Machine Learning Methods for Analysis of Metabolic Data and Metabolic Pathway Modeling

Abstract
1. Introduction
- Analyses of points or bins from the metabolomics measurements such as NMR spectra or MS spectrograms (examples of use of PCA and PLS methods are too many to reference). A recent example of an application of deep learning, as well as SVM, RF and several other machine learning algorithms was presented by Alakwaa et al. [20];
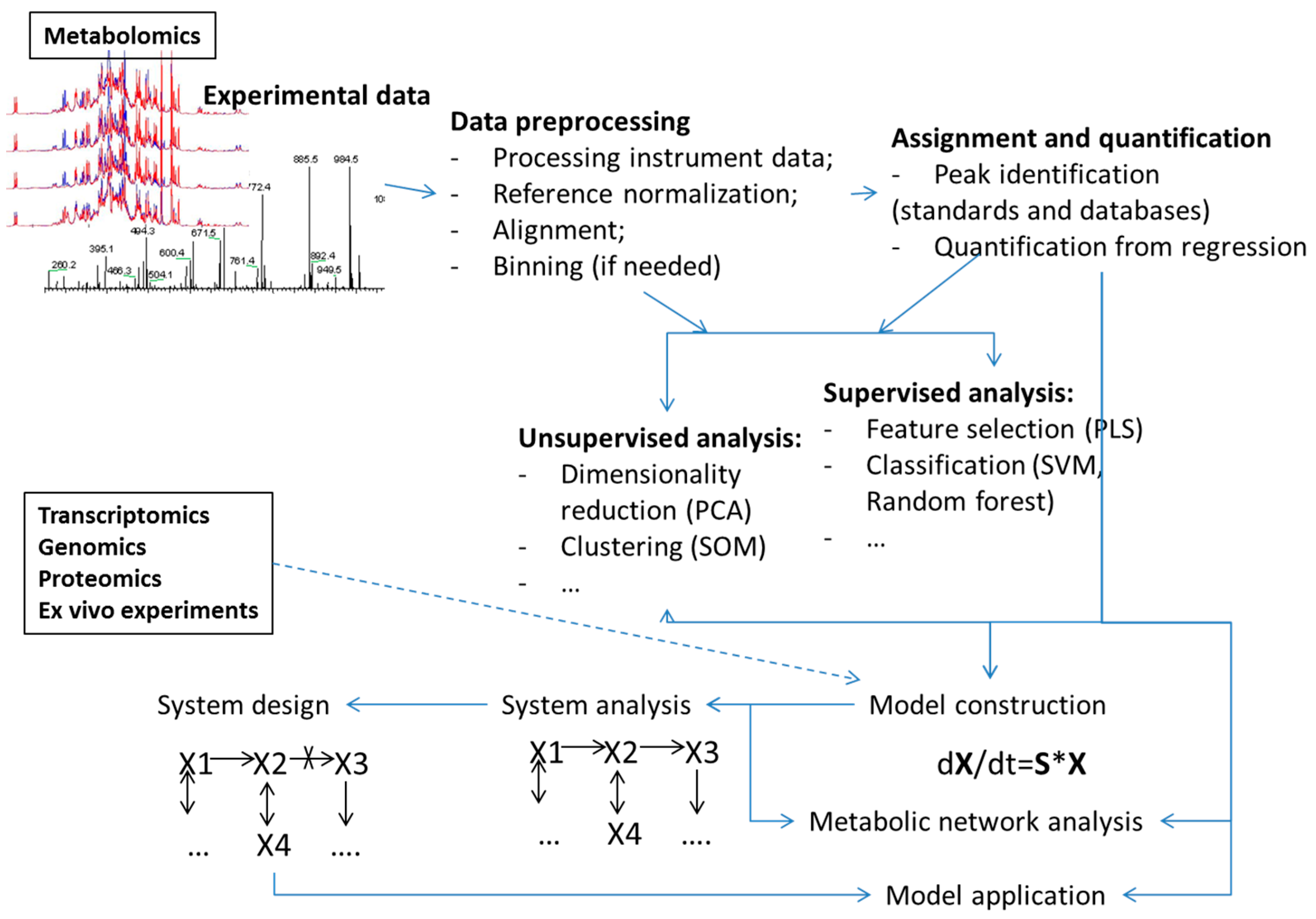
2. Metabolism Modeling
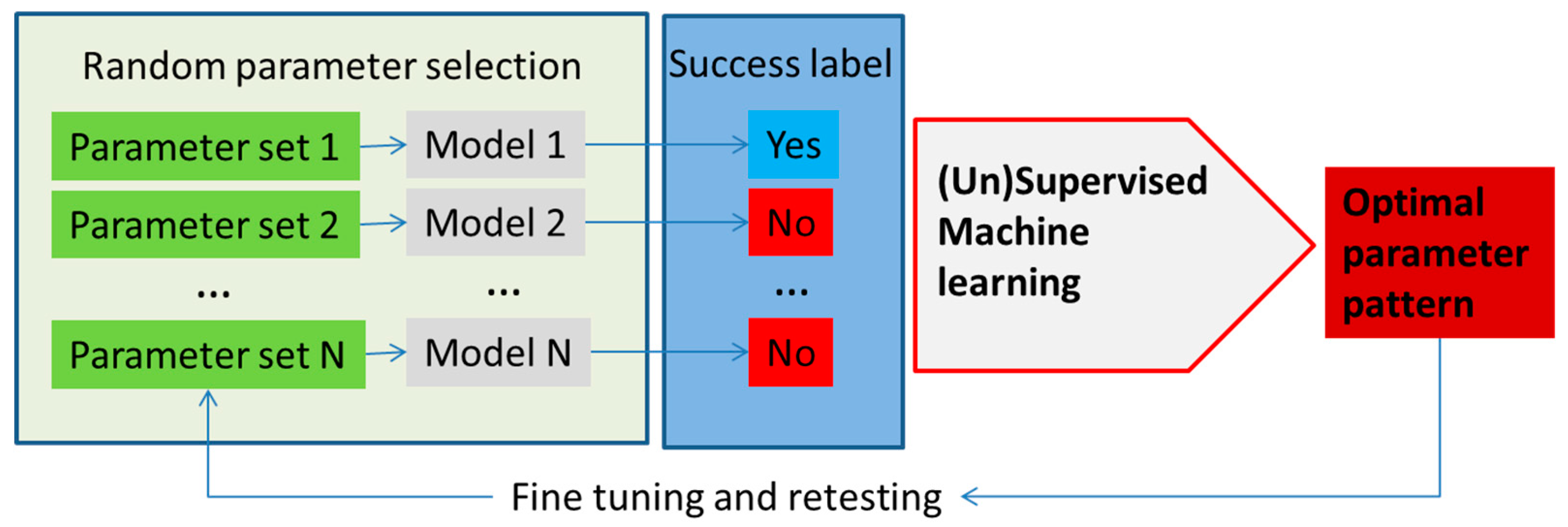
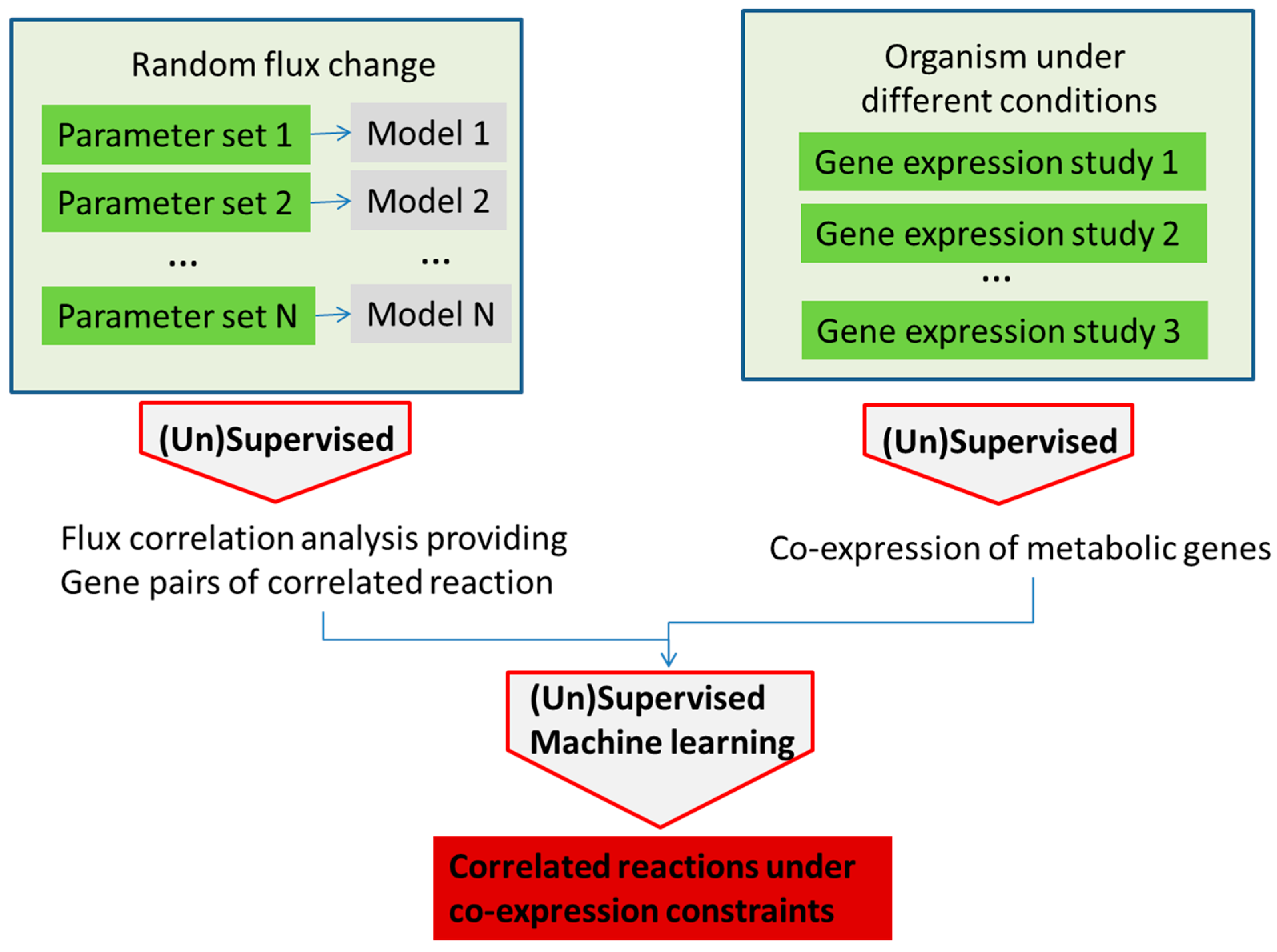
3. Machine Learning in Metabolism Modeling
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Guo, L.; Milburn, M.V.; Ryals, J.A.; Lonergan, S.C.; Mitchell, M.W.; Wulff, J.E.; Alexander, D.C.; Evans, A.M.; Bridgewater, B.; Miller, L.; et al. Plasma metabolomic profiles enhance precision medicine for volunteers of normal health. Proc. Natl. Acad. Sci. USA 2015, 112, E4901–E4910. [Google Scholar] [CrossRef] [PubMed]
- Samuel, A.L. Some studies in machine learning using the game of checkers. IBM J. Res. Dev. 1959, 3, 210–229. [Google Scholar] [CrossRef]
- Michell, T.M. Machine Learning; McGraw-Hill: New York, NY, USA, 1997. [Google Scholar]
- Brownlee, J. A Tour of Machine Learning Algorithms. Available online: https://machinelearningmastery.com/a-tour-of-machine-learning-algorithms/ (accessed on 8 January 2018).
- Kotsiantis, S.; Zaharakis, I.; Pintelas, P. Supervised machine learning: A review of classification techniques. Front. Artif. Intell. Appl. 2007, 160, 3–24. [Google Scholar]
- Forssen, H.; Patel, R.; Fitzpatrick, N.; Hingorani, A.; Timmis, A.; Hemingway, H.; Denaxas, S. Evaluation of Machine Learning Methods to Predict Coronary Artery Disease Using Metabolomic Data; IOS Press: Amsterdam, The Netherlands, 2017; pp. 1–5. [Google Scholar]
- Cuperlovic-Culf, M.; Ferguson, D.; Culf, A.; Morin, P., Jr.; Touaibia, M. 1H-NMR metabolomics analysis of glioblastoma subtypes: Correlation between metabolomics and gene expression characteristics. J. Biol. Chem. 2012, 287, 20164–20175. [Google Scholar] [CrossRef] [PubMed]
- Beckonert, O.; Monnerjahn, J.; Bonk, U.; Leibfritz, D. Visualizing metabolic changes in breast-cancer tissue using 1H-NMR spectroscopy and self-organizing maps. NMR Biomed. 2003, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, S.; Shah, S.L.; Marrie, T.J.; Slupsky, C.M. Analysis of metabolomic data using support vector machines. Anal. Chem. 2008, 80, 7562–7570. [Google Scholar] [CrossRef] [PubMed]
- Bujak, R.; Daghir-Wojtkowiak, E.; Kaliszan, R.; Markuszewski, M.J. PLS-based and regularization-based methods for the selection of relevant variables in non-targeted metabolomics data. Front. Mol. Biosci. 2016, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vaarhorst, A.A.; Verhoeven, A.; Weller, C.M.; Böhringer, S.; Göraler, S.; Meissner, A.; Deelder, A.M.; Henneman, P.; Gorgels, A.P.; van den Brandt, P.A.; et al. A metabolomic profile is associated with the risk of incident coronary heart disease. Am. Heart J. 2014, 168, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, C.; Böhm, C.; Baumgartner, D. Modelling of classification rules on metabolic patterns including machine learning and expert knowledge. J. Biomed. Inform. 2005, 38, 89–98. [Google Scholar] [CrossRef]
- Vehtari, A.; Makinen, V.P.; Soininen, P.; Ingman, P.; Makela, S.M.; Savolainen, M.J.; Hannuksela, M.L.; Kaski, K.; Ala-Korpela, M. A novel Bayesian approach to quantify clinical variables and to determine their spectroscopic counterparts in 1H NMR metabonomic data. BMC Bioinform. 2007, 8, S8. [Google Scholar] [CrossRef] [PubMed]
- Atluri, G.; Gupta, R.; Fang, G.; Pandey, G.; Steinbach, M.; Kumar, V. Association analysis techniques for bioinformatics problems. In Proceedings of the Bioinformatics and Computational Biology: First International Conference, BICoB 2009, New Orleans, LA, USA, 8–10 April 2009. [Google Scholar]
- Brougham, D.F.; Ivanova, G.; Gottschalk, M.; Collins, D.M.; Eustace, A.J.; O’Connor, R.; Havel, J. Artificial neural networks for classification in metabolomic studies of whole cells using 1H nuclear magnetic resonance. J. Biomed. Biotechnol. 2011, 2011, 158094. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.M.; Hill, D.W.; Menikarachchi, L.C.; Chen, M.H.; Hall, L.H.; Grant, D.F. Optimizing artificial neural network models for metabolomics and systems biology: An example using HPLC retention index data. Bioanalysis 2015, 7, 939–955. [Google Scholar] [CrossRef] [PubMed]
- Alsberg, B.K.; Kell, D.B.; Goodacre, R. Variable selection in discriminant partial least-squares analysis. Anal. Chem. 1998, 70, 4126–4133. [Google Scholar] [CrossRef] [PubMed]
- Coen, M.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. NMR-based metabolic profiling and metabonomic approaches to problems in molecular toxicology. Chem. Res. Toxicol. 2008, 21, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Grissa, D.; Pétéra, M.; Brandolini, M.; Napoli, A.; Comte, B.; Pujos-Guillot, E. Feature selection methods for early predictive biomarker discovery using untargeted metabolomic data. Front. Mol. Biosci. 2016, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Alakwaa, F.M.; Chaudhary, K.; Garmire, L.X. Deep Learning Accurately Predicts Estrogen Receptor Status in Breast Cancer Metabolomics Data. J. Proteom. Res. 2018, 17, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zamboni, N.; Heinonen, M.; Rousu, J. Metabolite identification through machine learning—Tackling CASMI challenge using fingerID. Metabolites 2013, 3, 484–505. [Google Scholar] [CrossRef] [PubMed]
- Ravanbakhsh, S.; Liu, P.; Bjorndahl, T.C.; Mandal, R.; Grant, J.R.; Wilson, M.; Eisner, R.; Sinelnikov, I.; Hu, X.; Luchinat, C.; et al. Accurate, fully-automated NMR spectral profiling for metabolomics. PLoS ONE 2015, 10, e0124219. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Astle, W.; De Iorio, M.; Ebbels, T. BATMAN—An R package for the automated quantification of metabolites from NMR spectra using a Bayesian model. Bioinformatics 2012, 28, 2088–2090. [Google Scholar] [CrossRef] [PubMed]
- Cavill, R.; Keun, H.C.; Holmes, E.; Lindon, J.C.; Nicholson, J.K.; Ebbels, T.M. Genetic algorithms for simultaneous variable and sample selection in metabonomics. Bioinformatics 2009, 25, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Worley, B.; Powers, R. Multivariate analysis in metabolomics. Curr. Metabol. 2013, 1, 92–107. [Google Scholar]
- Saccenti, E.; Hoefsloot, H.C.J.; Smilde, A.K.; Westerhuis, J.A.; Hendriks, M.M.W.B. Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics 2014, 10, 361–374. [Google Scholar] [CrossRef]
- D’Alche-Buc, F.; Wehenkel, L. Machine learning in systems biology. BMC Proc. 2008, 2, S1. Available online: https://bmcproc.biomedcentral.com/articles/10.1186/1753-6561-2-S4-S1 (accessed on 8 January 2018).
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Smolinska, A.; Hauschild, A.-C.; Fijten, R.R.R.; Dallinga, J.W.; Baumbach, J.; Schooten, F.J. Current breathomics—A review on data pre-processing techniques and machine learning in metabolomics breath analysis. J. Breath Res. 2014, 8, 27105. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B. Metabolomics, modelling and machine learning in systems biology—Towards an understanding of the languages of cells. FEBS J. 2006, 273, 873–894. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B. Understanding the languages of cells. Syst. Biol. 2011, 7, 4–7. [Google Scholar]
- Madsen, R.; Lundstedt, T.; Trygg, J. Chemometrics in metabolomics—A review in human disease diagnosis. Anal. Chim. Acta 2010, 659, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, D.K.; Hollywood, K.A.; Goodacre, R. Metabolomics for the masses: The future of metabolomics in a personalized world. New Horiz. Transl. Med. 2017, 3, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Acharjee, A.; Ament, Z.; West, J.A.; Stanley, E.; Griffin, J.L. Integration of metabolomics, lipidomics and clinical data using a machine learning method. BMC Bioinform. 2016, 17, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Metabolomics Software and Servers. Available online: http://metabolomicssociety.org/resources/metabolomics-software (accessed on 8 January 2018).
- Metabolomic Software. Available online: http://pmv.org.au/metabolomics/metabolomic-software/ (accessed on 8 January 2018).
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype–phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Bali Pulendran, B. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Guitton, Y.; Tremblay-Franco, M.; Le Corguillé, G.; Martin, J.F.; Pétéra, M.; Roger-Mele, P.; Delabrière, A.; Goulitquer, S.; Monsoor, M.; Duperier, C.; et al. Create, run, share, publish, and reference your LC–MS, FIA–MS, GC–MS, and NMR data analysis workflows with the Workflow4Metabolomics 3.0 Galaxy online infrastructure for metabolomics. Int. J. Biochem. Cell Biol. 2017, 93, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, M.; Shen, H.; Zamboni, N.; Rousu, J. Metabolite identification and molecular fingerprint prediction through machine learning. Bioinformatics 2012, 28, 2333–2341. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Böcker, S. Fragmentation trees reloaded. J. Cheminform. 2016, 8, 5. [Google Scholar]
- Xia, J.; Wishart, D.S. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2016, 55, 14.10.1–14.10.91. [Google Scholar]
- Kessler, N.; Dührkop, K. Learning to classify organic and conventional wheat—A machine learning driven approach using the MeltDB 2.0 metabolomics analysis platform. Front. Bioeng. Biotechnol. 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Frank, E.; Hall, M.A.; Witten, I.H. The WEKA Workbench. Online Appendix for “Data Mining: Practical Machine Learning Tools and Techniques”, 4th ed.; Morgan Kaufmann: Cambridge, MA, USA, 2016. [Google Scholar]
- Steuer, R.; Gross, T.; Selbig, J.; Blasius, B. Structural kinetic modeling of metabolic networks. Proc. Natl. Acad. Sci. USA 2006, 103, 11868–11873. [Google Scholar] [CrossRef] [PubMed]
- Nagele, T.; Mair, A.; Sun, X.; Fragner, L.; Teige, M.; Weckwerth, W. Solving the differential biochemical Jacobian from metabolomics covariance data. PLoS ONE 2014, 9, e92299. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.N.; Mavrovouniotis, M.L.; Liebman, M.N. Petri net representations in metabolic pathways. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1993, 1, 328–336. [Google Scholar] [PubMed]
- Materi, W.; Wishart, D.S. Computational systems biology in drug discovery and development: Methods and applications. Drug Discov. Today 2007, 12, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Baldan, P.; Cocco, N.; Marin, A.; Simeoni, M. Petri nets for modelling metabolic pathways: A survey. Nat. Comput. 2010, 9, 955–989. [Google Scholar] [CrossRef]
- Vijayakumar, S.; Conway, M.; Lió, P.; Angione, C. Seeing the wood for the trees: A forest of methods for optimization and omic-network integration in metabolic modelling. Brief. Bioinform. 2017, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.U.; Sohn, S.B.; Lee, S.Y. Metabolic network modeling and simulation for drug targeting and discovery. Biotechnol. J. 2012, 7, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.E.; Schramm, G.; Bordbar, A.; Schellenberger, J.; Andersen, M.P.; Cheng, J.K.; Patel, N.; Yee, A.; Lewis, R.A.; Eils, R.; et al. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat. Biotechnol. 2010, 28, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Sauro, H.M. Control and regulation of pathways via negative feedback—Supplementary. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef]
- Muggleton, S.H. Machine Learning for Systems Biology. In Proceedings of the 15th International Conference on Inductive Logic Programming, Bonn, Germany, 10–13 August 2005; pp. 416–423. [Google Scholar]
- Zhang, X.; Acencio, M.L.; Lemke, N. Predicting essential genes and proteins based on machine learning and network topological features: A comprehensive review. Front. Physiol. 2016, 7, 1–11. [Google Scholar]
- Saa, P.A.; Nielsen, L.K. Construction of feasible and accurate kinetic models of metabolism: A Bayesian approach. Sci. Rep. 2016, 6, 29635. [Google Scholar] [CrossRef] [PubMed]
- Sriyudthsak, K.; Shiraishi, F.; Hirai, M.Y. Mathematical modeling and dynamic simulation of metabolic reaction systems using metabolome time series data. Front. Mol. Biosci. 2016, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Saa, P.; Nielsen, L.K. A general framework for thermodynamically consistent parameterization and efficient sampling of enzymatic reactions. PLoS Comput. Biol. 2015, 11, e1004195. [Google Scholar] [CrossRef] [PubMed]
- Girbig, D.; Selbig, J.; Grimbs, S. A MATLAB toolbox for structural kinetic modeling. Bioinformatics 2012, 28, 2546–2547. [Google Scholar] [CrossRef] [PubMed]
- Girbig, D.; Grimbs, S.; Selbig, J. Systematic analysis of stability patterns in plant primary metabolism. PLoS ONE 2012, 7, e34686. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Cluett, W.R.; Mahadevan, R. Constructing kinetic models of metabolism at genome-scales: A review. Biotechnol. J. 2015, 10, 1345–1359. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Hrafnsdóttir, S.; Magnúsdóttir, M.; Fleming, R.M.; Thorlacius, S.; Palsson, B.Ø.; Thiele, I. Monitoring metabolites consumption and secretion in cultured cells using ultra-performance liquid chromatography quadrupole-time of flight mass spectrometry (UPLC-Q-TOF-MS). Anal. Bioanal. Chem. 2012, 402, 1183–1198. [Google Scholar] [CrossRef] [PubMed]
- Lerman, J.; Hyduke, D.R.; Latif, H.; Portnoy, V.A.; Lewis, N.E.; Orth, J.D.; Schrimpe-Rutledge, A.C.; Smith, R.D.; Adkins, J.N.; Zengler, K.; et al. In silico method for modelling metabolism and gene product expression at genome scale. Nat. Commun. 2012, 3, 929. [Google Scholar] [CrossRef] [PubMed]
- Bordbar, A.; Monk, J.M.; King, Z.A.; Palsson, B.O. Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 2014, 15, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, J.; Que, R.; Fleming, R.M.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef] [PubMed]
- Chindelevitch, L.; Trigg, J.; Regev, A.; Berger, B. An exact arithmetic toolbox for a consistent and reproducible structural analysis of metabolic network models. Nat. Commun. 2014, 5, 4893. [Google Scholar] [CrossRef] [PubMed]
- Puniya, B.L.; Kulshreshtha, D.; Mittal, I.; Mobeen, A.; Ramachandran, S. Integration of metabolic modeling with gene co-expression Reveals Transcriptionally programmed reactions explaining robustness in Mycobacterium tuberculosis. Sci. Rep. 2016, 6, 1–21. [Google Scholar]
- Colijn, C.; Brandes, A.; Zucker, J.; Lun, D.S.; Weiner, B.; Farhat, M.R.; Cheng, T.-Y.; Moody, D.B.; Murray, M.; Galagan, J.E. Interpreting expression data with metabolic flux models: Predicting Mycobacterium tuberculosis mycolic acid production. PLoS Comput. Biol. 2009, 5, e1000489. [Google Scholar] [CrossRef] [PubMed]
- Szappanos, B.; Kovács, K.; Szamecz, B.; Honti, F.; Costanzo, M.; Baryshnikova, A.; Gelius-Dietrich, G.; Lercher, M.J.; Jelasity, M.; Myers, C.L.; et al. An integrated approach to characterize genetic interaction networks in yeast metabolism. Nat. Genet. 2011, 43, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, S.; Miskovic, L.; Hatzimanikatis, V. ISCHRUNK—In silico approach to characterization and reduction of uncertainty in the kinetic models of genome-scale metabolic networks. Metab. Eng. 2016, 33, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Subramanian, A.; Sarkar, R. An integrative machine learning strategy for improved prediction of essential genes in Escherichia coli metabolism using flux-coupled features. Mol. BioSyst. 2017, 13, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Plaimas, K.; Mallm, J.-P.; Oswald, M.; Svara, F.; Sourjik, V.; Eils, R.; König, R. Machine learning based analyses on metabolic networks supports high-throughput knockout screens. BMC Syst. Boil. 2008, 2, 67. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rivera, J.G.; Liao, J.C. Ensemble modeling for robustness analysis in engineering non-native metabolic pathways. Metab. Eng. 2014, 25, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Henriques, D.; Villaverde, A.F.; Rocha, M.; Saez-Rodriguez, J.; Banga, J.R. Data-driven reverse engineering of signaling pathways using ensembles of dynamic models. PLoS Comput. Biol. 2017, 13, e1005379. [Google Scholar] [CrossRef] [PubMed]
- Tamaddoni-Nezhad, A.; Chaleil, R.; Kakas, A.; Muggleton, S. Application of abductive ILP to learning metabolic network inhibition from temporal data. Mach. Learn. 2006, 64, 209–230. [Google Scholar] [CrossRef]
- Tamaddoni-Nezhad, A.; Kakas, A.; Muggleton, S.; Pazos, F. Modelling inhibition in metabolic pathways through abduction and induction. Lect. Notes Artif. Intell. 2004, 3194, 305–322. [Google Scholar]
- Guo, W.; Xu, Y.; Feng, X. Deep metabolism: A deep learning system to predict phenotype from genome sequencing. Bioarxiv 2017, 1–7. [Google Scholar] [CrossRef]
- Dale, J.M.; Popescu, L.; Karp, P.D. Machine learning methods for metabolic pathway prediction. BMC Bioinform. 2010, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Shaked, I.; Oberhardt, M.A.; Atias, N.; Sharan, R.; Ruppin, E. Metabolic network prediction of drug side effects. Cell Syst. 2016, 2, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Lodhi, H.; Muggleton, S. Modelling metabolic pathways using stochastic logic programs-based ensemble methods. Lect. Notes Bioinform. 2005, 3082, 119–133. [Google Scholar]
- Chen, J.; Muggleton, S.; Santos, J. Learning probabilistic logic models from probabilistic examples. Mach. Learn. 2008, 73, 55–85. [Google Scholar] [CrossRef] [PubMed]
- Bongard, J.; Lipson, H. Automated reverse engineering of nonlinear dynamical systems. Proc. Natl. Acad. Sci. USA 2007, 104, 9943–9948. [Google Scholar] [CrossRef] [PubMed]
- Wanichthanarak, K.; Fahrmann, J.F.; Grapov, D. Genomic, proteomic, and metabolomic data integration strategies. Biomark. Insights 2015, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cambiaghi, A.; Ferrario, M.; Masseroli, M. Analysis of metabolomic data: Tools, current strategies and future challenges for omics data integration. Brief. Bioinform. 2017, 18, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Kelemen, A. Computational dynamic approaches for temporal omics data with applications to systems medicine. BioData Min. 2017, 10, 1–20. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Algorithm Group | Short Description | Methods and Some Metabolomics Uses |
|---|---|---|
| Regression algorithms [6,7] | Iteratively improve the model of the relationship between features and labels using the error measure | Ordinary Least Squares Regression (OLSR); linear regression; stepwise regression; Local Estimate Scatterplot Smoothing (LOESS) |
| Instance-based algorithms [8,9] | Compare new problem instances (e.g., samples) with examples seen in training. | k-Nearest Neighbors (kNN); Self-Organized Map (SOM) and Locally Weighted Learning (LWL); SVM |
| Regularization algorithms [10,11] | An extension to other models that penalize models based on their complexity generally favouring simpler models. | Least Absolute Shrinkage and Selection Operator (LASSO) and elastic net |
| Decision tree algorithms [6,12] | Trained on the data for classification and regression problems providing a flowchart-like structure model where nodes denote tests on an attribute with each branch representing the outcome of a test and each leaf node holding a class label. | Classification and regression tree (CART); C4.5 and C5.0; decision stump; regression tree |
| Bayesian algorithms [13] | Application of Bayes’ theorem for the probability of classification and regression. | Naive Bayes, Gaussian naive Bayes, Bayesian Belief Network (BBN); Bayesian Network (BN) |
| Association rule learning algorithms [14] | Methods aiming to extract rules that best explain the relationships between variables. | A priori algorithm; Eclat algorithm |
| Artificial neural network algorithms including deep learning [15,16] | Building of a neural network. | Perceptron back-propagation Hopfield network Radial Basis Function Network (RBFN) Deep Boltzmann Machine (DBM) Deep Belief Networks (DBN) Convolutional Neural Network (CNN) stacked auto-encoders |
| Dimensionality reduction algorithms [17,18] | Unsupervised and supervised methods seeking and exploiting inherent structures in the data in order to simplify data for easier visualization or selection of major characteristics. | Principal Component Analysis (PCA) Principal Component Regression (PCR) Partial Least Squares Regression (PLSR) Sammon mapping Multidimensional Scaling (MDS) projection pursuit Linear Discriminant Analysis (LDA) Mixture Discriminant Analysis (MDA) Quadratic Discriminant Analysis (QDA) Flexible Discriminant Analysis (FDA) |
| Ensemble algorithms [19] | Models composed of multiple weaker models that are independently trained leading to predictions that are combined in some way to provide greatly improved overall prediction. | boosting bootstrapped aggregation (bagging) AdaBoost stacked generalization (blending) Gradient Boosting Machines (GBM) Gradient Boosted Regression Trees (GBRT) random forest |
| Tool Name | Focus | Availability |
|---|---|---|
| FingerID [21,42] | Molecular fingerprinting | http://www.sourceforge.net/p/fingerid |
| SIRIUS [43] | Molecular fingerprinting | https://bio.informatik.uni-jena.de/software/sirius/ |
| Metaboanalyst [44] | General tool for metabolomics analysis | http://www.metaboanalyst.ca/ |
| MeltDB 2.0 [45] | General tool for metabolomics analysis | - |
| KNIME * | General machine learning tool | https://www.knime.com/ |
| Weka [46] | General machine learning tool | https://www.cs.waikato.ac.nz/ml/weka/ |
| Orange * | General machine learning tool | https://orange.biolab.si/ |
| TensorFlow | General machine learning tool | https://www.tensorflow.org/ |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuperlovic-Culf, M. Machine Learning Methods for Analysis of Metabolic Data and Metabolic Pathway Modeling. Metabolites 2018, 8, 4. https://doi.org/10.3390/metabo8010004
Cuperlovic-Culf M. Machine Learning Methods for Analysis of Metabolic Data and Metabolic Pathway Modeling. Metabolites. 2018; 8(1):4. https://doi.org/10.3390/metabo8010004
Chicago/Turabian StyleCuperlovic-Culf, Miroslava. 2018. "Machine Learning Methods for Analysis of Metabolic Data and Metabolic Pathway Modeling" Metabolites 8, no. 1: 4. https://doi.org/10.3390/metabo8010004
APA StyleCuperlovic-Culf, M. (2018). Machine Learning Methods for Analysis of Metabolic Data and Metabolic Pathway Modeling. Metabolites, 8(1), 4. https://doi.org/10.3390/metabo8010004