Monitoring for Response to Antineoplastic Drugs: The Potential of a Metabolomic Approach


Abstract
1. Introduction
2. Serial Scans: The Standard Approach to Response Estimation
3. “Response” as a Reflection of Therapeutic Benefit
4. Biomarker-Based Methods of Response
5. Serial Monitoring of the Metabolome
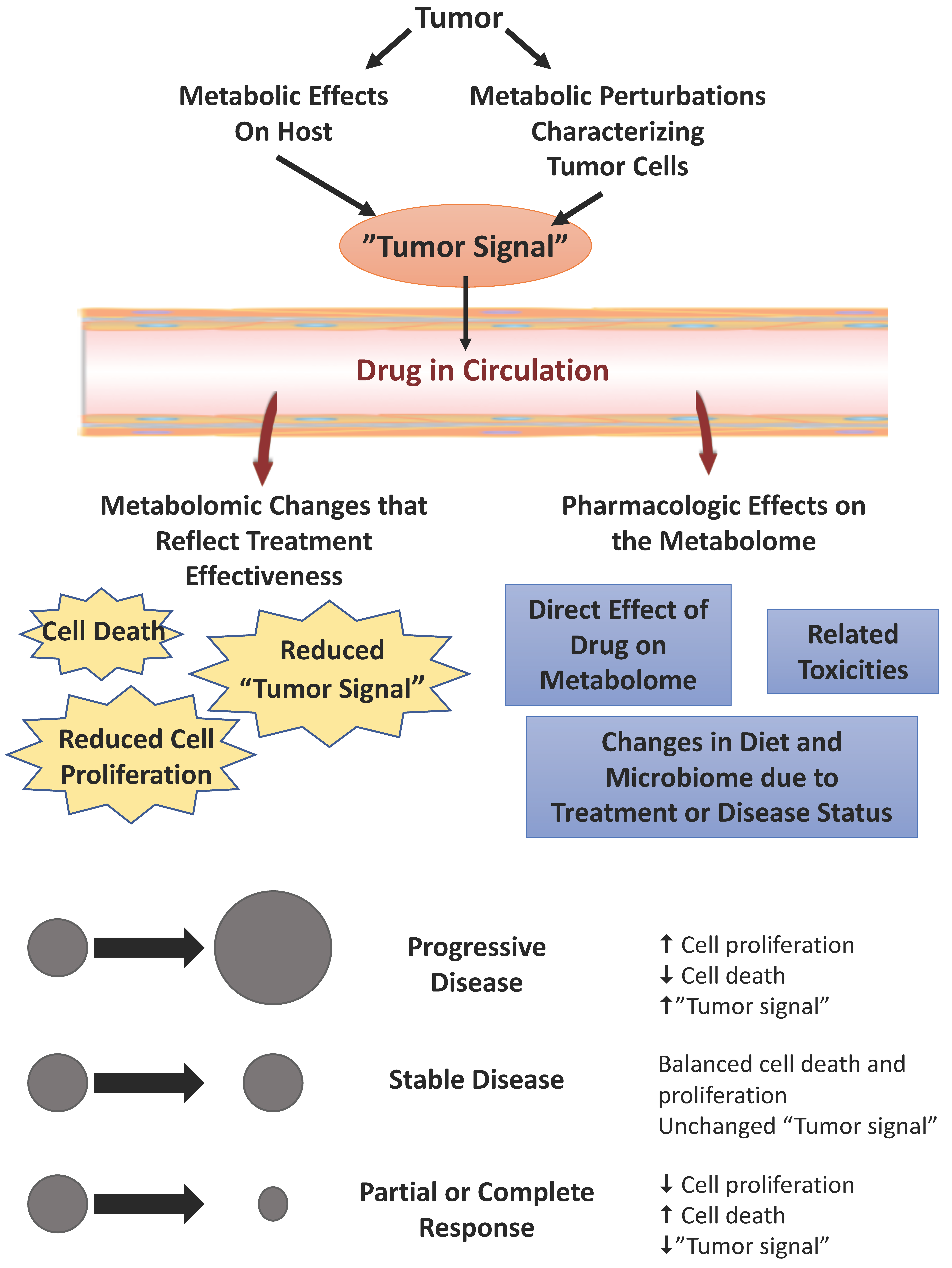
5.1. Putative Causes of Treatment-Related Changes in the Metabolome
5.2. In-Vitro Studies
5.3. In-Vivo Studies
6. Early Detection of Chemoresistance
7. Response Biomarkers as a Disruptive Influence on Oncologic Practice
8. Challenges in the Development of a Response Biomarker
8.1. Selecting the Best Analytical Platforms for Biomarker Discovery
8.2. Distinguishing Changes in the Metabolome that Reflect Treatment Effectiveness from Changes that Are Due to Pharmacological Effects
8.3. Accounting for Genetic Differences, Dietary Variations and Environmental Influences
8.4. Longitudinal Assessment of the Metabolome: Analytical Challenges
8.5. Response to Cytotoxic Agents vs. Cytostatic Agents
8.6. Assessment of Stable Disease
8.7. The Kinetics of Response
8.8. Drug-Specific vs. Generalizable Features of Response
9. Conclusions
Conflicts of Interest
Abbreviations
| CT | Computed tomography |
| FCH | Fluorocholine |
| FDG | Fluorodeoxyglucose |
| FLT | Fluorothymidine |
| GC | Gas chromatography |
| LC | Liquid chromatography |
| MALDI-IMS | Matrix-assisted laser desorption/ionization imaging mass spectrometry |
| MS | Mass spectrometry |
| NMR | Nuclear magnetic resonance |
| OS | Overall survival |
| PET | Positron emission tomography |
| PFS | Progression free survival |
References
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/search/browse?brwse=intr_cat_ANeo (accessed on 15 July 2017).
- Friboulet, L.; Olaussen, K.A.; Pignon, J.P.; Shepherd, F.A.; Tsao, M.S.; Graziano, S.; Kratzke, R.; Douillard, J.Y.; Seymour, L.; Pirker, R.; et al. ERCC1 isoform expression and DNA repair in non-small-cell lung cancer. N. Engl. J. Med. 2013, 368, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Vilmar, A.; Garcia-Foncillas, J.; Huarriz, M.; Santoni-Rugiu, E.; Sorensen, J.B. RT-PCR versus immunohistochemistry for correlation and quantification of ERCC1, BRCA1, TUBB3 and RRM1 in NSCLC. Lung Cancer 2012, 75, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Charnsangavej, C.; Faria, S.C.; Macapinlac, H.A.; Burgess, M.A.; Patel, S.R.; Chen, L.L.; Podoloff, D.A.; Benjamin, R.S. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: Proposal of new computed tomography response criteria. J. Clin. Oncol. 2007, 25, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Choi, H. Response evaluation of gastrointestinal stromal tumors. Oncologist 2008, 13, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Prior, J.O.; Montemurro, M.; Orcurto, M.V.; Michielin, O.; Luthi, F.; Benhattar, J.; Guillou, L.; Elsig, V.; Stupp, R.; Delaloye, A.B.; et al. Early prediction of response to sunitinib after imatinib failure by 18F-fluorodeoxyglucose positron emission tomography in patients with gastrointestinal stromal tumor. J. Clin. Oncol. 2009, 27, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Sohn, H.J.; Yang, Y.J.; Ryu, J.S.; Oh, S.J.; Im, K.C.; Moon, D.H.; Lee, D.H.; Suh, C.; Lee, J.S.; Kim, S.W. 18F Fluorothymidine positron emission tomography before and 7 days after gefitinib treatment predicts response in patients with advanced adenocarcinoma of the lung. Clin. Cancer Res. 2008, 14, 7423–7429. [Google Scholar] [CrossRef] [PubMed]
- Kenny, L.; Coombes, R.C.; Vigushin, D.M.; Al-Nahhas, A.; Shousha, S.; Aboagye, E.O. Imaging early changes in proliferation at 1 week post chemotherapy: A pilot study in breast cancer patients with 3′-deoxy-3′-18F fluorothymidine positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Parashar, B.; Wernicke, A.G.; Rice, S.; Osborne, J.; Singh, P.; Nori, D.; Vallabhajosula, S.; Goldsmith, S.; Chao, K.S. Early assessment of radiation response using a novel functional imaging modality—18F fluorocholine PET (FCH-PET): A pilot study. Discov. Med. 2012, 14, 13–20. [Google Scholar] [PubMed]
- De Giorgi, U.; Caroli, P.; Scarpi, E.; Conteduca, V.; Burgio, S.L.; Menna, C.; Moretti, A.; Galassi, R.; Rossi, L.; Amadori, D.; et al. 18F-Fluorocholine PET/CT for early response assessment in patients with metastatic castration-resistant prostate cancer treated with enzalutamide. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Shi, Y.; Wang, S.; Lu, Q. Meta-analysis showing that early response to neoadjuvant chemotherapy predicts better survival among cervical cancer patients. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Cloyd, J.M.; Wang, H.; Egger, M.E.; Tzeng, C.D.; Prakash, L.R.; Maitra, A.; Varadhachary, G.R.; Shroff, R.; Javle, M.; Fogelman, D.; et al. association of clinical factors with a major pathologic response following preoperative therapy for pancreatic ductal adenocarcinoma. JAMA Surg. 2017. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.; Pascal, G.; Castaing, D.; Azoulay, D.; Delvart, V.; Paule, B.; Levi, F.; Bismuth, H. Tumor progression while on chemotherapy: A contraindication to liver resection for multiple colorectal metastases? Ann. Surg. 2004, 240, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Tannir, N.M.; Figlin, R.A.; Gore, M.E.; Michaelson, M.D.; Motzer, R.J.; Porta, C.; Rini, B.I.; Hoang, C.; Lin, X.; Escudier, B. Long-term response to sunitinib treatment in metastatic renal cell carcinoma: A pooled analysis of clinical trials. Clin. Genitourin. Cancer 2017. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Pietrantonio, F.; Cremolini, C.; Di Bartolomeo, M.; Coinu, A.; Lonati, V.; de Braud, F.; Barni, S. Early tumour shrinkage as a prognostic factor and surrogate end-point in colorectal cancer: A systematic review and pooled-analysis. Eur. J. Cancer 2015, 51, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Fukada, I.; Araki, K.; Kobayashi, K.; Shibayama, T.; Takahashi, S.; Gomi, N.; Kokubu, Y.; Oikado, K.; Horii, R.; Akiyama, F.; et al. Pattern of tumor shrinkage during neoadjuvant chemotherapy is associated with prognosis in low-grade luminal early breast cancer. Radiology 2017. [Google Scholar] [CrossRef] [PubMed]
- Osumi, H.; Takahari, D.; Shinozaki, E.; Chin, K.; Ogura, M.; Wakatsuki, T.; Ichimura, T.; Nakayama, I.; Matsushima, T.; Yamaguchi, K. Associations between early tumor shrinkage and depth of response and clinical outcomes in patients treated with 1st-line chemotherapy for advanced gastric cancer. Gastric Cancer 2017. [Google Scholar] [CrossRef] [PubMed]
- Modest, D.P.; Stintzing, S.; Fischer von Weikersthal, L.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Lerchenmuller, C.; Kahl, C.; et al. Relation of early tumor shrinkage (ETS) observed in first-line treatment to efficacy parameters of subsequent treatment in FIRE-3 (AIOKRK0306). Int. J. Cancer 2017, 140, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Sun, Y.; Chen, L.; Mao, Y.P.; Tang, L.L.; Li, W.F.; Liu, X.; Zhang, W.N.; Zhou, G.Q.; Guo, R.; et al. Surrogate endpoints for overall survival in combined chemotherapy and radiotherapy trials in nasopharyngeal carcinoma: Meta-analysis of randomised controlled trials. Radiother. Oncol. 2015, 116, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Coinu, A.; Borgonovo, K.; Cabiddu, M.; Barni, S. Progression-free survival as surrogate endpoint in advanced pancreatic cancer: Meta-analysis of 30 randomized first-line trials. Hepatobiliary Pancreat Dis. Int. 2015, 14, 124–131. [Google Scholar] [CrossRef]
- Ciani, O.; Buyse, M.; Garside, R.; Peters, J.; Saad, E.D.; Stein, K.; Taylor, R.S. Meta-analyses of randomized controlled trials show suboptimal validity of surrogate outcomes for overall survival in advanced colorectal cancer. J. Clin. Epidemiol. 2015, 68, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Cartier, S.; Zhang, B.; Rosen, V.M.; Zarotsky, V.; Bartlett, J.B.; Mukhopadhyay, P.; Wagner, S.; Davis, C. Relationship between treatment effects on progression-free survival and overall survival in multiple myeloma: A systematic review and meta-analysis of published clinical trial data. Oncol. Res. Treat. 2015, 38, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Rotolo, F.; Pignon, J.P.; Bourhis, J.; Marguet, S.; Leclercq, J.; Tong Ng, W.; Ma, J.; Chan, A.T.; Huang, P.Y.; Zhu, G.; et al. Surrogate end points for overall survival in loco-regionally advanced nasopharyngeal carcinoma: An individual patient data meta-analysis. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Michiels, S.; Saad, E.D.; Buyse, M. Progression-free survival as a surrogate for overall survival in clinical trials of targeted therapy in advanced solid tumors. Drugs 2017, 77, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Nakamura, K.; Mizusawa, J.; Kato, K.; Eba, J.; Katayama, H.; Shibata, T.; Fukuda, H. Surrogacy of progression-free survival (PFS) for overall survival (OS) in esophageal cancer trials with preoperative therapy: Literature-based meta-analysis. Eur. J. Surg. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S.; Ahn, S.J.; Nah, B.S.; Chung, W.K.; Song, J.Y.; Jeong, J.U.; Nam, T.K. The metabolic response using 18F-fluorodeoxyglucose-positron emission tomography/computed tomography and the change in the carcinoembryonic antigen level for predicting response to pre-operative chemoradiotherapy in patients with rectal cancer. Radiother. Oncol. 2011, 98, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Mundle, S.D.; Marathe, A.S.; Chelladurai, M. Transient therapy-related surge in serum tumor biomarkers: Characterizing behavior and postulating its biologic role. Crit. Rev. Oncol. Hematol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, K.; Kawamoto, H.; Hirao, K.; Sakakihara, I.; Yamamoto, N.; Noma, Y.; Fujii, M.; Kato, H.; Ogawa, T.; Ishida, E.; et al. Monitoring of CA19-9 and SPan-1 can facilitate the earlier confirmation of progressing pancreatic cancer during chemotherapy. Pancreatology 2012, 12, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, J.; Brown, C.; Coeffic, D.; Raban, N.; Pfisterer, J.; Maenpaa, J.; Chalchal, H.; Fitzharris, B.; Volgger, B.; Vergote, I.; et al. CA-125 can be part of the tumour evaluation criteria in ovarian cancer trials: Experience of the GCIG CALYPSO trial. Br. J. Cancer 2012, 106, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Pavel, M.; Phan, A.T.; Kulke, M.H.; Hoosen, S.; St Peter, J.; Cherfi, A.; Oberg, K.E. Chromogranin A and neuron-specific enolase as prognostic markers in patients with advanced pNET treated with everolimus. J. Clin Endocrinol. Metab. 2011, 96, 3741–3749. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.E.; Sim, S. Evolving role of bone biomarkers in castration-resistant prostate cancer. Neoplasia 2010, 12, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Smerage, J.B.; Barlow, W.E.; Hortobagyi, G.N.; Winer, E.P.; Leyland-Jones, B.; Srkalovic, G.; Tejwani, S.; Schott, A.F.; O’Rourke, M.A.; Lew, D.L.; et al. Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J. Clin. Oncol. 2014, 32, 3483–3489. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Kirkwood, A.A.; Tsigani, T.; Lowe, H.; Goldstein, R.; Hartley, J.A.; Caplin, M.E.; Meyer, T. Early changes in circulating tumor cells are associated with response and survival following treatment of metastatic neuroendocrine neoplasms. Clin. Cancer Res. 2016, 22, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine metabolism in cancer: Understanding the heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, M.; Nakamoto, Y.; Shinohara, S.; Fujiwara, K.; Yamazaki, H.; Kanazawa, Y.; Kurihara, R.; Kishimoto, I.; Harada, H.; Naito, Y. Early evaluation of neoadjuvant chemotherapy response using FDG-PET/CT predicts survival prognosis in patients with head and neck squamous cell carcinoma. Int. J. Clin. Oncol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Duch, J.; Fuster, D.; Munoz, M.; Fernandez, P.L.; Paredes, P.; Fontanillas, M.; Skaltsa, K.; Domenech, B.; Lomena, F.; Pons, F. PET/CT with 18F fluorodeoxyglucose in the assessment of metabolic response to neoadjuvant chemotherapy in locally advanced breast cancer. Q. J. Nucl. Med. Mol. Imaging 2012, 56, 291–298. [Google Scholar] [PubMed]
- Herrmann, K.; Benz, M.R.; Czernin, J.; Allen-Auerbach, M.S.; Tap, W.D.; Dry, S.M.; Schuster, T.; Eckardt, J.J.; Phelps, M.E.; Weber, W.A.; et al. 18F-FDG-PET/CT imaging as an early survival predictor in patients with primary high grade soft tissue sarcomas undergoing neoadjuvant therapy. Clin. Cancer Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, K.; Benz, M.R.; Krause, B.J.; Pomykala, K.L.; Buck, A.K.; Czernin, J. 18F-FDG-PET/CT in evaluating response to therapy in solid tumors: Where we are and where we can go. Q. J. Nucl. Med. Mol. Imaging 2011, 55, 620–632. [Google Scholar] [PubMed]
- Decker, T.; Lohmann-Matthes, M.L. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods 1988, 115, 61–69. [Google Scholar] [CrossRef]
- Bayet-Robert, M.; Loiseau, D.; Rio, P.; Demidem, A.; Barthomeuf, C.; Stepien, G.; Morvan, D. Quantitative two-dimensional HRMAS 1H-NMR spectroscopy-based metabolite profiling of human cancer cell lines and response to chemotherapy. Magn. Reson. Med. 2010, 63, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Tiziani, S.; Kang, Y.; Choi, J.S.; Roberts, W.; Paternostro, G. Metabolomic high-content nuclear magnetic resonance-based drug screening of a kinase inhibitor library. Nat. Commun. 2011, 2, 545. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Wilson, M.; Mirbahai, L.; McConville, C.; Arvanitis, T.N.; Griffin, J.L.; Kauppinen, R.A.; Peet, A.C. In vitro metabonomic study detects increases in UDP-GlcNAc and UDP-GalNAc, as early phase markers of cisplatin treatment response in brain tumor cells. J. Proteome Res. 2011, 10, 3493–3500. [Google Scholar] [CrossRef] [PubMed]
- Sasada, S.; Miyata, Y.; Tsutani, Y.; Tsuyama, N.; Masujima, T.; Hihara, J.; Okada, M. Metabolomic analysis of dynamic response and drug resistance of gastric cancer cells to 5-fluorouracil. Oncol. Rep. 2013, 29, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Lamego, I.; Duarte, I.F.; Marques, M.P.; Gil, A.M. Metabolic markers of MG-63 osteosarcoma cell line response to doxorubicin and methotrexate treatment: Comparison to cisplatin. J. Proteome Res. 2014, 13, 6033–6045. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, H.; Yang, L.; Jiang, H.; Guo, S.; Li, Y.; Tao, S. Construction of a metabolomics profile of arsenic trioxide effect in gastric carcinoma cell line SGC7901. Acta Biochim. Biophys. Sin. 2016, 48, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Alonezi, S.; Tusiimire, J.; Wallace, J.; Dufton, M.J.; Parkinson, J.A.; Young, L.C.; Clements, C.J.; Park, J.K.; Jeon, J.W.; Ferro, V.A.; et al. Metabolomic profiling of the synergistic effects of melittin in combination with cisplatin on ovarian cancer cells. Metabolites 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Kather, M.; Willmann, L.; Schlimpert, M.; Bauer, C.; Lagies, S.; Schmidtkunz, K.; Eisenhardt, S.U.; Jung, M.; Gunther, S.; et al. Metabolic response to XD14 treatment in human breast cancer cell line MCF-7. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Lodi, A.; Ronen, S.M. Magnetic resonance spectroscopy detectable metabolomic fingerprint of response to antineoplastic treatment. PLoS ONE 2011, 6, e26155. [Google Scholar] [CrossRef] [PubMed]
- Halama, A.; Riesen, N.; Moller, G.; Hrabe de Angelis, M.; Adamski, J. Identification of biomarkers for apoptosis in cancer cell lines using metabolomics: Tools for individualized medicine. J. Intern. Med. 2013, 274, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Tiziani, S.; Lodi, A.; Khanim, F.L.; Viant, M.R.; Bunce, C.M.; Gunther, U.L. Metabolomic profiling of drug responses in acute myeloid leukaemia cell lines. PLoS ONE 2009, 4, e4251. [Google Scholar] [CrossRef]
- Cao, B.; Li, M.; Zha, W.; Zhao, Q.; Gu, R.; Liu, L.; Shi, J.; Zhou, J.; Zhou, F.; Wu, X.; et al. Metabolomic approach to evaluating adriamycin pharmacodynamics and resistance in breast cancer cells. Metabolomics 2013, 9, 960–973. [Google Scholar] [CrossRef] [PubMed]
- Farshidfar, F.; Weljie, A.M.; Kopciuk, K.; Buie, W.D.; Maclean, A.; Dixon, E.; Sutherland, F.R.; Molckovsky, A.; Vogel, H.J.; Bathe, O.F. Serum metabolomic profile as a means to distinguish stage of colorectal cancer. Genome Med. 2012, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Farshidfar, F.; Weljie, A.M.; Kopciuk, K.A.; Hilsden, R.; McGregor, S.E.; Buie, W.D.; MacLean, A.; Vogel, H.J.; Bathe, O.F. A validated metabolomic signature for colorectal cancer: Exploration of the clinical value of metabolomics. Br. J. Cancer 2016, 115, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Leichtle, A.B.; Nuoffer, J.M.; Ceglarek, U.; Kase, J.; Conrad, T.; Witzigmann, H.; Thiery, J.; Fiedler, G.M. Serum amino acid profiles and their alterations in colorectal cancer. Metabolomics 2012, 8, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Borgan, E.; Lindholm, E.M.; Moestue, S.; Maelandsmo, G.M.; Lingjaerde, O.C.; Gribbestad, I.S.; Borresen-Dale, A.L.; Engebraaten, O.; Sorlie, T. Subtype-specific response to bevacizumab is reflected in the metabolome and transcriptome of breast cancer xenografts. Mol. Oncol. 2013, 7, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Jobard, E.; Blanc, E.; Negrier, S.; Escudier, B.; Gravis, G.; Chevreau, C.; Elena-Herrmann, B.; Tredan, O. A serum metabolomic fingerprint of bevacizumab and temsirolimus combination as first-line treatment of metastatic renal cell carcinoma. Br. J. Cancer 2015, 113, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Yang, J.Y.; Kwack, S.J.; Kim, H.S.; Ryu, D.H.; Kim, Y.J.; Bae, J.Y.; Lim, D.S.; Choi, S.M.; Kwon, M.J.; et al. Potential metabolomic biomarkers for evaluation of adriamycin efficacy using a urinary 1H-NMR spectroscopy. J. Appl. Toxicol. 2013, 33, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Morvan, D.; Demidem, A. Metabolomics by proton nuclear magnetic resonance spectroscopy of the response to chloroethylnitrosourea reveals drug efficacy and tumor adaptive metabolic pathways. Cancer Res. 2007, 67, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Weaver, Z.; Difilippantonio, S.; Carretero, J.; Martin, P.L.; El Meskini, R.; Iacovelli, A.J.; Gumprecht, M.; Kulaga, A.; Guerin, T.; Schlomer, J.; et al. Temporal molecular and biological assessment of an erlotinib-resistant lung adenocarcinoma model reveals markers of tumor progression and treatment response. Cancer Res. 2012, 72, 5921–5933. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.D.; Giskeodegard, G.F.; Bathen, T.F.; Sitter, B.; Bofin, A.; Lonning, P.E.; Lundgren, S.; Gribbestad, I.S. Prognostic value of metabolic response in breast cancer patients receiving neoadjuvant chemotherapy. BMC Cancer 2012, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Liu, X.; Jiao, L.; Xu, C.; Zhang, Z.; Wang, L.; Li, Y.; Yang, C.; Zhang, W.; Sun, Y. Metabolomic evaluation of the response to endocrine therapy in patients with prostate cancer. Eur. J. Pharmacol. 2014, 729, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Gruner, B.M.; Winkelmann, I.; Feuchtinger, A.; Sun, N.; Balluff, B.; Teichmann, N.; Herner, A.; Kalideris, E.; Steiger, K.; Braren, R.; et al. Modeling therapy response and spatial tissue distribution of erlotinib in pancreatic cancer. Mol. Cancer Ther. 2016, 15, 1145–1152. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Beneficiary | Benefits |
|---|---|
| Benefits to the Patient |
|
| Effects on Clinical Practice |
|
| Socioeconomic Benefits |
|
| Benefits to Industry |
|
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rattner, J.; Bathe, O.F. Monitoring for Response to Antineoplastic Drugs: The Potential of a Metabolomic Approach. Metabolites 2017, 7, 60. https://doi.org/10.3390/metabo7040060
Rattner J, Bathe OF. Monitoring for Response to Antineoplastic Drugs: The Potential of a Metabolomic Approach. Metabolites. 2017; 7(4):60. https://doi.org/10.3390/metabo7040060
Chicago/Turabian StyleRattner, Jodi, and Oliver F. Bathe. 2017. "Monitoring for Response to Antineoplastic Drugs: The Potential of a Metabolomic Approach" Metabolites 7, no. 4: 60. https://doi.org/10.3390/metabo7040060
APA StyleRattner, J., & Bathe, O. F. (2017). Monitoring for Response to Antineoplastic Drugs: The Potential of a Metabolomic Approach. Metabolites, 7(4), 60. https://doi.org/10.3390/metabo7040060