Metabolic Profile of the Cellulolytic Industrial Actinomycete Thermobifida fusca

Abstract
1. Introduction
2. Results
2.1. Coverage Polar and Non-Polar Metabolites
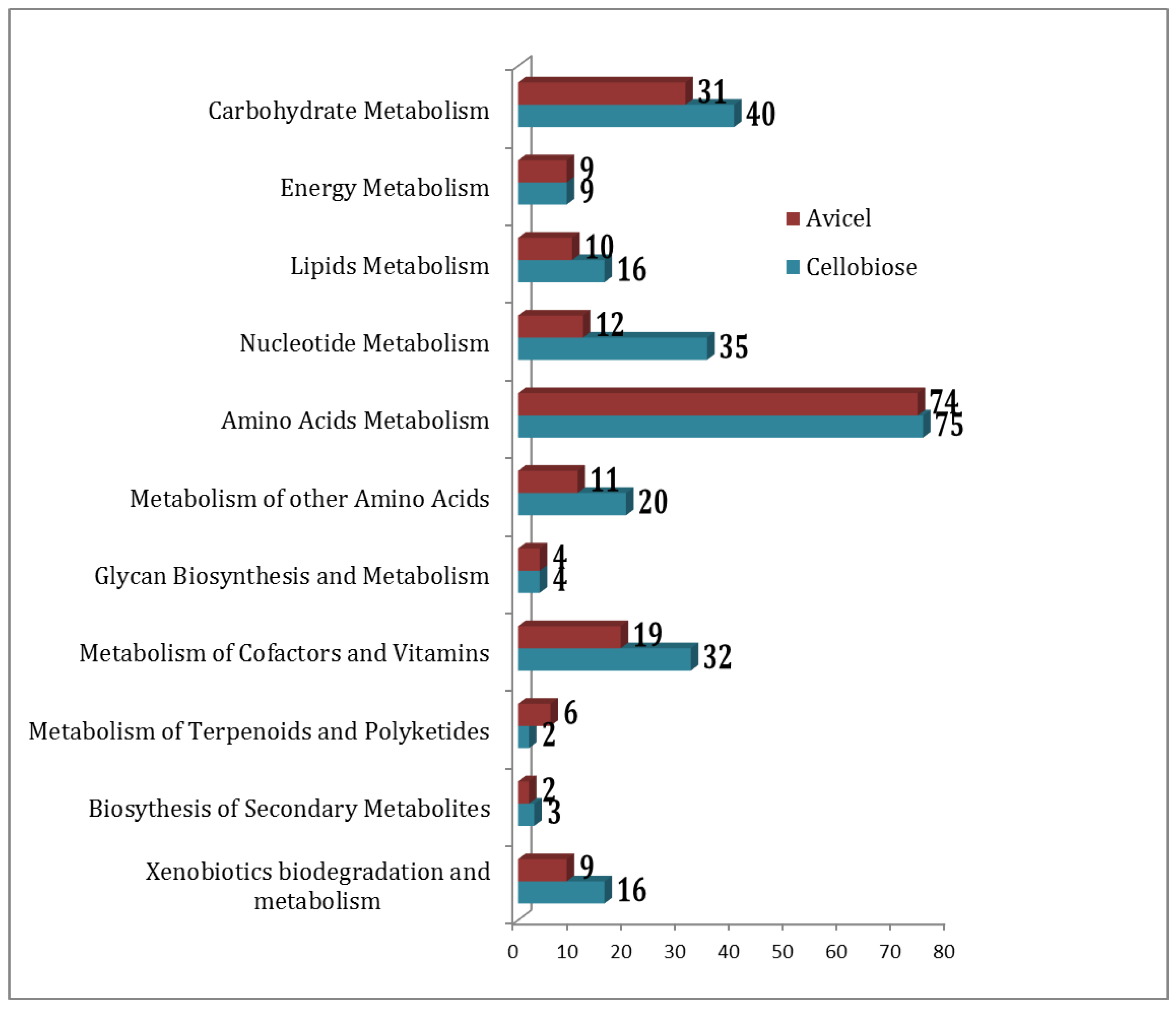
2.2. Functional Association of Metabolites
2.3. Metabolites for Cellobiose Growth
2.4. Metabolites for Avicel Growth
2.5. Comparing Cellobiose vs Avice
2.6. Metabolites in Secondary Metabolism
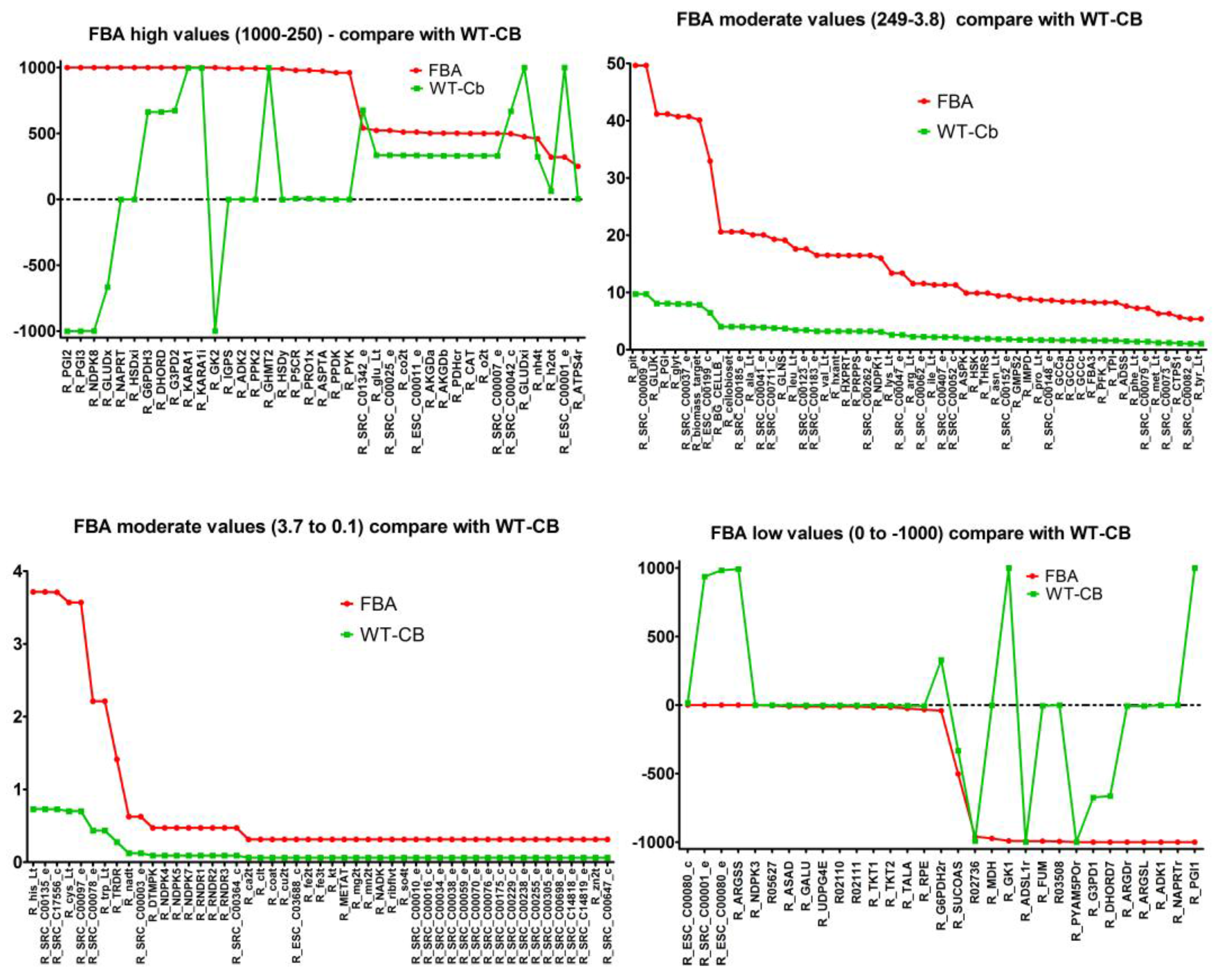
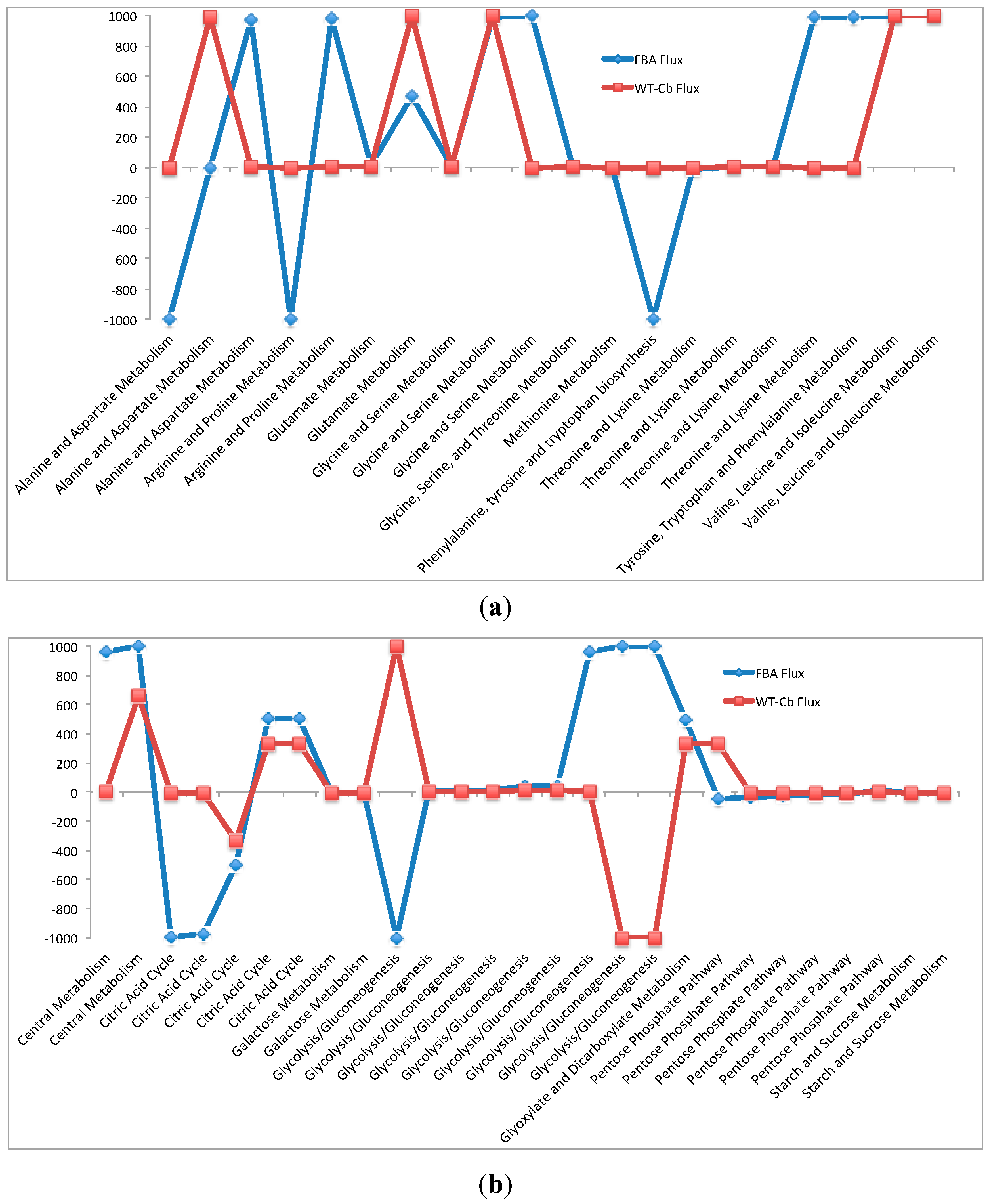
2.7. Integrating Metabolomic Data with a Metabolic Model
3. Discussion
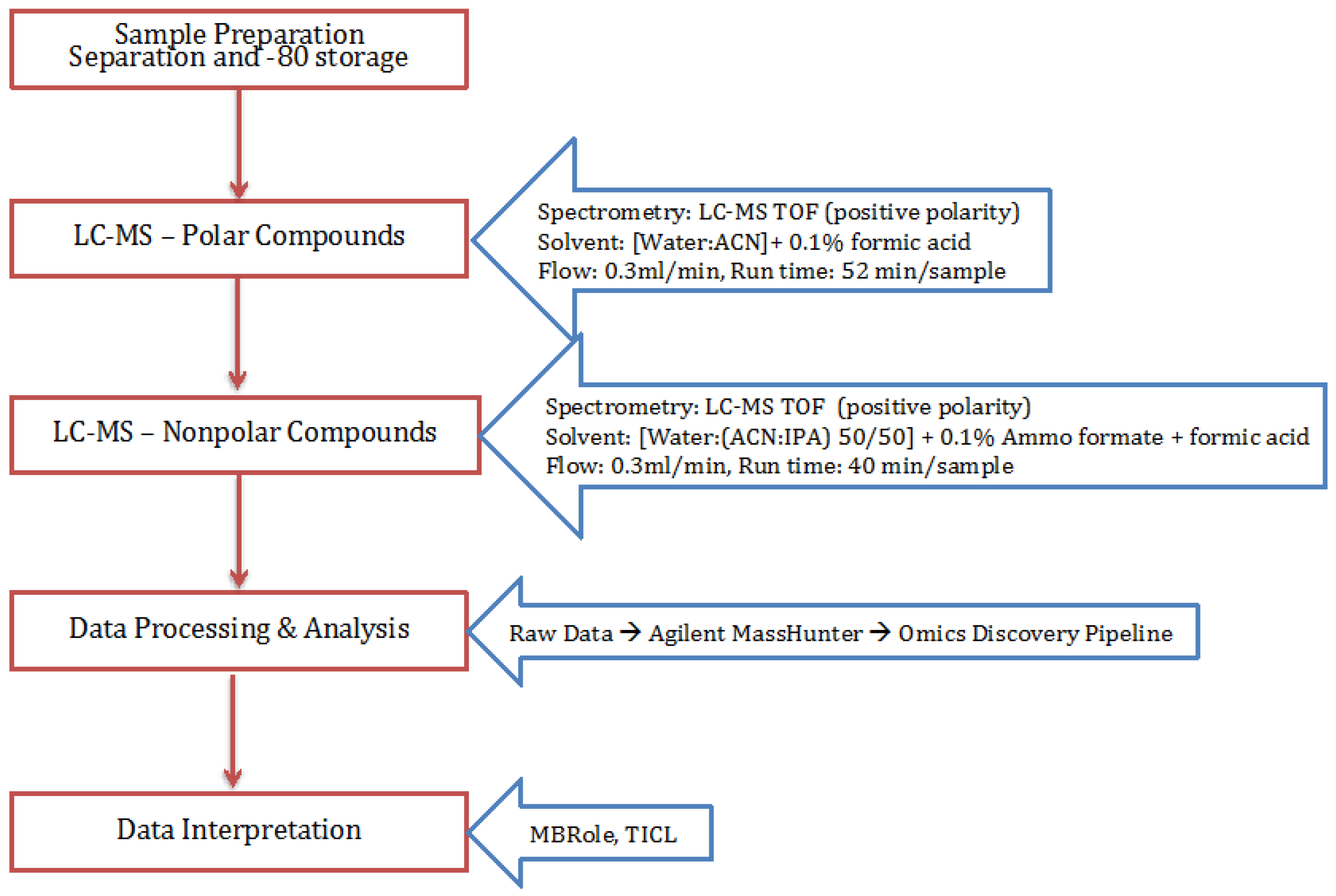
4. Materials and Methods
4.1. Cell Growth
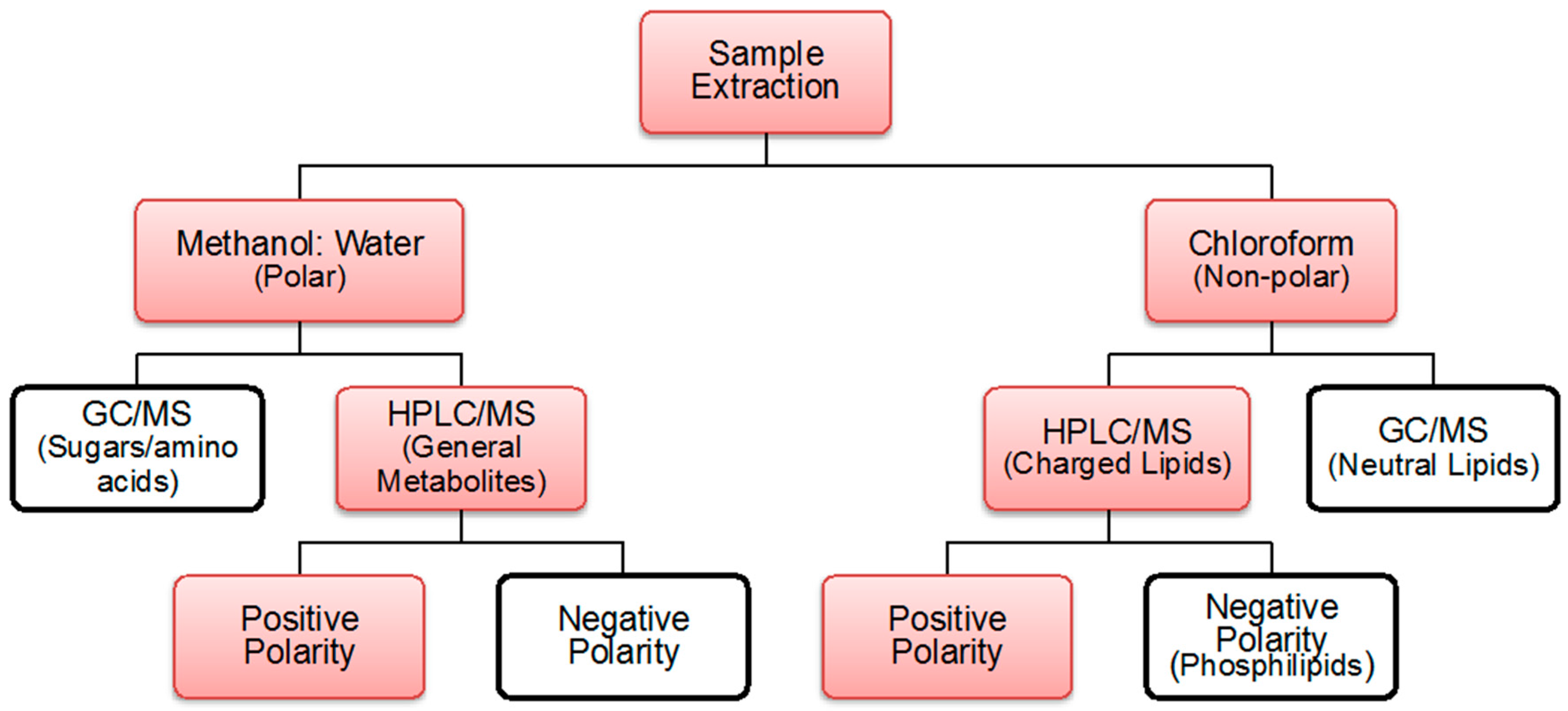
4.2. Extraction Process
4.3. Polar and Non-Polar General Metabolites Isolation
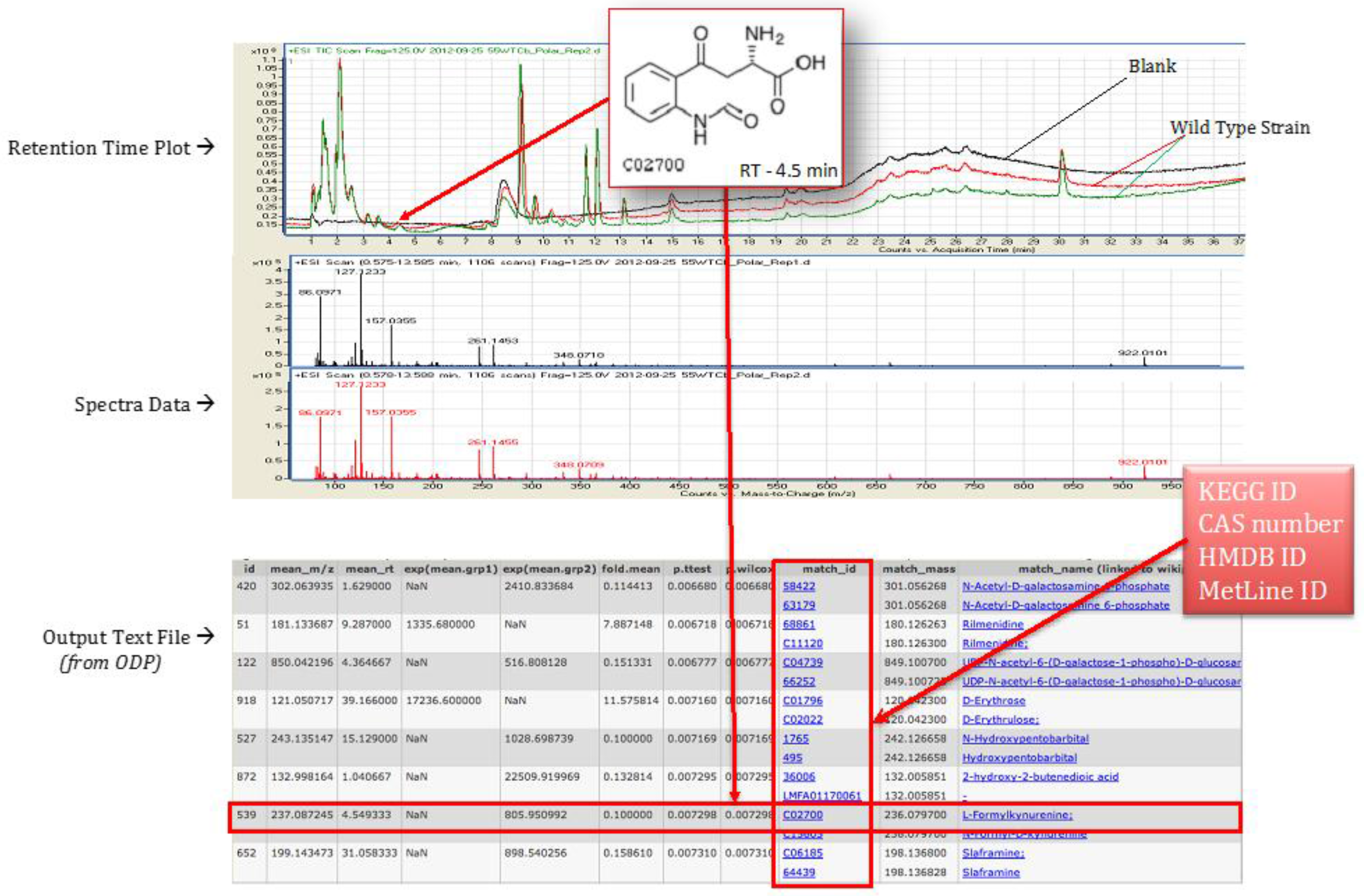
4.4. Statistical Analysis Using Omics Discovery Pipeline
4.5. Metabolite Identification and Pathway Association
4.6. Metabolic Model and Metabolomics Integration
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, J.; Postmaster, A.; Soon, H.P.; Keast, D.; Carson, K.C. Siderophore production by actinomycetes isolates from two soil sites in Western Australia. Biometals 2012, 25, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Niraula, N.P.; Kim, S.-H.; Sohng, J.K.; Kim, E.-S. Biotechnological doxorubicin production: Pathway and regulation engineering of strains for enhanced production. Appl. Microbiol. Biotechnol. 2010, 87, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Toyoda, A.; Sekiyama, Y.; Takagi, H.; Nogawa, T.; Uramoto, M.; Suzuki, R.; Koshino, H.; Kumano, T.; Panthee, S.; et al. Reveromycin A biosynthesis uses RevG and RevJ for stereospecific spiroacetal formation. Nat. Chem. Biol. 2011, 7, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Citron, C.A.; Gleitzmann, J.; Laurenzano, G.; Pukall, R.; Dickschat, J.S. Terpenoids are widespread in actinomycetes: A correlation of secondary metabolism and genome data. Chembiochem 2012, 13, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Cane, D.E.; Ikeda, H. Exploration and mining of the bacterial terpenome. Acc. Chem. Res. 2012, 45, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Vanee, N.; Brooks, J.P.; Spicer, V.; Shamshurin, D.; Krokhin, O.; Wilkins, J.A.; Deng, Y.; Fong, S.S. Proteomics-based metabolic modeling and characterization of the cellulolytic bacterium Thermobifida fusca. BMC Syst. Biol. 2014, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wilson, D.B. Proteomic and transcriptomic analysis of extracellular proteins and mRNA levels in Thermobifida fusca grown on cellobiose and glucose. J. Bacteriol. 2007, 189, 6260–6265. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Fong, S.S. Influence of culture aeration on the cellulase activity of Thermobifida fusca. Appl. Microbiol. Biotechnol. 2010, 85, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Fong, S.S. Development and application of a PCR-targeted gene disruption method for studying CelR function in Thermobifida fusca. Appl. Environ. Microbiol. 2010, 76, 2098–2106. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deng, Y.; Fong, S.S. Metabolic engineering of Thermobifida fusca for direct aerobic bioconversion of untreated lignocellulosic biomass to 1-propanol. Metab. Eng. 2011, 13, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Fong, S.S. Laboratory evolution and multi-platform genome re-sequencing of the cellulolytic actinobacterium Thermobifida fusca. J. Biol. Chem. 2011, 286, 39958–39966. [Google Scholar] [CrossRef] [PubMed]
- Spiridonov, N.A.; Wilson, D.B. Regulation of biosynthesis of individual cellulases in Thermomonospora fusca. J. Bacteriol. 1998, 180, 3529–3532. [Google Scholar] [PubMed]
- Spiridonov, N.A.; Wilson, D.B. Characterization and cloning of celR, a transcriptional regulator of cellulase genes from Thermomonospora fusca. J. Biol. Chem. 1999, 274, 13127–13132. [Google Scholar] [CrossRef] [PubMed]
- Chagoyen, M.; Pazos, F. MBRole: Enrichment analysis of metabolomic data. Bioinformatics 2011, 27, 730–731. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef] [PubMed]
- Ger, M.-F.; Rendon, G.; Tilson, J.L.; Jakobsson, E. Domain-based identification and analysis of glutamate receptor ion channels and their relatives in prokaryotes. PLoS ONE 2010, 5, e12827. [Google Scholar] [CrossRef] [PubMed]
- Adav, S.S.; Ng, C.S.; Sze, S.K. iTRAQ-based quantitative proteomic analysis of Thermobifida fusca reveals metabolic pathways of cellulose utilization. J. Proteom. 2011, 74, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, S.N.; Graham, R.L.J.; McMullan, G.; Ternan, N.G. A role for carbon catabolite repression in the metabolism of phosphonoacetate by Agromyces fucosus Vs2. FEMS Microbiol. Lett. 2006, 261, 133–140. [Google Scholar] [PubMed]
- Kuzma, J.; Nemecek-Marshall, M.; Pollock, W.H.; Fall, R. Bacteria produce the volatile hydrocarbon isoprene. Curr. Microbiol. 1995, 30, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, T.D.; Wiberley, A.E.; Donohue, A.R. Isoprene emission from plants: Why and how. Ann. Bot. 2008, 101, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Griffiths, W.J. Capillary liquid chromatography combined with tandem mass spectrometry for the study of neurosteroids and oxysterols in brain. Neurochem. Int. 2008, 52, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Riley, C.P.; Gough, E.S.; He, J.; Jandhyala, S.S.; Kennedy, B.; Orcun, S.; Ouzzani, M.; Buck, C.; Roumani, A.M.; Zhang, X. The proteome discovery pipeline—A data analysis pipeline for mass spectrometry-based differential proteomics discovery. Open Proteom. J. 2010, 3, 8–19. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Growth Condition | Total Detected Metabolites | Polar | Non-Polar | In Both |
|---|---|---|---|---|
| Cellobiose | 667 | 618 | 64 | 15 |
| Avicel | 461 | 402 | 82 | 23 |
| Category | Total Relations | Number of Compounds | No Annotation * | |||
|---|---|---|---|---|---|---|
| Pathways | 72 | 71 | 191 | 129 | 476 | 332 |
| Enzyme Interactions | 299 | 213 | 71 | 45 | 595 | 416 |
| Biological Roles | 71 | 63 | 67 | 45 | 600 | 416 |
| Chemical Groups | 62 | 56 | 186 | 123 | 481 | 338 |
| Other Interactions | 95 | 74 | 31 | 25 | 636 | 436 |
| KEGG Pathway ID | Pathway | Detected | Compounds |
|---|---|---|---|
| Unique to Cellobiose | |||
| tfu00230 | Purine metabolism | 15 | C00362 C04376 C00360 C06196 C00130 C00035 C00242 C00655 C00330 C00262 C05515 C03794 C00212 C00048 C00385 |
| tfu00240 | Pyrimidine metabolism | 8 | C00364 C00099 C00112 C00015 C00380 C00055 C02376 C03997 |
| tfu00270 | Cysteine and methionine metabolism | 7 | C00109 C02989 C00041 C00073 C01005 C00021 C01180 |
| tfu00330 | Arginine and proline metabolism | 6 | C00791 C00555 C05931 C00062 C00437 C00048 |
| tfu00450 | Selenoamino acid metabolism | 5 | C05696 C05692 C00041 C05335 C05708 |
| tfu00770 | Pantothenate and CoA biosynthesis | 4 | C00099 C01088 C03492 C00864 |
| tfu00740 | Riboflavin metabolism | 3 | C05775 C00472 C00061 |
| tfu00190 | Oxidative phosphorylation | 3 | C00004 C00061 C00003 |
| Unique to Avicel | |||
| tfu00300 | Lysine biosynthesis | 5 | C00047 C00322 C03340 C00049 C01251 |
| tfu00330 | Arginine and proline metabolism | 5 | C03440 C01043 C00049 C18174 C00327 |
| tfu00906 | Carotenoid biosynthesis | 5 | C16280 C08585 C08606 C15892 C08583 |
| tfu00260 | Glycine, serine and threonine metabolism | 5 | C03283 C06231 C00049 C00188 C00576 |
| tfu02010 | ABC transporters | 4 | C00049 C00047 C03557 C00188 |
| tfu00970 | Aminoacyl-tRNA biosynthesis | 4 | C00188 C00049 C00047 C00152 |
| tfu00340 | Histidine metabolism | 3 | C05575 C05131 C00049 |
| tfu00250 | Alanine, aspartate and glutamate metabolism | 3 | C00152 C00940 C00049 |
| tfu00310 | Lysine degradation | 3 | C00322 C00047 C00739 |
| tfu00910 | Nitrogen metabolism | 2 | C00152 C00049 |
| tfu00641 | 3-Chloroacrylic acid degradation | 2 | C16348 C06613 |
| KEGG Compound IDs | Description | Pathway ID | Name |
|---|---|---|---|
| Higher Concentration in Cellobiose | |||
| C05763 | (2E)-Hexadecenoyl-[acp] | tfu00061 | Fatty acid biosynthesis |
| C03871 | L-2-Amino-6-oxopimelate | tfu00300 | Lysine biosynthesis |
| C12986 | N2-Acetyl-L-aminoadipate | tfu00300 | Lysine biosynthesis |
| C00624 | N-Acetyl-L-glutamate | tfu00330 | Arginine and proline metabolism |
| C03415 | Succinyl-L-ornithine | tfu00330 | Arginine and proline metabolism |
| C00513 | CDP-glycerol | tfu00564 | Glycerophospholipid metabolism |
| Higher Concentration in Avicel | |||
| C00811 | 4-Coumarate | tfu00130 | Ubiquinone & other terpenoid-quinone biosynthesis |
| C03955 | N6-Acetyl-N6-hydroxy-L-lysine | tfu00310 | Lysine degradation |
| C00811 | 4-Coumarate | tfu00360 | Phenylalanine metabolism |
| C12836 | 2-Chloro-3-oxoadipate | tfu00361 | gamma-Hexachlorocyclohexane degradation |
| C05699 | L-Selenocystathionine | tfu00450 | Selenoamino acid metabolism |
| Both | Cb | Av | Total | |
|---|---|---|---|---|
| Alkaloids | 21 | 17 | 3 | 41 |
| Alkaloids derived by amination reactions | 6 | 1 | 1 | 8 |
| Alkaloids derived from lysine | 2 | 4 | 0 | 6 |
| Alkaloids derived from nicotinic acid | 2 | 2 | 0 | 4 |
| Alkaloids derived from ornithine | 7 | 1 | 1 | 9 |
| Alkaloids derived from tryptophan and anthranilic acid | 1 | 3 | 1 | 5 |
| Alkaloids derived from tyrosine | 3 | 3 | 0 | 6 |
| Others | 0 | 3 | 0 | 3 |
| Amino acid related compounds | 1 | 2 | 1 | 4 |
| Betalains | 1 | 2 | 1 | 4 |
| Fatty acids related compounds | 0 | 2 | 0 | 2 |
| Fatty acids | 0 | 2 | 0 | 2 |
| Flavonoids | 0 | 1 | 1 | 2 |
| Flavonoids | 0 | 1 | 1 | 2 |
| Phenylpropanoids | 3 | 4 | 1 | 8 |
| Coumarins | 1 | 2 | 1 | 4 |
| Lignans | 1 | 2 | 0 | 3 |
| Monolignols | 1 | 0 | 0 | 1 |
| Polyketides | 4 | 0 | 0 | 4 |
| Others | 2 | 0 | 0 | 2 |
| Pyrones | 2 | 0 | 0 | 2 |
| Terpenoids | 6 | 12 | 7 | 25 |
| Carotenoids and apocarotenoids | 0 | 0 | 3 | 3 |
| Diterpenoids (C20) | 0 | 4 | 1 | 5 |
| Hemiterpenoids (C5) | 1 | 0 | 0 | 1 |
| Monoterpenoids (C10) | 2 | 2 | 1 | 5 |
| Sesquiterpenoids (C15) | 3 | 3 | 1 | 7 |
| Steroids | 0 | 3 | 0 | 3 |
| Triterpenoids (C30) | 0 | 0 | 1 | 1 |
| Others | 2 | 0 | 0 | 2 |
| Others | 1 | 0 | 0 | 1 |
| Tannins and galloyl derivatives | 1 | 0 | 0 | 1 |
| Total | 37 | 38 | 13 | 88 |
| (a) Flux through Amino Acid Pathway before and after Metabolomics Data Integration | ||||
| Pathways | Flux−FBA | Flux−WT-Cb | ECs | Equation |
| Alanine and Aspartate Metabolism | −1000.00 | −7.55 | 4.3.2.1 | [c]: C03406 <==> C00062 + C00122 |
| 0.00 | 992.45 | 6.3.4.5 | [c]: C00049 + C00002 + C00327 → C00020 + C03406 + C00080 + C00013 | |
| 973.09 | 2.27 | 2.6.1.1 | [c]: C00026 + C00049 <==> C00025 + C00036 | |
| Arginine and Proline Metabolism | −1000.00 | −7.55 | 3.5.3.6 | [c]: C00062 + C00001 <==> C00327 + C01342 |
| 978.81 | 6.61 | 1.5.1.2 | [c]: C00003 + C00148 → C03912 + 2 C00080 + C00004 | |
| Glutamate Metabolism | 19.12 | 3.75 | 6.3.1.2 | [c]: C00002 + C00025 + C01342 → C00008 + C00064 + C00080 + C00009 |
| 475.06 | 1000.00 | 1.4.1.2 | [c]: C00025 + C00001 + C00003 → C00026 + C00080 + C00004 + C01342 | |
| Glycine and Serine Metabolism | 8.41 | 1.65 | 1.8.1.4 | [c]: C02972 + C00003 → C00080 + C02051 + C00004 |
| 991.59 | 998.35 | 2.1.2.1 | [c]: C00716 + C00101 → C00037 + C00001 + C00143 | |
| 1000.00 | 0.00 | 1.1.1.3 | [c]: C00441 + C00080 + C00004 → C00263 + C00003 | |
| Glycine, Serine, and Threonine Metabolism | 9.89 | 1.94 | 4.2.3.1 | [c]: C00001 + C01102 → C00009 + C00188 |
| Methionine Metabolism | 0.31 | 0.06 | 2.5.1.6 | [c]: C00002 + C00001 + C00073 → C00019 + C00009 + C00013 |
| Phenylalanine, Tyrosine, and Tryptophan Biosynthesis | −994.44 | 0.00 | 4.1.1.48 | [c]: C01302 <==> C03506 + C00011 + C00001 |
| Threonine and Lysine Metabolism | −9.89 | −1.94 | 1.2.1.11 | [c]: C00441 + C00006 + C00009 <==> C03082 + C00080 + C00005 |
| 9.89 | 1.94 | 2.7.2.4 | [c]: C00049 + C00002 <==> C03082 + C00008 | |
| 9.89 | 1.94 | 2.7.1.39 | [c]: C00002 + C00263 → C00008 + C00080 + C01102 | |
| 990.11 | −1.94 | 1.1.1.3 | [c]: C00263 + C00006 <==> C00441 + C00080 + C00005 | |
| Tyrosine, Tryptophan, and Phenylalanine Metabolism | 994.44 | 0.00 | 4.1.1.48 | [c]: C01302 + C00080 → C03506 + C00011 + C00001 |
| Valine, Leucine, and Isoleucine Metabolism | 1000.00 | 998.08 | 1.1.1.86 | [c]: C04039 + C00006 <==> C06010 + C00080 + C00005 |
| 1000.00 | 998.08 | 1.1.1.86 | [c]: C06010 + C00080 + C00005 → C04039 + C00006 | |
| (b) Flux Through Carbohydrate Pathway before and after Metabolomics Data Integration | ||||
| Pathways | Flux−FBA | Flux−WT-Cb | ECs | Equation |
| Central Metabolism | 961.51 | 0.00 | 2.7.9.1 | [c]: C00002 + C00009 + C00022 → C00020 + C00080 + C00074 + C00013 |
| 1000.00 | 663.00 | 1.1.1.49 | [c]: f420-2 + C00668 → C01236 + f420-2h2 | |
| Citric Acid Cycle | −992.39 | −6.06 | 4.2.1.2 | [c]: C00122 + C00001 <==> C00711 |
| −973.09 | −2.27 | 1.1.1.37 | [c]: C00711 + C00003 <==> C00080 + C00004 + C00036 | |
| −501.96 | −331.89 | 6.2.1.5 | [c]: C00002 + C00010 + C00042 <==> C00008 + C00009 + C00091 | |
| 501.96 | 331.89 | 1.2.4.2 | [c]: C00026 + C00080 + C15972 <==> C00011 + C16254 | |
| 501.96 | 331.89 | 2.3.1.61 | [c]: C00010 + C16254 → C00579 + C00091 | |
| Galactose Metabolism | −11.30 | −2.22 | 2.7.7.9 | [c]: C00103 + C00080 + C00075 <==> C00013 + C00029 |
| −11.30 | −2.22 | 5.1.3.2 | [c]: C00029 <==> C00052 | |
| Glycolysis/Gluconeogenesis | −1000.00 | 1000.00 | 5.3.1.9 | [x]: g6p-a <==> C05345 |
| 8.23 | 1.62 | 4.1.2.13 | [c]: C00447 <==> C00111 + C00279 | |
| 8.23 | 1.62 | 2.7.1.11 | [c]: C00002 + C05382 → C00008 + C00080 + C00447 | |
| 8.23 | 1.62 | 5.3.1.1 | [c]: C00111 <==> C00118 | |
| 41.16 | 8.08 | 2.7.1.2 | [c]: C00002 + C00267 → C00008 + C01172 + C00080 | |
| 41.16 | 8.08 | 5.3.1.9 | [c]: C00668 <==> C05345 | |
| 961.51 | 0.00 | 2.7.1.40 | [c]: C00008 + C00080 + C00074 → C00002 + C00022 | |
| 1000.00 | −1000.00 | 5.3.1.9 | [x] : C01172 <==> C05345 | |
| 1000.00 | −1000.00 | 5.3.1.9 | [x] : g6p-a <==> C01172 | |
| Glyoxylate and Dicarboxylate Metabolism | 500.16 | 331.53 | 1.11.1.6 | [c]: 2 C00027 → 2 C00001 + C00007 |
| Pentose Phosphate Pathway | −41.16 | 328.92 | 1.1.1.49 | [c]: C00668 + C00006 <==> C01236 + C00080 + C00005 |
| −32.93 | −6.46 | 5.1.3.1 | [c]: C00199 <==> C00231 | |
| −24.69 | −4.85 | 2.2.1.2 | [c]: C00118 + C05382 <==> C00279 + C05345 | |
| −16.46 | −3.23 | 2.2.1.1 | [c]: C03736 + C00231 <==> C00118 + C05382 | |
| −16.46 | −3.23 | 2.2.1.1 | [c]: C00279 + C00231 <==> C05345 + C00118 | |
| 16.46 | 3.23 | 2.7.6.1 | [c]: C00002 + C03736 <==> C00020 + C00080 + C00119 | |
| Starch and Sucrose Metabolism | −11.30 | −2.22 | 2.4.1.18 | [c]: C00718 <==> strch2_strch1 |
| −11.30 | −2.22 | 2.4.1.1 | [c]: strch2_strch1 + C00009 <==> C00718 + C00103 | |
| Pathways | Flux−FBA | Flux−WT-Cb | ECs | Equation |
|---|---|---|---|---|
| NAD Biosynthesis | −1000.00 | 0.00 | 2.4.2.11 | [c]: C00080 + C11486 + C00119 <==> C01185 + C00013 |
| Phenylalanine, Tyrosine, and Tryptophan Biosynthesis | −994.44 | 0.00 | 4.1.1.48 | [c]: C01302 <==> C03506 + C00011 + C00001 |
| Central Metabolism | 961.51 | 0.00 | 2.7.9.1 | [c]: C00002 + C00009 + C00022 → C00020 + C00080 + C00074 + C00013 |
| Glycolysis/Gluconeogenesis | 961.51 | 0.00 | 2.7.1.40 | [c]: C00008 + C00080 + C00074 → C00002 + C00022 |
| Nucleotide Metabolism | 994.44 | 0.00 | 2.7.4.3 | [c]: C00020 + C00536 <==> C00008 + C00013 |
| Oxidative Phosphorylation | 994.44 | 0.00 | 2.7.4.1 | [c]: C00002 + C00013 → C00008 + C00536 |
| Tyrosine, Tryptophan, and Phenylalanine Metabolism | 994.44 | 0.00 | 4.1.1.48 | [c]: C01302 + C00080 → C03506 + C00011 + C00001 |
| NAD Biosynthesis | 1000.00 | 0.00 | 2.4.2.11 | [c]: C00080 + C11486 + C00119 → C01185 + C00013 |
| Glycine and Serine Metabolism | 1000.00 | 0.00 | 1.1.1.3 | [c]: C00441 + C00080 + C00004 → C00263 + C00003 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanee, N.; Brooks, J.P.; Fong, S.S. Metabolic Profile of the Cellulolytic Industrial Actinomycete Thermobifida fusca. Metabolites 2017, 7, 57. https://doi.org/10.3390/metabo7040057
Vanee N, Brooks JP, Fong SS. Metabolic Profile of the Cellulolytic Industrial Actinomycete Thermobifida fusca. Metabolites. 2017; 7(4):57. https://doi.org/10.3390/metabo7040057
Chicago/Turabian StyleVanee, Niti, J. Paul Brooks, and Stephen S. Fong. 2017. "Metabolic Profile of the Cellulolytic Industrial Actinomycete Thermobifida fusca" Metabolites 7, no. 4: 57. https://doi.org/10.3390/metabo7040057
APA StyleVanee, N., Brooks, J. P., & Fong, S. S. (2017). Metabolic Profile of the Cellulolytic Industrial Actinomycete Thermobifida fusca. Metabolites, 7(4), 57. https://doi.org/10.3390/metabo7040057