
Fully Automated Trimethylsilyl (TMS) Derivatisation Protocol for Metabolite Profiling by GC-MS



Abstract
:1. Introduction
2. Results and Discussion
2.1. Comparison between Automated and Manual TMS Derivsation
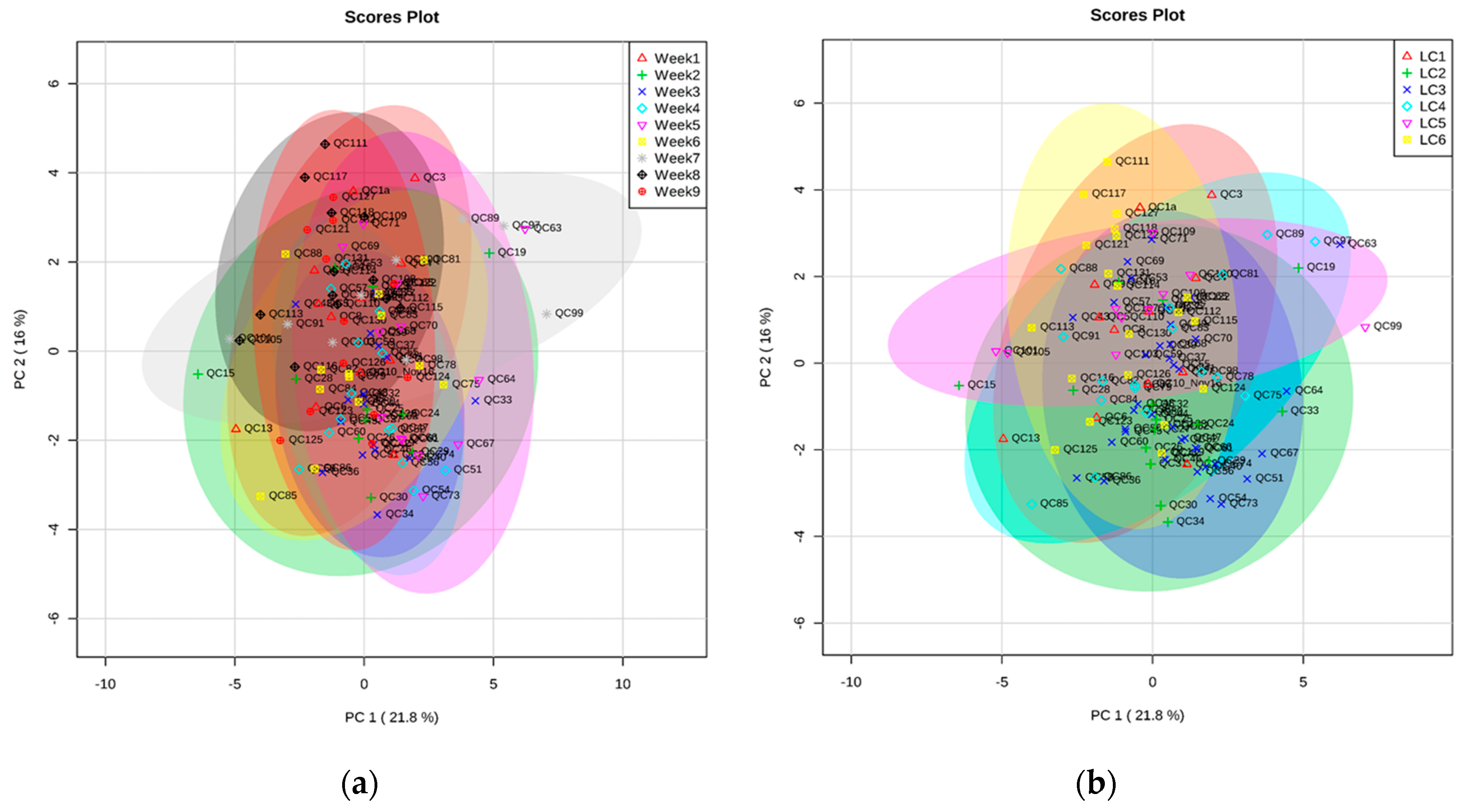
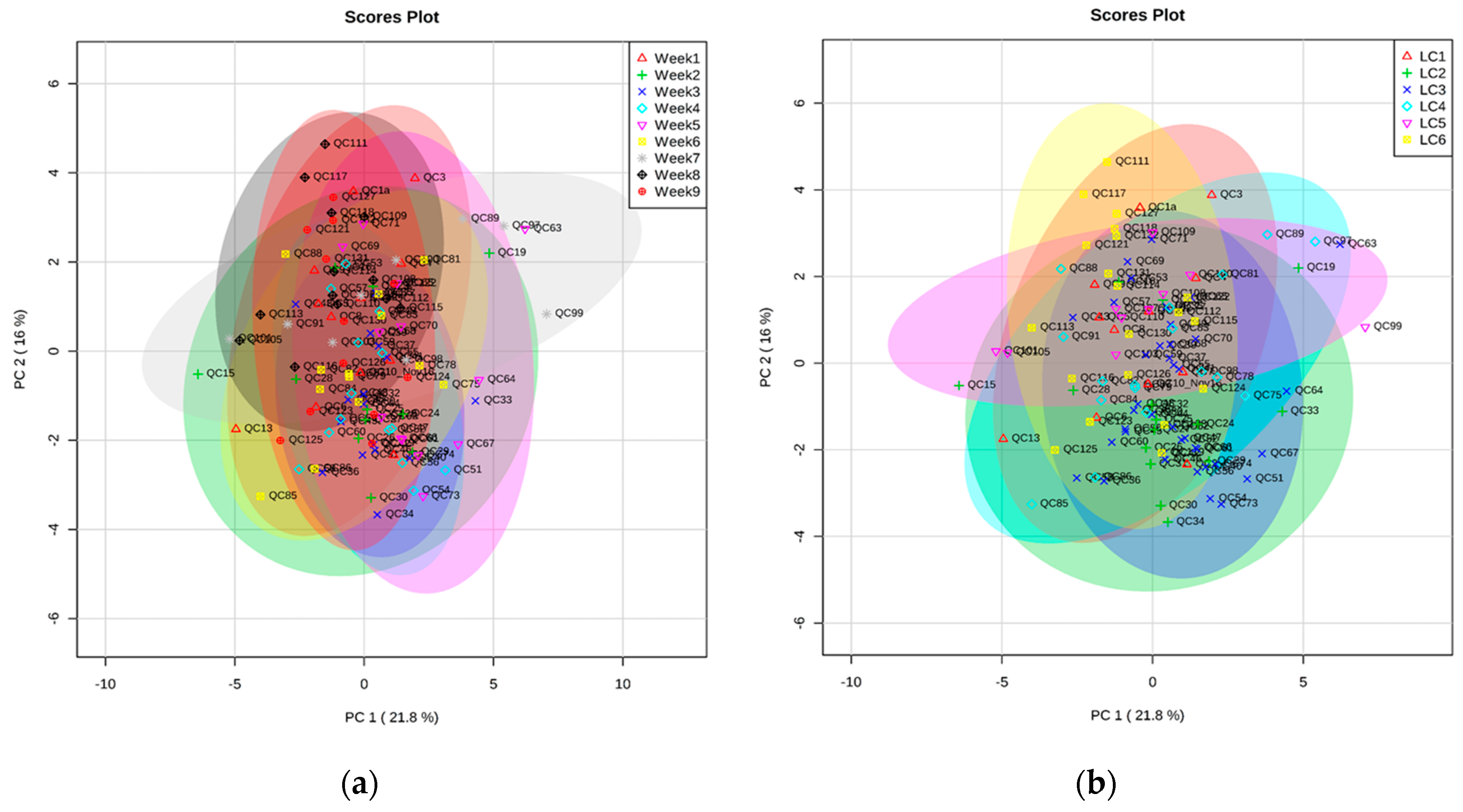
2.2. Validation of Automated TMS Method Using Plasma QC Samples over Nine Weeks
3. Materials and Methods
3.1. Chemicals
3.2. Sample Preparation
3.2.1. Preparation of Standard Mixes and Wine Samples
3.2.2. Extraction of Metabolites from Plasma Samples and Preparation of Quality Control (QC) Samples
3.3. Trimethylsilyl (TMS) Derivatisation Protocols
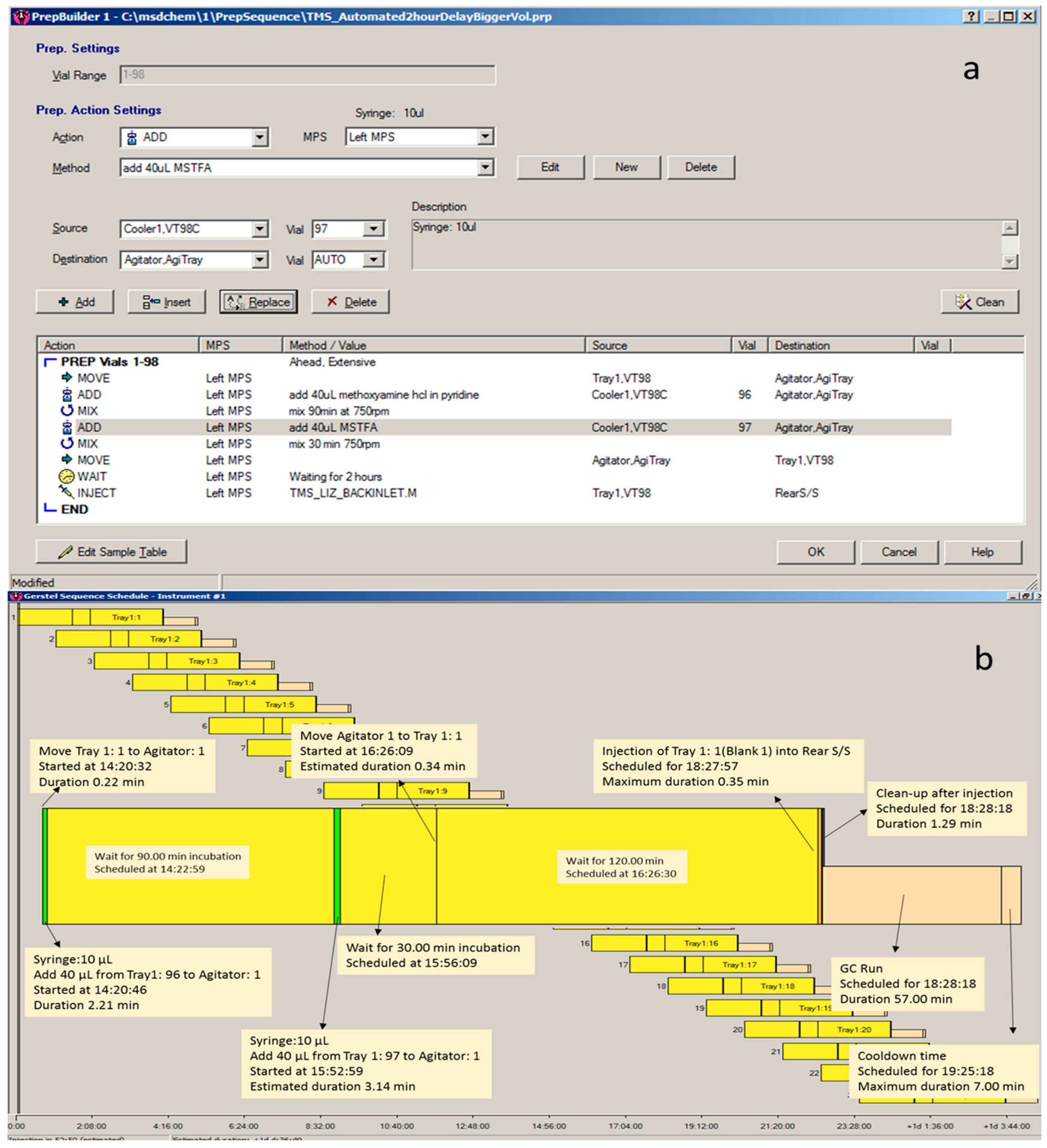
3.3.1. Automated TMS Derivatisation
3.3.2. Manual TMS derivatisation
3.4. Data Mining and Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dunn, W.B.; Hankemeier, T. Mass spectrometry and metabolomics: Past, present and future. Metabolomics 2013, 9 (Suppl. 1), 1–3. [Google Scholar] [CrossRef]
- Raterink, R.J.; Lindenburg, P.W.; Vreeken, R.J.; Ramautar, R.; Hankemeier, T. Recent developments in sample-pretreatment techniques for mass spectrometry-based metabolomics. TrAc Trends Anal. Chem. 2014, 61, 157–167. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef] [PubMed]
- Sana, T.; Fischer, S.; Tichy, S.E. Metabolomics Workflows: Combining Untargeted Discovery-Based and Targeted Confirmation Approaches for Mining Metabolomics Data. Spectrosc. 2011, 9, 12–17. [Google Scholar]
- Bachlechner, U.; Floegel, A.; Steffen, A.; Prehn, C.; Adamski, J.; Pischon, T.; Boeing, H. Associations of anthropometric markers with serum metabolites using a targeted metabolomics approach: Results of the EPIC-potsdam study. Nutr. Diabetes 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Tschirner, S.K.; Bahre, H.; Kaever, A.; Schneider, E.H.; Seifert, R.; Kaever, V. Non-targeted metabolomics by high resolution mass spectrometry in HPRT knockout mice. Life Sci. 2016, 156, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Wolfender, J.L.; Marti, G.; Thomas, A.; Bertrand, S. Current approaches and challenges for the metabolite profiling of complex natural extracts. J. Chromatogr. A 2015, 1382, 136–164. [Google Scholar] [CrossRef] [PubMed]
- Horning, E.C.; Horning, M.G. Human metabolic profiles obtained by GC and GC/MS. J. Chromatogr. Sci. 1971, 9, 129–140. [Google Scholar] [CrossRef]
- Lisec, J.; Schauer, N.; Kopka, J.; Willmitzer, L.; Fernie, A.R. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 2006, 1, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Smart, K.F.; Aggio, R.B.M.; van Houtte, J.R.; Villas-Boas, S.G. Analytical platform for metabolome analysis of microbial cells using methyl chloroformate derivatization followed by gas chromatography-mass spectrometry. Nat. Protoc. 2010, 5, 1709–1729. [Google Scholar] [CrossRef] [PubMed]
- Pinu, F.R.; Edwards, P.J.B.; Gardner, R.C.; Villas-Boas, S.G. Nitrogen and carbon assimilation by Saccharomyces cerevisiae during Sauvignon blanc juice fermentation. FEMS Yeast Res. 2014, 14, 1206–1222. [Google Scholar] [CrossRef] [PubMed]
- Abbiss, H.; Rawlinson, C.; Maker, G.L.; Trengove, R. Assessment of automated trimethylsilyl derivatization protocols for GC-MS-based untargeted metabolomic analysis of urine. Metabolomics 2015, 11, 1908–1921. [Google Scholar] [CrossRef]
- Bruce, S.J.; Breton, I.; Decombaz, J.; Boesch, C.; Scheurer, E.; Montoliu, I.; Rezzi, S.; Kochhar, S.; Guy, P.A. A plasma global metabolic profiling approach applied to an exercise study monitoring the effects of glucose, galactose and fructose drinks during post-exercise recovery. J. Chromatogr. B 2010, 878, 3015–3023. [Google Scholar] [CrossRef] [PubMed]
- Villas-Bôas, S.G.; Noel, S.; Lane, G.A.; Attwood, G.; Cookson, A. Extracellular metabolomics: A metabolic footprinting approach to assess fiber degradation in complex media. Anal. Biochem. 2006, 349, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Fernie, A.R.; Trethewey, R.N.; Krotzky, A.J.; Willmitzer, L. Innovation—Metabolite profiling: From diagnostics to systems biology. Nat. Rev. Mol. Cell Biol. 2004, 5, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Koek, M.M.; Jellema, R.H.; van der Greef, J.; Tas, A.C.; Hankemeier, T. Quantitative metabolomics based on gas chromatography mass spectrometry: Status and perspectives. Metabolomics 2011, 7, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Villas-Boas, S.G.; Mas, S.; Akesson, M.; Smedsgaard, J.; Nielsen, J. Mass spectrometry in metabolome analysis. Mass Spectrom. Rev. 2005, 24, 613–646. [Google Scholar] [CrossRef] [PubMed]
- Villas-Bôas, S.G.; Smart, K.F.; Sivakumaran, S.; Lane, G.A. Alkylation or Silylation for Analysis of Amino and Non-Amino Organic Acids by GC-MS? Metabolites 2011, 1, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Söderholm, S.L.; Damm, M.; Kappe, C.O. Microwave-assisted derivatization procedures for gas chromatography/mass spectrometry analysis. Mol. Divers. 2010, 14, 869–888. [Google Scholar] [CrossRef] [PubMed]
- Villas-Bôas, S.G.; Sivakumaran, S.; Attwood, G.; Lane, G.A.; Cookson, A. Rapid estimation of polysaccharide content in complex microbial culture media. World J. Microbiol. Biotechnol. 2007, 23, 873–876. [Google Scholar] [CrossRef]
- Pinu, F.R.; Edwards, P.J.B.; Jouanneau, S.; Kilmartin, P.A.; Gardner, R.C.; Villas-Boas, S.G. Sauvignon blanc metabolomics: Grape juice metabolites affecting the development of varietal thiols and other aroma compounds in wines. Metabolomics 2014, 10, 556–573. [Google Scholar] [CrossRef]
- Gu, Q.; David, F.; Lynen, F.; Rumpel, K.; Dugardeyn, J.; van Der Straeten, D.; Xu, G.W.; Sandra, P. Evaluation of automated sample preparation, retention time locked gas chromatography-mass spectrometry and data analysis methods for the metabolomic study of Arabidopsis species. J. Chromatogr. A 2011, 1218, 3247–3254. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- Trygg, J.; Gullberg, J.; Johansson, A.I.; Jonsson, P.; Antti, H.; Marklund, S.L.; Moritz, T. Extraction and GC/MS analysis of the human blood plasma metabolome. Anal. Chem. 2005, 77, 8086–8094. [Google Scholar]
- Harvey, N.C.; Javaid, K.; Bishop, N.; Kennedy, S.; Papageorghiou, A.T.; Fraser, R.; Gandhi, S.V.; Schoenmakers, I.; Prentice, A.; Cooper, C. MAVIDOS Maternal Vitamin D Osteoporosis Study: Study protocol for a randomized controlled trial. The MAVIDOS Study Group. Trials 2012, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggio, R.; Villas-Boas, S.G.; Ruggiero, K. Metab: An R package for high-throughput analysis of metabolomics data generated by GC-MS. Bioinformatics 2011, 27, 2316–2318. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.G.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0-making metabolomics more meaningful. Nucl. Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef] [PubMed]
- Koek, M.M.; van der Kloet, F.M.; Kleemann, R.; Kooistra, T.; Verheij, E.R.; Hankemeier, T. Semi-automated non-target processing in GC × GC-MS metabolomics analysis: Applicability for biomedical studies. Metabolomics 2011, 7. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Wine (n = 8) | Plasma (n = 4) | |||
|---|---|---|---|---|
| Automated | Manual | Automated | Manual | |
| Average No. of Detection | 240 ± 25 b | 157 ± 18 | 167 ± 17 a | 114 ± 17 |
| Average TIC | 112,058 ± 15,245 b | 40,907 ± 3125 | 12,717 ± 3015 | 10,424 ± 2255 |
| Standard Mix (RSD%, n = 5) | Wine (RSD%, n = 8) | Plasma (RSD%, n = 3) | |||||
|---|---|---|---|---|---|---|---|
| Metabolites | Automated | Manual | Automated | Manual | Automated | Manual | |
| Internal Standard | Ribitol | 2 | 3 | 7 | 15 | 3 | 5 |
| Amino Acids | Alanine | 9 | 19 | 13 | 11 | 10 | 3 |
| Glycine | NI | NI | 10 | 19 | 19 | 7 | |
| Leucine | 7 | 21 | 23 | 20 | 45 | 18 | |
| Isoleucine | NI | NI | ND | ND | 54 | 12 | |
| Lysine | 11 | ND | ND | ND | ND | ND | |
| Proline | NI | NI | 13 | 10 | 23 | 11 | |
| Serine | NI | NI | ND | ND | 9 | 22 | |
| Threonine | NI | NI | ND | ND | 13 | 13 | |
| Tryptophan | 14 | ND | ND | ND | ND | ND | |
| Valine | NI | NI | 16 | 15 | 18 | 28 | |
| Sugars and Sugar Alcohols | Allose | NI | NI | 8 | 12 | 7 | 22 |
| Arabinose | 2 | 5 | 9 | 13 | ND | ND | |
| Cellobiose | NI | NI | 5 | 13 | ND | ND | |
| Fructose 1 | 3 | 5 | 7 | 8 | ND | ND | |
| Fructose 2 | 3 | 4 | 7 | 9 | ND | ND | |
| Galactose | 3 | 3 | 8 | 9 | 7 | 4 | |
| Glycerol | 5 | 8 | 7 | 13 | 4 | 4 | |
| Glucose 1 | 3 | 2 | ND | ND | 4 | 2 | |
| Glucose 2 | 5 | 3 | ND | ND | 5 | 4 | |
| Mannose 1 | 3 | 6 | 8 | 9 | ND | ND | |
| Mannose 2 | 6 | 5 | 8 | 12 | ND | ND | |
| Meso-erythritol | NI | NI | 6 | 17 | ND | ND | |
| Meso-inositol | NI | NI | 9 | 6 | 12 | 21 | |
| Sorbose | NI | NI | 7 | 8 | ND | ND | |
| Sucrose | 6 | 6 | ND | ND | ND | ND | |
| Xylose | 5 | 5 | 2 | 3 | ND | ND | |
| Organic Acids | 2-hydroxybutyric acid | 5 | 11 | ND | ND | ND | ND |
| Glucaric acid | NI | NI | 9 | 6 | ND | ND | |
| Gluconic acid | NI | NI | 27 | 8 | ND | ND | |
| Others | Ferulic acid | 5 | 9 | ND | ND | ND | ND |
| Lactic acid | NI | NI | 23 | 17 | 5 | 4 | |
| Malic acid | NI | NI | 8 | 9 | ND | ND | |
| Oxalic acid | NI | NI | 38 | 27 | 22 | 8 | |
| Butane-2,3-diol | NI | NI | 7 | 14 | ND | ND | |
| Phosphate | NI | NI | 11 | 10 | 6 | 7 | |
| Urea | 8 | 13 | ND | ND | 9 | 11 | |
| Metabolite | Average Relative Abundance in Plasma Samples (n = 114) | Average RSD over Nine Weeks (%; n = 114) |
|---|---|---|
| Sugars and sugar alcohols | ||
| Galactose | 0.011 ± 0.002 | 14 |
| Glucose peak 1 | 1.368 ± 0.134 | 10 |
| Glucose peak 2 | 0.204 ± 0.030 | 15 |
| Glycerol | 0.038 ± 0.011 | 28 |
| Meso-inositol | 0.010 ± 0.001 | 12 |
| Amino acids | ||
| Alanine | 0.108 ± 0.027 | 25 |
| Glycine | 0.076 ± 0.018 | 23 |
| Lysine | 0.011 ± 0.003 | 29 |
| Proline | 0.079 ± 0.022 | 27 |
| Serine | 0.006 ± 0.001 | 20 |
| Threonine | 0.012 ± 0.002 | 20 |
| Valine | 0.062 ± 0.016 | 26 |
| Organic acids | ||
| 2-hydroxybutyic acid | 0.018 ± 0.004 | 24 |
| Isocitric acid | 0.028 ± 0.004 | 14 |
| Lactic acid | 0.701 ± 0.099 | 14 |
| Fatty acids | ||
| Linoleic acid | 0.004 ± 0.0016 | 42 |
| Oleic acid | 0.007 ± 0.0031 | 43 |
| Palmitoleic acid | 0.012 ± 0.0064 | 56 |
| Stearic acid | 0.001 ± 0.0005 | 51 |
| Others | ||
| Cholesterol | 0.076 ± 0.036 | 47 |
| Phosphate | 0.869 ± 0.122 | 14 |
| Urea | 0.887 ± 0.121 | 14 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarate, E.; Boyle, V.; Rupprecht, U.; Green, S.; Villas-Boas, S.G.; Baker, P.; Pinu, F.R. Fully Automated Trimethylsilyl (TMS) Derivatisation Protocol for Metabolite Profiling by GC-MS. Metabolites 2017, 7, 1. https://doi.org/10.3390/metabo7010001
Zarate E, Boyle V, Rupprecht U, Green S, Villas-Boas SG, Baker P, Pinu FR. Fully Automated Trimethylsilyl (TMS) Derivatisation Protocol for Metabolite Profiling by GC-MS. Metabolites. 2017; 7(1):1. https://doi.org/10.3390/metabo7010001
Chicago/Turabian StyleZarate, Erica, Veronica Boyle, Udo Rupprecht, Saras Green, Silas G. Villas-Boas, Philip Baker, and Farhana R. Pinu. 2017. "Fully Automated Trimethylsilyl (TMS) Derivatisation Protocol for Metabolite Profiling by GC-MS" Metabolites 7, no. 1: 1. https://doi.org/10.3390/metabo7010001
APA StyleZarate, E., Boyle, V., Rupprecht, U., Green, S., Villas-Boas, S. G., Baker, P., & Pinu, F. R. (2017). Fully Automated Trimethylsilyl (TMS) Derivatisation Protocol for Metabolite Profiling by GC-MS. Metabolites, 7(1), 1. https://doi.org/10.3390/metabo7010001