh, P.J.; Borenstein, E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2012, 109, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.; Kim, S.; Cabatbat, J.J.T.; Jang, S.; Jin, Y.-S.; Jung, G.Y.; Chia, N.; Kim, P.-J. Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis. arXiv, 2017; arXiv:1706.01787. [Google Scholar] [PubMed]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, M.I.; Jamshidi, N.; Steingrimsson, E.; Thiele, I.; Palsson, B.Ø. A detailed genome-wide reconstruction of mouse metabolism based on human recon 1. BMC Syst. Biol. 2010, 4, 140. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Swainston, N.; Fleming, R.M.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Eddy, J.A.; Price, N.D. Reconstruction of genome-scale metabolic models for 126 human tissues using mcadre. BMC Syst. Biol. 2012, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using init. PLoS Comput. Biol. 2012, 8, e1002518. [Google Scholar] [CrossRef] [PubMed]
- Borenstein, E.; Kupiec, M.; Feldman, M.W.; Ruppin, E. Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proc. Natl. Acad. Sci. USA 2008, 105, 14482–14487. [Google Scholar] [CrossRef] [PubMed]
- Steinway, S.N.; Biggs, M.B.; Loughran, T.P., Jr.; Papin, J.A.; Albert, R. Inference of network dynamics and metabolic interactions in the gut microbiome. PLoS Comput. Biol. 2015, 11, e1004338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hua, Q. Applications of genome-scale metabolic models in biotechnology and systems medicine. Front. Physiol. 2016, 6, 413. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.E.; Collart, F.R.; Field, D.; Meyer, F.; Keegan, K.P.; Henry, C.S.; McGrath, J.; Quinn, J.; Gilbert, J.A. Predicted relative metabolomic turnover (PRMT): Determining metabolic turnover from a coastal marine metagenomic dataset. Microb. Inform. Exp. 2011, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Noecker, C.; Eng, A.; Srinivasan, S.; Theriot, C.M.; Young, V.B.; Jansson, J.K.; Fredricks, D.N.; Borenstein, E. Metabolic model-based integration of microbiome taxonomic and metabolomic profiles elucidates mechanistic links between ecological and metabolic variation. mSystems 2016, 1, e00013-15. [Google Scholar] [CrossRef] [PubMed]
- McGeachie, M.J.; Sordillo, J.E.; Gibson, T.; Weinstock, G.M.; Liu, Y.-Y.; Gold, D.R.; Weiss, S.T.; Litonjua, A. Longitudinal prediction of the infant gut microbiome with dynamic bayesian networks. Sci. Rep. 2016, 6, 20359. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sova, P.; Xu, Q.; Dombek, K.M.; Xu, E.Y.; Vu, H.; Tu, Z.; Brem, R.B.; Bumgarner, R.E.; Schadt, E.E. Stitching together multiple data dimensions reveals interacting metabolomic and transcriptomic networks that modulate cell regulation. PLoS Biol. 2012, 10, e1001301. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Shoaie, S.; Karlsson, F.; Mardinoglu, A.; Nookaew, I.; Bordel, S.; Nielsen, J. Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci. Rep. 2013, 3, 2532. [Google Scholar] [CrossRef] [PubMed]
- El-Semman, I.E.; Karlsson, F.H.; Shoaie, S.; Nookaew, I.; Soliman, T.H.; Nielsen, J. Genome-scale metabolic reconstructions of Bifidobacterium adolescentis L2–32 and Faecalibacterium prausnitzii A2–165 and their interaction. BMC Syst. Biol. 2014, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Shoaie, S.; Ghaffari, P.; Kovatcheva-Datchary, P.; Mardinoglu, A.; Sen, P.; Pujos-Guillot, E.; de Wouters, T.; Juste, C.; Rizkalla, S.; Chilloux, J.; et al. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 2015, 22, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Harcombe, W.R.; Riehl, W.J.; Dukovski, I.; Granger, B.R.; Betts, A.; Lang, A.H.; Bonilla, G.; Kar, A.; Leiby, N.; Mehta, P.; et al. Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 2014, 7, 1104–1115. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chong, J.; Xia, J. Computational Approaches for Integrative Analysis of the Metabolome and Microbiome. Metabolites 2017, 7, 62. https://doi.org/10.3390/metabo7040062
Chong J, Xia J. Computational Approaches for Integrative Analysis of the Metabolome and Microbiome. Metabolites. 2017; 7(4):62. https://doi.org/10.3390/metabo7040062
Chicago/Turabian StyleChong, Jasmine, and Jianguo Xia. 2017. "Computational Approaches for Integrative Analysis of the Metabolome and Microbiome" Metabolites 7, no. 4: 62. https://doi.org/10.3390/metabo7040062
APA StyleChong, J., & Xia, J. (2017). Computational Approaches for Integrative Analysis of the Metabolome and Microbiome. Metabolites, 7(4), 62. https://doi.org/10.3390/metabo7040062