Metabolomic Modularity Analysis (MMA) to Quantify Human Liver Perfusion Dynamics

Abstract
1. Introduction
2. Results
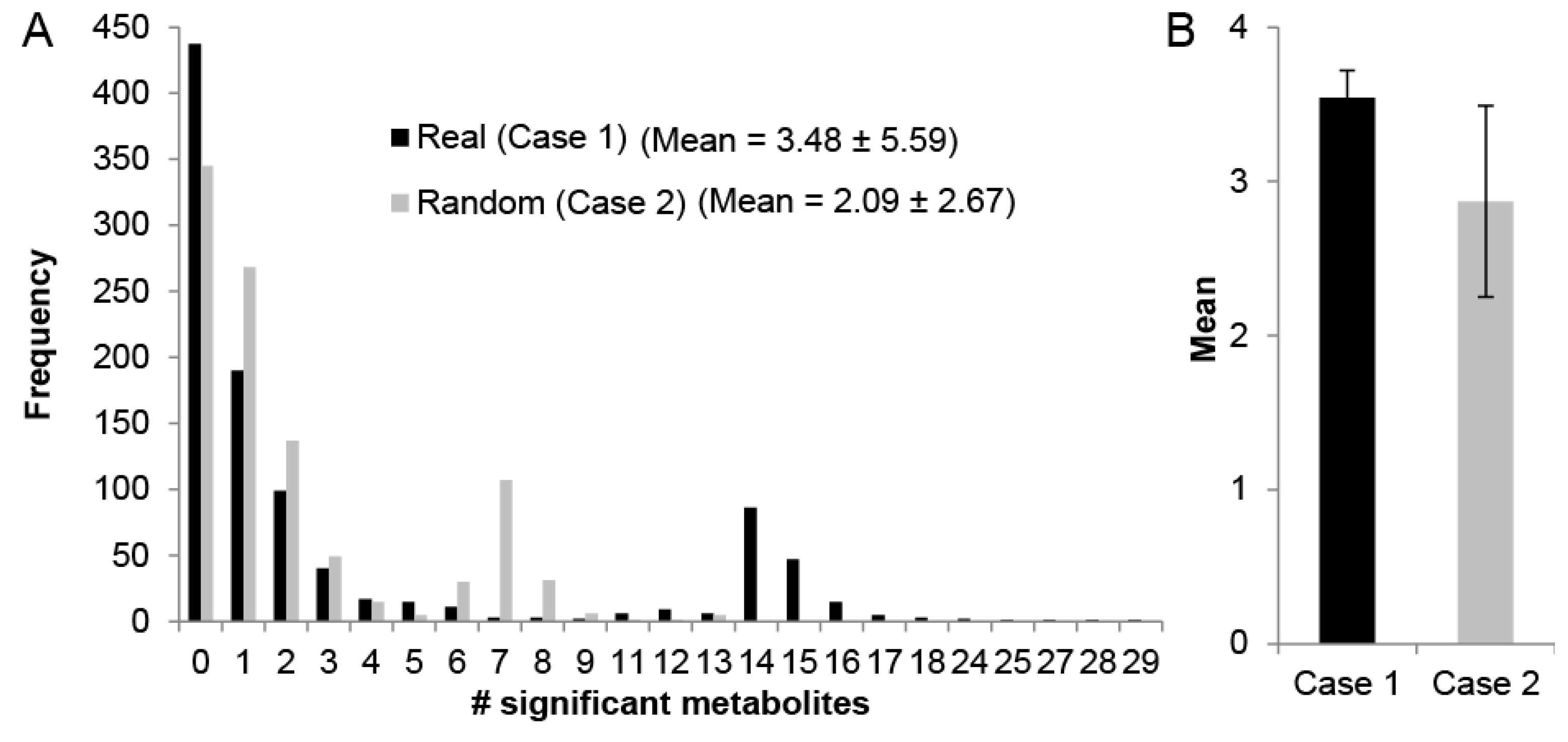
2.1. Human Recon 2.1 Network Topology Offers Insight into Metabolomics Data
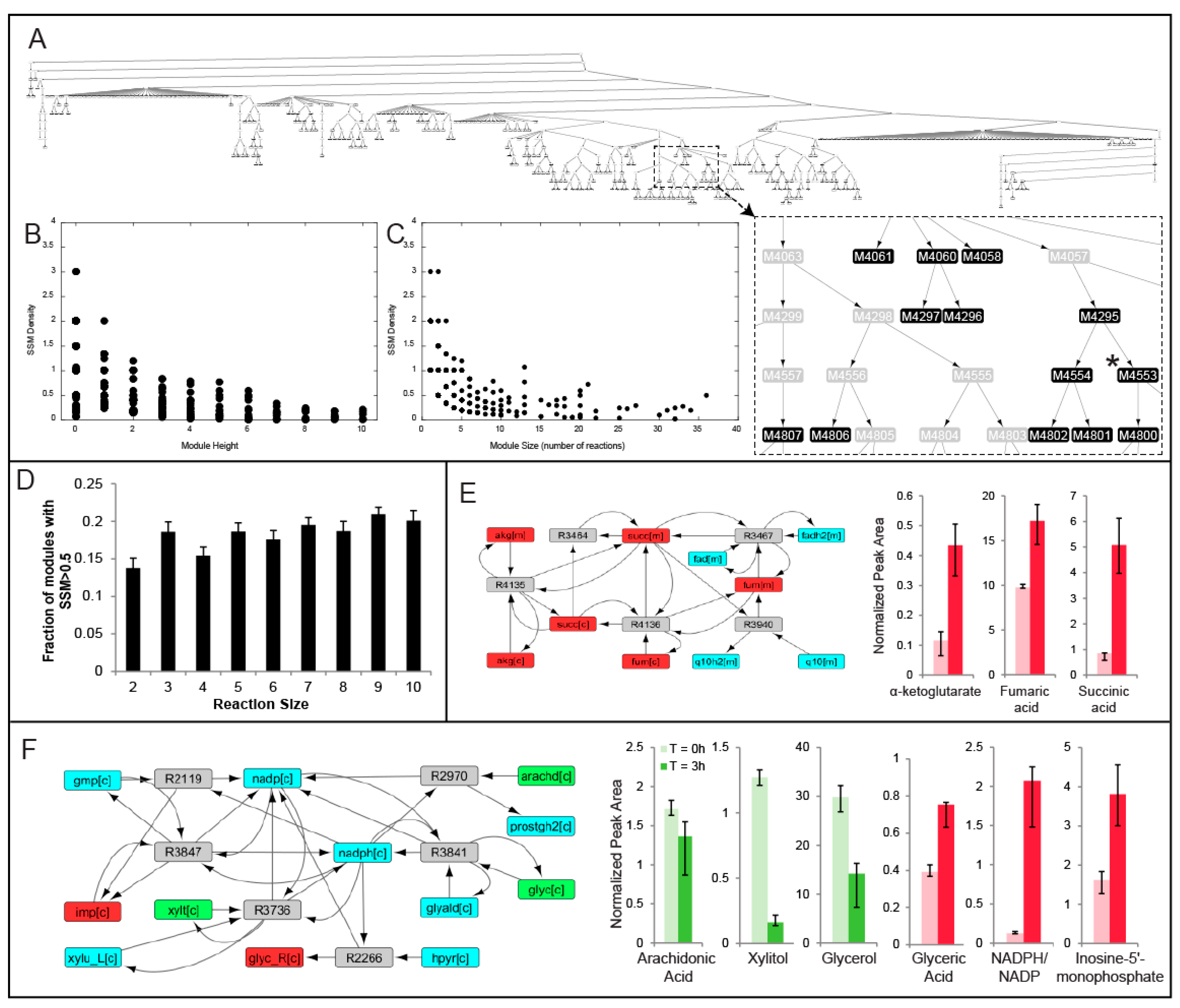
2.2. MMA Reveals Non-Intuitive Pathway Modules Engaged during Human Liver Perfusion
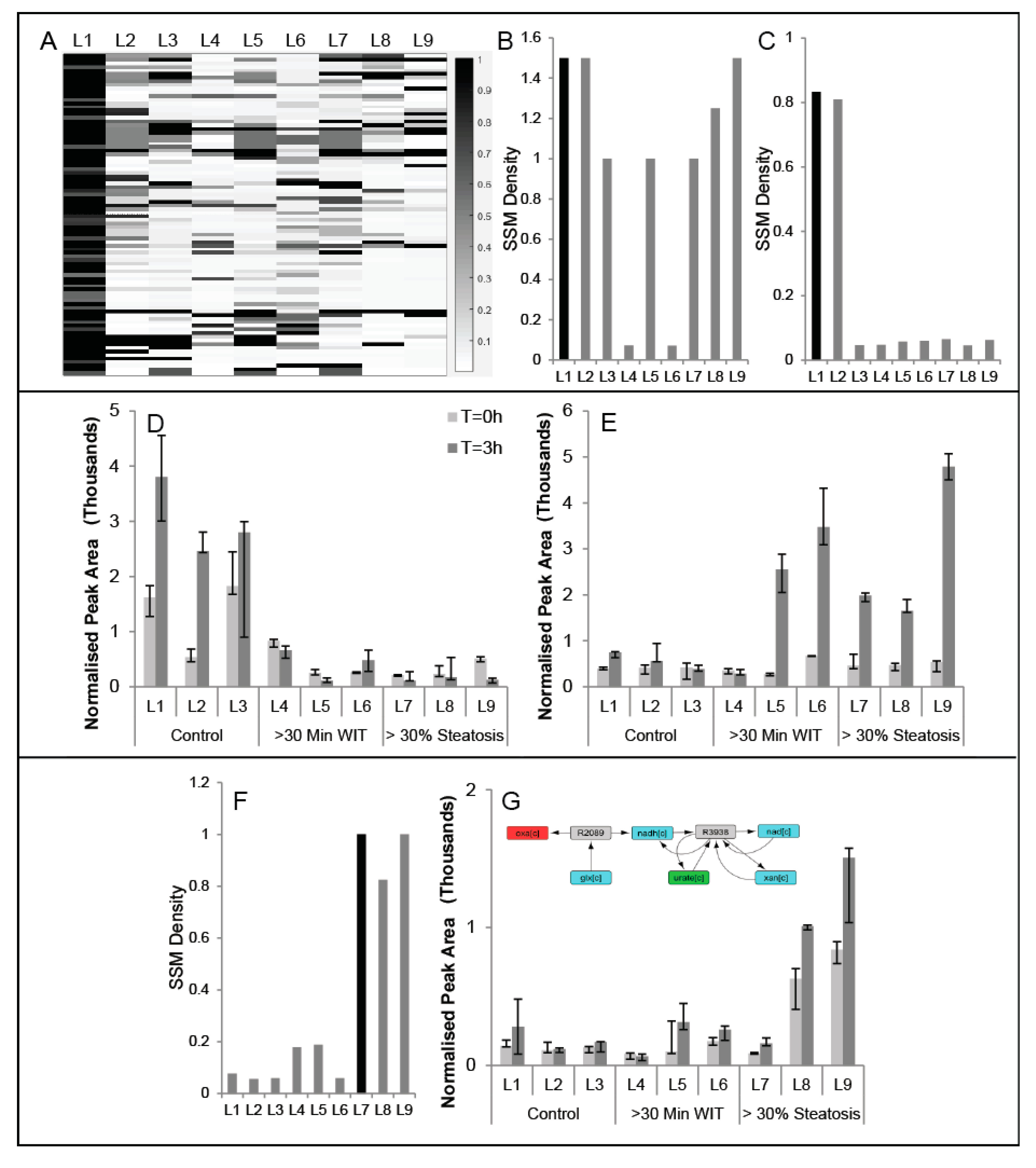
2.3. Comparison of Conserved Modules across Livers
2.4. Impact of Edge-Weights on Identifying Cofactor Modules
3. Discussion
4. Methods
4.1. Human Liver Perfusion
4.2. Metabolomics Analysis
4.3. Statistically Significant Metabolites (SSM)
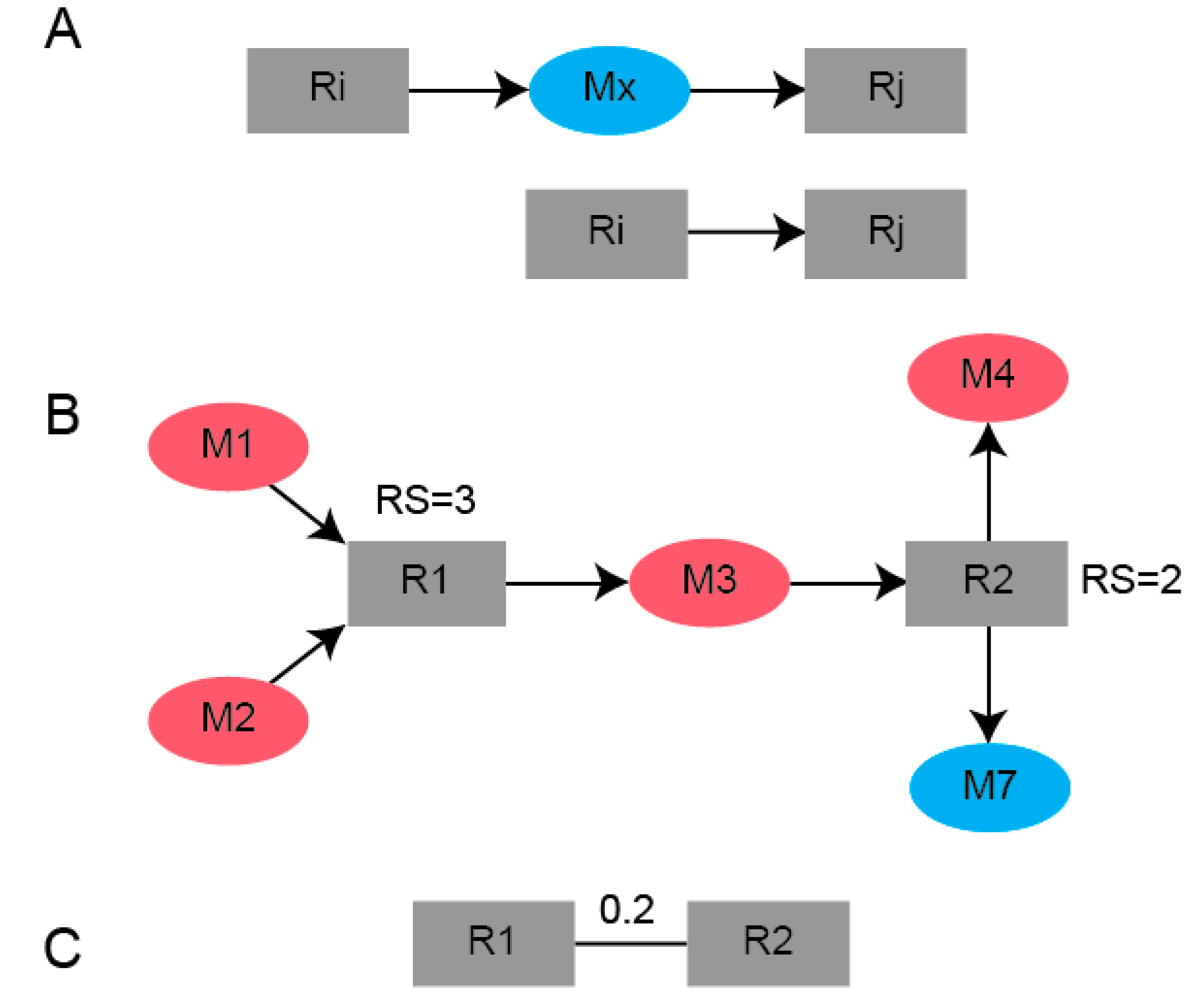
4.4. Bipartite Graph Construction
4.5. Reaction-Centric Adjacency Matrix Computation
4.6. B-Matrix Construction
4.7. Network Partitioning Using Newman’s Algorithm
4.8. Random Connected Subnetworks Computation
4.9. Pathway Enrichment Analysis (PEA)
4.10. Conserved Modules
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guo, T.; Kouvonen, P.; Koh, C.C.; Gillet, L.C.; Wolski, W.E.; Röst, H.L.; Rosenberger, G.; Collins, B.C.; Blum, L.C.; Gillessen, S.; et al. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat. Med. 2015, 21, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, G.; Koh, C.C.; Guo, T.; Röst, H.L.; Kouvonen, P.; Collins, B.C.; Heusel, M.; Liu, Y.; Caron, E.; Vichalkovski, A.; et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data 2014, 1. [Google Scholar] [CrossRef] [PubMed]
- Fahrmann, J.; Grapov, D.; Yang, J.; Hammock, B.; Fiehn, O.; Bell, G.I.; Hara, M. Systemic alterations in the metabolome of diabetic nod mice delineate increased oxidative stress accompanied by reduced inflammation and hypertriglyceridemia. Am. J. Physiol. Metab. 2015, 308, E978–E989. [Google Scholar]
- Yuan, M.; Breitkopf, S.B.; Yang, X.; Asara, J.M. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 2012, 7, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef] [PubMed]
- Nkuipou-Kenfack, E.; Duranton, F.; Gayrard, N.; Argilés, À.; Lundin, U.; Weinberger, K.M.; Dakna, M.; Delles, C.; Mullen, W.; Husi, H.; et al. Assessment of metabolomic and proteomic biomarkers in detection and prognosis of progression of renal function in chronic kidney disease. PLoS ONE 2014, 9, e96955. [Google Scholar] [CrossRef] [PubMed]
- Milreu, P.V.; Klein, C.C.; Cottret, L.; Acuña, V.; Birmelé, E.; Borassi, M.; Junot, C.; Marchetti-Spaccamela, A.; Marino, A.; Stougie, L.; et al. Telling metabolic stories to explore metabolomics data: A case study on the yeast response to cadmium exposure. Bioinformatics 2013, 30, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Kastenmüller, G.; Römisch-Margl, W.; Wägele, B.; Altmaier, E.; Suhre, K. metaP-server: A web-based metabolomics data analysis tool. Biomed. Res. Int. 2010, 2011. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.-C.; Tian, T.-F.; Tseng, Y.J. 3Omics: A web-based systems biology tool for analysis, integration and visualization of human transcriptomic, proteomic and metabolomic data. BMC Syst. Biol. 2013, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, S.; Dinh, N.-T.; Skolnick, J.; Styczynski, M.P. Metabolomics identifies the intersection of phosphoethanolamine with menaquinone-triggered apoptosis in an in vitro model of leukemia. Mol. Biosyst. 2015, 11, 2406–2416. [Google Scholar] [CrossRef] [PubMed]
- Weng, R.; Shen, S.; Tian, Y.; Burton, C.; Xu, X.; Liu, Y.; Chang, C.; Bai, Y.; Liu, H. Metabolomics approach reveals integrated metabolic network associated with serotonin deficiency. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Larson, M.G.; McCabe, E.L.; Murabito, J.M.; Rhee, E.P.; Ho, J.E.; Jacques, P.F.; Ghorbani, A.; Magnusson, M.; Souza, A.L.; et al. Distinct metabolomic signatures are associated with longevity in humans. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, B.G.; Yarmush, M.L.; Uygun, K. Organomatics and organometrics: Novel platforms for long-term whole-organ culture. Technology 2014, 2, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Stegemann, J.; Minor, T. Energy charge restoration, mitochondrial protection and reversal of preservation induced liver injury by hypothermic oxygenation prior to reperfusion. Cryobiology 2009, 58, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, B.G.; Sridharan, G.V.; Weeder, P.D.; Avruch, J.H.; Saeidi, N.; Özer, S.; Geerts, S.; Porte, R.J.; Heger, M.; van Gulik, T.M.; et al. Metabolic profiling during ex vivo machine perfusion of the human liver. Sci. Rep. 2016, 6, 22415. [Google Scholar] [CrossRef] [PubMed]
- Rajeev, K.G.; Nair, J.K.; Jayaraman, M.; Charisse, K.; Taneja, N.; O’Shea, J.; Willoughby, J.L.; Yucius, K.; Nguyen, T.; Shulga-Morskaya, S.; et al. Hepatocyte-specific delivery of sirnas conjugated to novel non-nucleosidic trivalent N-acetylgalactosamine elicits robust gene silencing in vivo. Chembiochem 2015, 16, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Dupont, P.; Callut, J.; Van Helden, J. Pathway discovery in metabolic networks by subgraph extraction. Bioinformatics 2010, 26, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Croes, D.; Couche, F.; Wodak, S.J.; van Helden, J. Inferring meaningful pathways in weighted metabolic networks. J. Mol. Biol. 2006, 356, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef] [PubMed]
- Kotze, H.L.; Armitage, E.G.; Sharkey, K.J.; Allwood, J.W.; Dunn, W.B.; Williams, K.J.; Goodacre, R. A novel untargeted metabolomics correlation-based network analysis incorporating human metabolic reconstructions. BMC Syst. Biol. 2013, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.; Datta, S.; Datta, S. A statistical framework for differential network analysis from microarray data. BMC Bioinform. 2010, 11, 95. [Google Scholar] [CrossRef] [PubMed]
- Frainay, C.; Jourdan, F. Computational methods to identify metabolic sub-networks based on metabolomic profiles. Brief. Bioinform. 2017, 18, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Deo, R.C.; Hunter, L.; Lewis, G.D.; Pare, G.; Vasan, R.S.; Chasman, D.; Wang, T.J.; Gerszten, R.E.; Roth, F.P. Interpreting metabolomic profiles using unbiased pathway models. PLoS Comput. Biol. 2010, 6, e1000692. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, G.V.; Hassoun, S.; Lee, K. Identification of biochemical network modules based on shortest retroactive distances. PLoS Comput. Biol. 2011, 7, e1002262. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Swainston, N.; Fleming, R.M.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Schadt, E.E.; Linderman, M.D.; Sorenson, J.; Lee, L.; Nolan, G.P. Computational solutions to large-scale data management and analysis. Nat. Rev. Genet. 2010, 11, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, I.; Chang, A.; Ebeling, C.; Gremse, M.; Heldt, C.; Huhn, G.; Schomburg, D. BRENDA, the enzyme database: Updates and major new developments. Nucleic Acids Res. 2004, 32, D431–D433. [Google Scholar] [CrossRef] [PubMed]
- Haugaa, H.; Thorgersen, E.B.; Pharo, A.; Boberg, K.M.; Foss, A.; Line, P.D.; Sanengen, T.; Almaas, R.; Grindheim, G.; Pischke, S.E.; et al. Early bedside detection of ischemia and rejection in liver transplants by microdialysis. Liver Transplant. 2012, 18, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.; Richards, D.A.; Silva, M.A.; Ahmed, N.; Neil, D.A.; Murphy, N.; Mirza, D.F. Comparison of energy metabolism in liver grafts from donors after circulatory death and donors after brain death during cold storage and reperfusion. Br. J. Surg. 2014, 101, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R. 13C metabolic flux analysis: Optimal design of isotopic labeling experiments. Curr. Opin. Biotechnol. 2013, 24, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, A.; Hoffmann, S.; Holzhütter, H.-G. Including metabolite concentrations into flux balance analysis: Thermodynamic realizability as a constraint on flux distributions in metabolic networks. BMC Syst. Biol. 2007, 1, 23. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, B.G.; Yeh, H.; Ozer, S.; Martins, P.N.; Farmer, A.; Wu, W.; Saeidi, N.; Op den Dries, S.; Berendsen, T.A.; Smith, R.N.; et al. Subnormothermic machine perfusion for ex vivo preservation and recovery of the human liver for transplantation. Am. J. Transplant 2014, 14, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Liver | Classification | WIT | % Steatosis | Age | Gender |
|---|---|---|---|---|---|
| 1 | Control | 19 | 0 | 54 | M |
| 2 | Control | 19 | 0 | 42 | F |
| 3 | Control | 23 | 0 | 46 | M |
| 4 | WI | 36 | 0 | 35 | M |
| 5 | WI | 44 | 0 | 66 | F |
| 6 | WI | 54 | 0 | 50 | M |
| 7 | Steatotic | 16 | >33% | 44 | M |
| 8 | Steatotic | 24 | >33% | 69 | F |
| 9 | Steatotic | 27 | <33% | 68 | F |
| Liver | Num. SSM | Num. MMA Modules | Num. Modules Sig. Met. Density >0.5 | MMA Run Time (Hours) |
|---|---|---|---|---|
| 1 | 73 | 577 | 88 | 2.36 |
| 2 | 75 | 762 | 143 | 2.96 |
| 3 | 92 | 716 | 147 | 2.40 |
| 4 | 65 | 673 | 117 | 2.60 |
| 5 | 87 | 1061 | 191 | 2.33 |
| 6 | 77 | 544 | 102 | 2.76 |
| 7 | 65 | 748 | 143 | 2.51 |
| 8 | 94 | 894 | 170 | 2.53 |
| 9 | 79 | 790 | 141 | 2.67 |
| Liver | Total Modules | Counter-Intuitive Modules | % Total |
|---|---|---|---|
| 1 | 88 | 20 | 22.73 |
| 2 | 143 | 52 | 36.36 |
| 3 | 147 | 67 | 45.58 |
| 4 | 117 | 41 | 35.04 |
| 5 | 191 | 57 | 29.84 |
| 6 | 102 | 30 | 29.41 |
| 7 | 170 | 91 | 53.53 |
| 8 | 143 | 90 | 62.94 |
| 9 | 141 | 68 | 48.23 |
| Average | 40.41 | ||
| Stdev | 13.03 |
| Liver | Total Modules | Number of Corresponding Un-Weighted Modules | % Total |
|---|---|---|---|
| 1(25) | 88 | 13 | 14.77 |
| 2(35) | 143 | 9 | 6.29 |
| 3(28) | 147 | 7 | 4.76 |
| 4(34) | 117 | 2 | 1.71 |
| 5(27) | 191 | 9 | 4.71 |
| 6(8) | 102 | 4 | 3.92 |
| 7(30) | 170 | 3 | 1.76 |
| 8(23) | 143 | 5 | 3.50 |
| 9(31) | 141 | 8 | 5.67 |
| Average | 5.23 | ||
| Stdev | 3.91 |
| Liver | Total Modules | NADPH | ATP | NADH | FADH2 |
|---|---|---|---|---|---|
| 1 | 88 | 8 | 15 | 10 | 3 |
| 2 | 143 | 20 | 30 | 32 | 0 |
| 3 | 147 | 21 | 47 | 18 | 2 |
| 4 | 117 | 16 | 34 | 6 | 2 |
| 5 | 191 | 21 | 57 | 8 | 1 |
| 6 | 102 | 6 | 31 | 23 | 1 |
| 7 | 170 | 23 | 74 | 45 | 7 |
| 8 | 143 | 17 | 61 | 32 | 1 |
| 9 | 141 | 21 | 50 | 19 | 1 |
| Average | 17.00 | 44.33 | 21.44 | 2.00 | |
| Stdev | 6.08 | 18.40 | 12.98 | 2.06 | |
| Unweighted | 192 | 0 | 3 | 6 | 0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sridharan, G.V.; Bruinsma, B.G.; Bale, S.S.; Swaminathan, A.; Saeidi, N.; Yarmush, M.L.; Uygun, K. Metabolomic Modularity Analysis (MMA) to Quantify Human Liver Perfusion Dynamics. Metabolites 2017, 7, 58. https://doi.org/10.3390/metabo7040058
Sridharan GV, Bruinsma BG, Bale SS, Swaminathan A, Saeidi N, Yarmush ML, Uygun K. Metabolomic Modularity Analysis (MMA) to Quantify Human Liver Perfusion Dynamics. Metabolites. 2017; 7(4):58. https://doi.org/10.3390/metabo7040058
Chicago/Turabian StyleSridharan, Gautham Vivek, Bote Gosse Bruinsma, Shyam Sundhar Bale, Anandh Swaminathan, Nima Saeidi, Martin L. Yarmush, and Korkut Uygun. 2017. "Metabolomic Modularity Analysis (MMA) to Quantify Human Liver Perfusion Dynamics" Metabolites 7, no. 4: 58. https://doi.org/10.3390/metabo7040058
APA StyleSridharan, G. V., Bruinsma, B. G., Bale, S. S., Swaminathan, A., Saeidi, N., Yarmush, M. L., & Uygun, K. (2017). Metabolomic Modularity Analysis (MMA) to Quantify Human Liver Perfusion Dynamics. Metabolites, 7(4), 58. https://doi.org/10.3390/metabo7040058