
Cancer Metabolism and Drug Resistance

Abstract
:1. Introduction
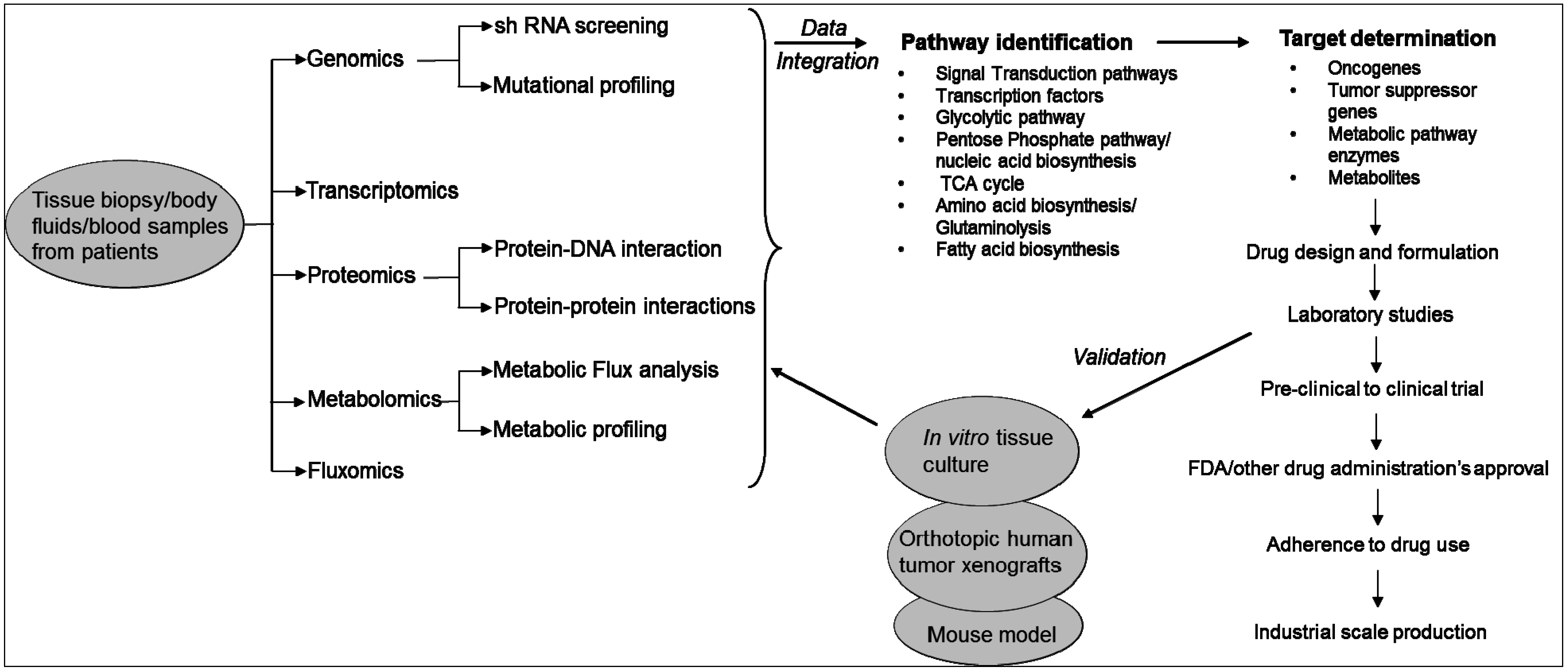
2. Technologies Used to Study the Metabolic Pathways of Cancer Cells
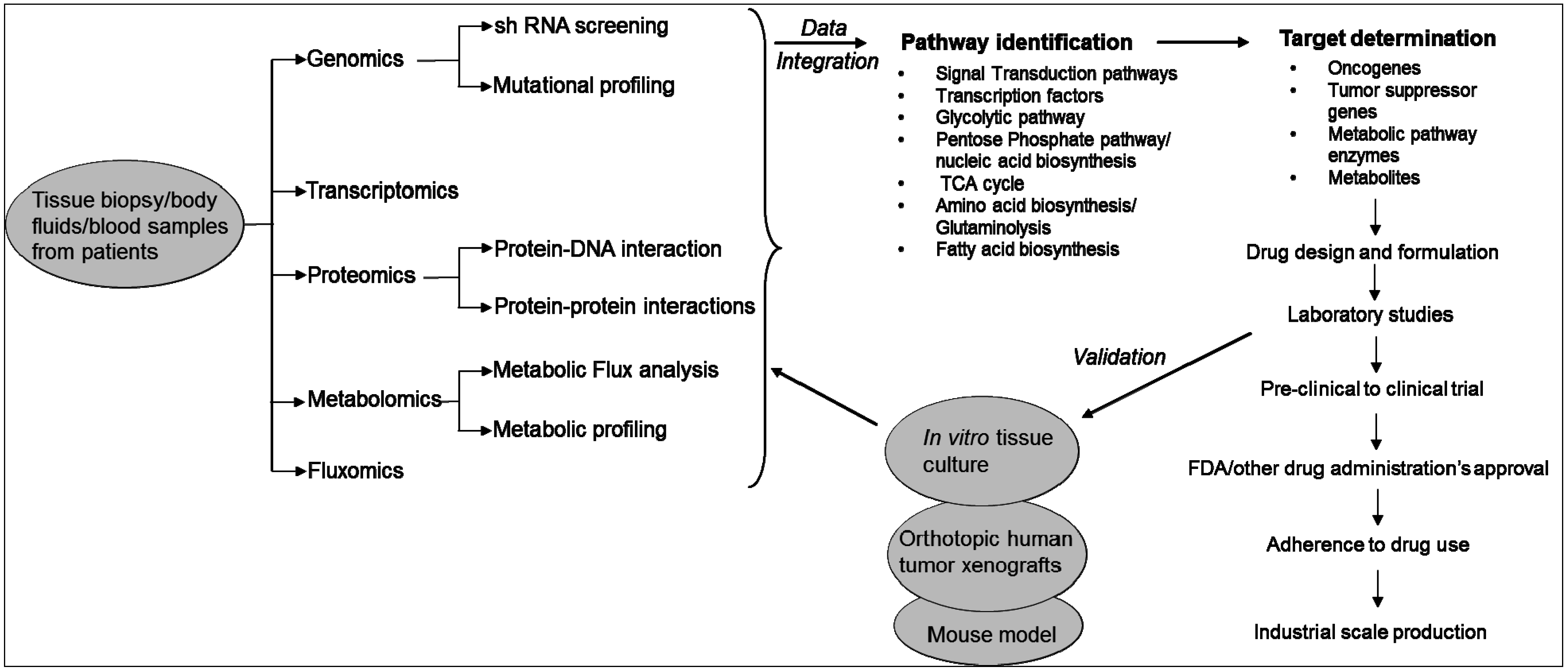
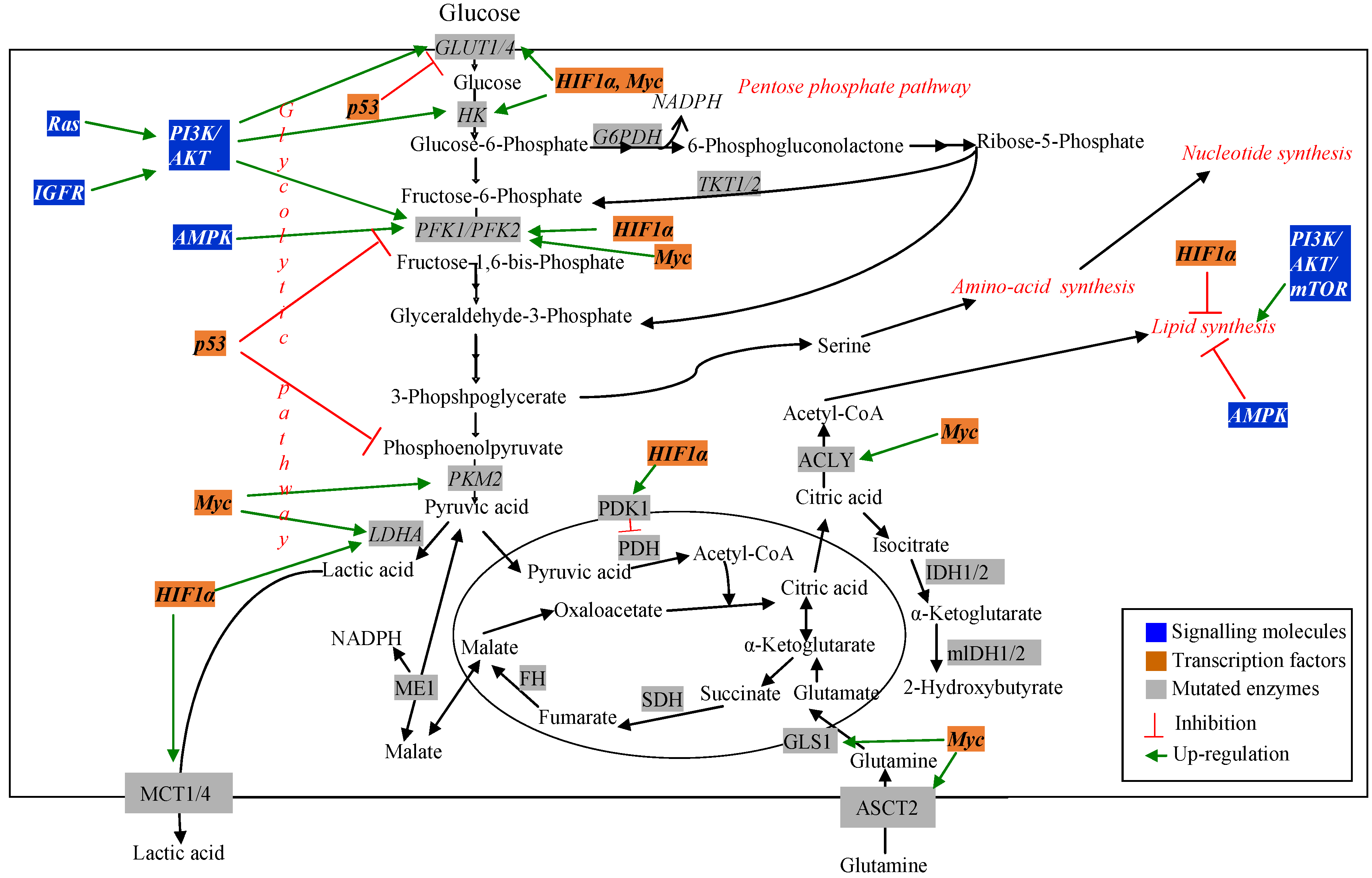
3. Metabolic Pathways Associated with Cancer and Drug Resistance
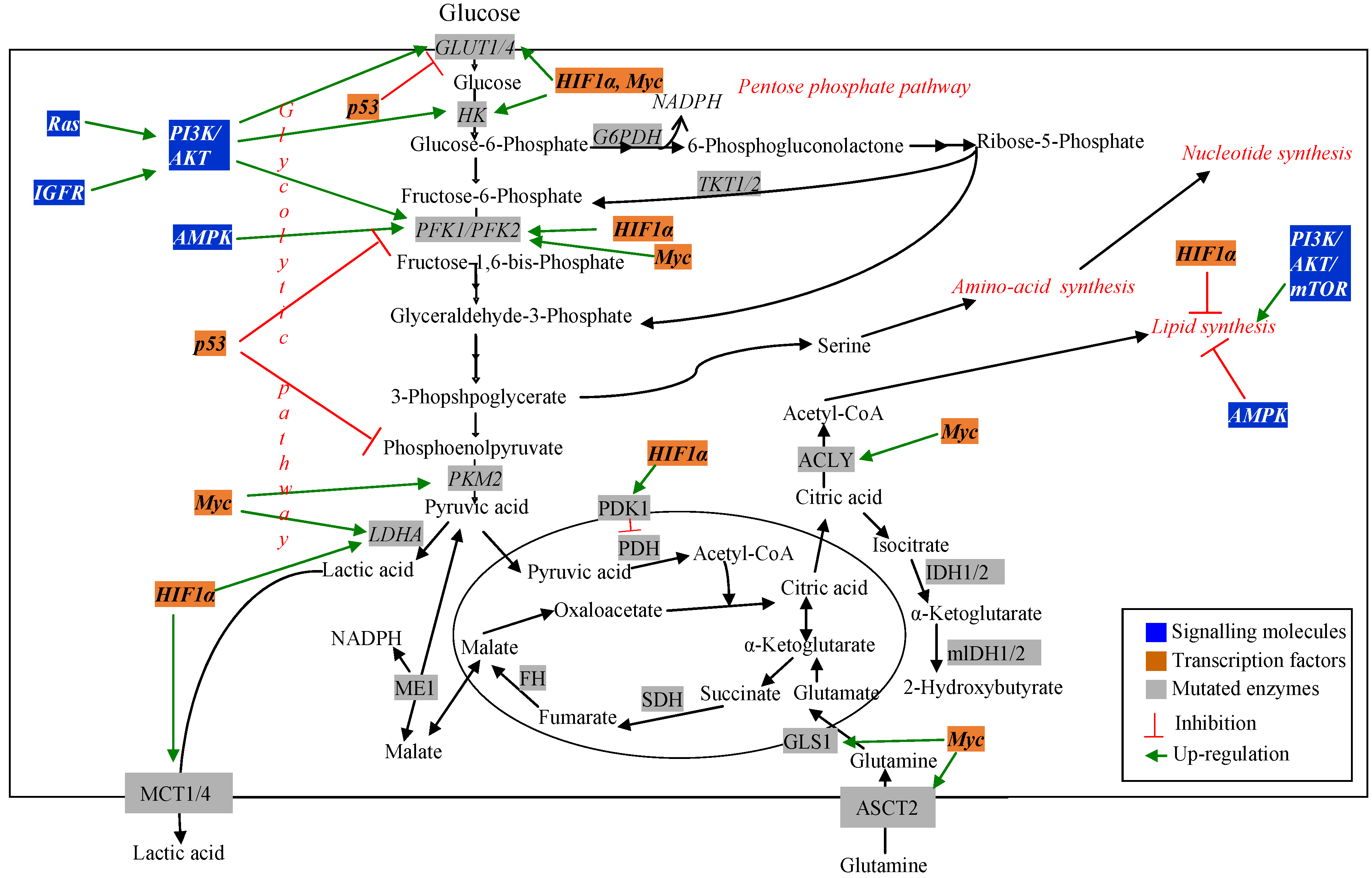
3.1. Signalling and Regulatory Molecules Associated with Metabolic Reprogramming in Cancer Cells
3.1.1. Signaling Molecules
3.1.2. Transcription Factors
3.2. Metabolic Pathway Genes Associated with Altered Metabolism in Cancer Cells
3.2.1. Glucose Metabolism Pathways in Cancer Cells
3.2.2. Glucose Transporters
3.2.3. Glycolytic Pathway
3.2.4. Pentose Phosphate Pathway
3.2.5. Tri-Carboxylic Acid (TCA) Cycle
3.2.6. Amino Acid Metabolism Pathways
3.2.7. Lipid Metabolism Pathways
4. Anticancer Agents that Target Metabolic Pathways and Their Regulators

{kind=link}
{kind=link}
| Pathways | Target Proteins/Enzymes or Metabolites | Drugs/Compounds | Cancer/Tumor Type | Clinical Trial Status |
|---|---|---|---|---|
| Signalling proteins and transcription factors | ||||
| mTORC1 | Temsirolimus and Everolimus | Metastatic/non-metastatic solid tumors | US FDA approved | |
| Ridaforolimus and other rapalogues | Pancreatic, endometrial and glioblastoma; lymphoma. | Phase I/II | ||
| mTORC1 and mTORC2 | Torin1 and PP242 | - | Preclinical | |
| HIF1α | PX-478 | Advanced solid tumor and lymphoma | Phase I | |
| Acriflavine | - | Preclinical | ||
| Hypoxia | Tirapazamine and other bioreductive compounds | Cervical, SCLC, NSCLC | Phase III | |
| Hypoxia, VEGF and VEGFR | Bevacizumab | Malignant glioma, NSCLC, ovarian and colorectal | US FDA approved | |
| IGF1R | Dalotuzumab (MK-0646), BIIB022, AVE1642 etc. | Solid tumors of NSCLC, pancreatic, hepatocellular carcinoma (HCC) and metastatic breast cancer | Phase I/II | |
| PI3K and mTOR | BEZ235, XL765, SF1126 and BGT226 | Malignant glioma and NSCLC | Phase I/II | |
| PI3K | GDC-0941 and PX866 | Metastatic breast cancer and non-Hodgkin’s lymphoma | Phase I | |
| AKT | Perifosine and GSK690693 | Renal cancer, NSCLC and lymphoma | Phase I/II | |
| AMPK and Complex I (mitochondrial) | Metformin | Solid tumors and lymphoma | US FDA approved | |
| Metabolic pathway enzymes | ||||
| Nucleotide biosynthesis pathway | DNA and RNA synthesis | 5-FU, cytarabine and methotrexate | Different types of tumors | US FDA approved |
| DNA synthesis | Folate, choline, methionine, | Lab studies | ||
| Methyltransferases | Betaine, selected B vitamins, Flavonoids, EGCG, genistein | Lab studies | ||
| Histone deacetylases (HDAC) | Butyrate, sulforaphane, Allylmercaptan, 3,3-Diindolylmethane | Lab studies | ||
| Histone acetyltranferase | Anacardic acid, garcinol, Curcumin, EGCG, Genistein | |||
| Acetylation of non-histone proteins | Butyrate, cambinol, Dihydrocoumarin, genistein | |||
| Glycolysis pathway | GLUT1 | Phloretin | Colon cancer and leukemia | - |
| GLUT1 | WZB117 | Lung cancer and breast cancer | - | |
| GLUT4 | Ritonavir | Multiple myeloma | - | |
| Hexokinase | 2-deoxyglucose (2-DG) | Leukemia, cervical cancer, hepatocarcinoma, breast cancer, small lung cancer, lymphoma and prostate cancer | PhaseI/II | |
| Hexokinase Hexokinase | Lonidamine (LND) | Benign prostatic hyperplasia, leukemia and lymphoma | Phase III | |
| 3-bromopyruvate (3-BrPA) | Leukemia, multiple myeloma, colon cancer and leukemia | Preclinical | ||
| Pyruvate kinase M2 (PKM2) | shRNA | Lung cancer | - | |
| Pyruvate kinase (PK) | TLN-232 | Metastatic melanoma and renal cell carcinoma | Phase II | |
| Lactate dehydrogenase (LDHA) | Oxamate | Breast cancer | ||
| Pentose phosphate pathway (PPP) | Glucose-6-phosphate dehydrogenase (G6PDH) | Resveratrol | Colon cancer | |
| Transketolase (TK) | Oxythiamine (OT) | Colon cancer | ||
| G6PDH and TK | Avemar | Jurkat T cells (Leukemia) | ||
| G6PDH, 6PGDH and Transaldolase TA | Combination of arginine and ascorbic acid | Human hepatoma cell lines (HA2T/VGH) | ||
| G6PDH, also depletion of ribose-5-phosphate (R-5P) | Dehydroepiandrosterone (DHEA) | Indirect study on polycystic ovary syndrome | ||
| TCA cycle | Pyruvate dehydrogenase kinase (PDK3) | siRNA | Cervical cancer and breast cancer | |
| Pyruvate dehydrogenase kinase (PDK1) | Dichloroacetate (DCA) | Fibrosarcoma, colon cnacer, lung cancer, squamous cell carcinoma and prostate cancer | ||
| Fatty acid synthesis | FASN | Cerulenin and C75 | Breast cancer | |
| Orlistat | Breast and pancreatic cancer | Preclinical | ||
| ATP-citrate lyase | SB-204990 | - | Preclinical | |
| Amino acid metabolism pathway | Glutamine | Phenylacetate | Brain tumors | Phase II |
| Asparagine | Asparaginase and pegasparaginase | Acute lymphoblastic leukaemia (ALL), T-cell lymphoma (TCL) and B-cell lymphoma (BCL) | Phase II/III | |
| Arginine | Arginine deiminase | Metastatic melanoma and hepatocellular carcinoma | Phase I/II | |
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Luengo-Fernandez, R.; Leal, J.; Gray, A.; Sullivan, R. Economic burden of cancer across the european union: A population-based cost analysis. Lancet Oncol. 2013, 14, 1165–1174. [Google Scholar] [CrossRef]
- Henderson, D.; Ogilvie, L.A.; Hoyle, N.; Keilholz, U.; Lange, B.; Lehrach, H.; OncoTrack, C. Personalized medicine approaches for colon cancer driven by genomics and systems biology: Oncotrack. Biotechnol. J. 2014, 9, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- IARC. Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. Available online: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx (accessed on 21 September 2015).
- Liu, H.; Lv, L.; Yang, K. Chemotherapy targeting cancer stem cells. Am. J. Cancer Res. 2015, 5, 880–893. [Google Scholar] [PubMed]
- De Souza, R.; Zahedi, P.; Badame, R.M.; Allen, C.; Piquette-Miller, M. Chemotherapy dosing schedule influences drug resistance development in ovarian cancer. Mol. Cancer Ther. 2011, 10, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Persidis, A. Cancer multidrug resistance. Nat. Biotechnol. 1999, 17, 94–95. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S. Is it time for a new paradigm for systemic cancer treatment? Lessons from a century of cancer chemotherapy. Front. Pharmacol. 2013, 4, 68. [Google Scholar] [CrossRef] [PubMed]
- Cimino, G.D.; Pan, C.X.; Henderson, P.T. Personalized medicine for targeted and platinum-based chemotherapy of lung and bladder cancer. Bioanalysis 2013, 5, 369–391. [Google Scholar] [CrossRef] [PubMed]
- Joo, W.D.; Visintin, I.; Mor, G. Targeted cancer therapy—Are the days of systemic chemotherapy numbered? Maturitas 2013, 76, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Duckwall, C.S.; Murphy, T.A.; Young, J.D. Mapping cancer cell metabolism with(13)c flux analysis: Recent progress and future challenges. J. Carcinog. 2013, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Warmoes, M.O.; Shen, X.; Locasale, J.W. Epigenetics and cancer metabolism. Cancer Lett. 2015, 356, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, E.; Bucher, J.; Ryll, A.; Niklas, J.; Mauch, K.; Klamt, S.; Rocha, M.; Saez-Rodriguez, J. Bridging the layers: Towards integration of signal transduction, regulation and metabolism into mathematical models. Mol. Biosyst. 2013, 9, 1576–1583. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oliveira, A.P.; Jewett, M.C.; Nielsen, J. From Gene Expression to Metabolic Fluxes. In Introduction to Systems Biology; Choi, S., Ed.; The Humana Press Inc.: Totowa, NJ, USA, 2007. [Google Scholar]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Wilson, R.K. Cancer genome sequencing: A review. Hum. Mol. Genet. 2009, 18, R163–R168. [Google Scholar] [CrossRef] [PubMed]
- Kitano, H. Systems biology: A brief overview. Science 2002, 295, 1662–1664. [Google Scholar] [CrossRef] [PubMed]
- Wang, E. A Roadmap of Cancer Systems Biology; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- You, L.; Zhang, B.; Tang, Y.J. Application of stable isotope-assisted metabolomics for cell metabolism studies. Metabolites 2014, 4, 142–165. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Franklin, R.B. Tumor cell metabolism: The marriage of molecular genetics and proteomics with cellular intermediary metabolism; proceed with caution! Mol. Cancer 2006, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Raamsdonk, L.M.; Teusink, B.; Broadhurst, D.; Zhang, N.; Hayes, A.; Walsh, M.C.; Berden, J.A.; Brindle, K.M.; Kell, D.B.; Rowland, J.J.; et al. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotechnol. 2001, 19, 45–50. [Google Scholar] [PubMed]
- Artati, A.; Prehn, C.; Möller, G.; Adamski, J. Assay tools for metabolomics. In Genetics Meets Metabolomics; Suhre, K., Ed.; Springer International Publishing AG: Gewerbestrasse, Switzerland, 2012. [Google Scholar]
- Wolfe, R.R.; Chinkes, D.L. Isotope Tracers in Metabolic Research: Principles and Practice of Kinetic Analysis, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Kreeger, P.K.; Lauffenburger, D.A. Cancer systems biology: A network modeling perspective. Carcinogenesis 2010, 31, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.S.; Baty, J.W.; Berridge, M.V. The role of mitochondrial electron transport in tumorigenesis and metastasis. Biochim. Biophys. Acta 2014, 1840, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.I.; Cravatt, B.F.; Nomura, D.K. Global profiling strategies for mapping dysregulated metabolic pathways in cancer. Cell Metab. 2012, 16, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Nagrath, D.; Caneba, C.; Karedath, T.; Bellance, N. Metabolomics for mitochondrial and cancer studies. Biochim. Biophys. Acta 2011, 1807, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.E.; Abdel-Haleem, A.M. The evolution of genome-scale models of cancer metabolism. Front. Physiol. 2013, 4, 237. [Google Scholar] [CrossRef] [PubMed]
- Khazaei, T.; McGuigan, A.; Mahadevan, R. Ensemble modeling of cancer metabolism. Front. Physiol. 2012, 3, 135. [Google Scholar] [CrossRef] [PubMed]
- Guhathakurta, D.; Sheikh, N.A.; Meagher, T.C.; Letarte, S.; Trager, J.B. Applications of systems biology in cancer immunotherapy: From target discovery to biomarkers of clinical outcome. Expert Rev. Clin. Pharmacol. 2013, 6, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Coller, H.A. Is cancer a metabolic disease? Am. J. Pathol. 2014, 184, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef] [PubMed]
- Prasasya, R.D.; Tian, D.; Kregger, P.K. Analysis of cancer signaling networks by systems biology to develop therapies. Semin. Cancer Biol. 2011, 21, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Pinedo, C.; El Mjiyad, N.; Ricci, J.E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Levitzki, A.; Klein, S. Signal transduction therapy of cancer. Mol. Asp. Med. 2010, 31, 287–329. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Cantley, L.C. Ras, pi(3)k and mtor signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Cantley, L.C. Pi3k pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase akt pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Vander Heiden, M.G.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Gunther, U.L.; Chong, M.G.; Volpari, T.; Koczula, K.M.; Atkins, K.; Bunce, C.M.; Khanim, F.L. Metabolic fluxes in cancer metabolism. In Tumor Cell Metabolism; Mazurek, S., Shoshan, M., Eds.; Springer-Verlag Wien: Birmingham, UK, 2015. [Google Scholar]
- Jones, N.P.; Schulze, A. Targeting cancer metabolism—Aiming at a tumour’s sweet-spot. Drug Discov. Today 2012, 17, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. Ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar] [PubMed]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic k-ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The ampk signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor lkb1 kinase directly activates amp-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Huang, C.; Wei, Y. The metabolic switch and its regulation in cancer cells. Sci. China Life Sci. 2010, 53, 942–958. [Google Scholar] [CrossRef] [PubMed]
- Soga, T. Cancer metabolism: Key players in metabolic reprogramming. Cancer Sci. 2013, 104, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hif-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, M.S.; Jurgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Horstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of hif-2alpha in distinct cell populations of different organs. FASEB J. 2003, 17, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, P.A.; Yang, J.; Metelo, A.M.; Perez-Carro, R.; Baker, R.; Wang, Z.; Arreola, A.; Rathmell, W.K.; Olumi, A.; Lopez-Larrubia, P.; et al. In vivo hif-mediated reductive carboxylation is regulated by citrate levels and sensitizes vhl-deficient cells to glutamine deprivation. Cell Metab. 2013, 17, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase m2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [PubMed]
- Sermeus, A.; Michiels, C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011, 2, e164. [Google Scholar] [CrossRef] [PubMed]
- Cascante, M.; Benito, A.; Zanuy, M.; Vizan, P.; Marin, S.; de Atauri, P. Metabolic network adaptations in cancer as targets for novel therapies. Biochem. Soc. Trans. 2010, 38, 1302–1306. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of ldh-a expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase a induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.; Adekola, K.U.; Rosen, S.T.; Shanmugam, M. Cancer metabolism as a therapeutic target. Oncology 2013, 27, 460–467. [Google Scholar] [PubMed]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed]
- Hulleman, E.; Kazemier, K.M.; Holleman, A.; VanderWeele, D.J.; Rudin, C.M.; Broekhuis, M.J.; Evans, W.E.; Pieters, R.; Den Boer, M.L. Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood 2009, 113, 2014–2021. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. Idh1 and idh2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- McBrayer, S.K.; Cheng, J.C.; Singhal, S.; Krett, N.L.; Rosen, S.T.; Shanmugam, M. Multiple myeloma exhibits novel dependence on glut4, glut8, and glut11: Implications for glucose transporter-directed therapy. Blood 2012, 119, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhang, Y.; Chen, T.; Wang, Y.; Xue, J.; Zhang, Y.; Xiao, W.; Mo, X.; Lu, Y. Efficacy of rnai targeting of pyruvate kinase m2 combined with cisplatin in a lung cancer model. J. Cancer Res. Clin. Oncol. 2011, 137, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, S.N.; Sethuraman, S.; Krishnan, U.M. Metabolic pathways in cancers: Key targets and implications in cancer therapy. RSC Adv. 2015, 5, 41751–41762. [Google Scholar] [CrossRef]
- Bustamante, E.; Pedersen, P.L. High aerobic glycolysis of rat hepatoma cells in culture: Role of mitochondrial hexokinase. Proc. Natl. Acad. Sci. USA 1977, 74, 3735–3739. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, E.; Morris, H.P.; Pedersen, P.L. Energy metabolism of tumor cells. Requirement for a form of hexokinase with a propensity for mitochondrial binding. J. Biol. Chem. 1981, 256, 8699–8704. [Google Scholar] [PubMed]
- Cheng, Y.; Diao, D.; Zhang, H.; Guo, Q.; Wu, X.; Song, Y.; Dang, C. High glucose-induced resistance to 5-fluorouracil in pancreatic cancer cells alleviated by 2-deoxy-d-glucose. Biomed. Rep. 2014, 2, 188–192. [Google Scholar] [PubMed]
- Pilkis, S.J.; Claus, T.H. Hepatic gluconeogenesis/glycolysis: Regulation and structure/function relationships of substrate cycle enzymes. Annu. Rev. Nutr. 1991, 11, 465–515. [Google Scholar] [CrossRef] [PubMed]
- Staal, G.E.; Kalff, A.; Heesbeen, E.C.; van Veelen, C.W.; Rijksen, G. Subunit composition, regulatory properties, and phosphorylation of phosphofructokinase from human gliomas. Cancer Res. 1987, 47, 5047–5051. [Google Scholar] [PubMed]
- Ashizawa, K.; Willingham, M.C.; Liang, C.M.; Cheng, S.Y. In vivo regulation of monomer-tetramer conversion of pyruvate kinase subtype m2 by glucose is mediated via fructose 1,6-bisphosphate. J. Biol. Chem. 1991, 266, 16842–16846. [Google Scholar] [PubMed]
- Griffin, J.L.; Shockcor, J.P. Metabolic profiles of cancer cells. Nat. Rev. Cancer 2004, 4, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated idh1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of krebs cycle intermediates and over-expression of hif1alpha in tumours which result from germline fh and sdh mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mtor and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Tomita, M.; Kami, K. Cancer. Systems biology, metabolomics, and cancer metabolism. Science 2012, 336, 990–991. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R.; Colomer, R. Inhibition of tumor-associated fatty acid synthase hyperactivity induces synergistic chemosensitization of her -2/ neu -overexpressing human breast cancer cells to docetaxel (taxotere). Breast Cancer Res. Treat. 2004, 84, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Types of Chemotherapy Drugs. Available online: http://www.cancer.org/treatment/treatmentsandsideeffects/treatmenttypes/chemotherapy/chemotherapyprinciplesanin-depthdiscussionofthetechniquesanditsroleintreatment/chemotherapy-principles-types-of-chemo-drugs (accessed on 26 August 2015).
- Bensaad, K.; Harris, A.L. Cancer metabolism as a therapeutic target: Metabolic synthetic lethality. Oncology 2013, 27, 467, 473–474. [Google Scholar] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Grimaldi, C.; Ricci, F.; Tazzari, P.L.; Evangelisti, C.; Ognibene, A.; Battistelli, M.; Falcieri, E.; Melchionda, F.; Pession, A.; et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor nvp-bez235 against t-cell acute lymphoblastic leukemia. Cancer Res. 2010, 70, 8097–8107. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (dca) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Yangzom, D.; Bhutia, E.B.; Prasad, P.D.; Ganapathy, V. The amino acid transporter slc6a14 in cancer and its potential use in chemotherapy. Asian J. Pharm. Sci. 2014, 9, 293–303. [Google Scholar]
- Rahman, M. Pentose phosphate pathway in disease and therapy. Adv. Mater. Res. 2014, 995, 1–27. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.; Hasan, M.R. Cancer Metabolism and Drug Resistance. Metabolites 2015, 5, 571-600. https://doi.org/10.3390/metabo5040571
Rahman M, Hasan MR. Cancer Metabolism and Drug Resistance. Metabolites. 2015; 5(4):571-600. https://doi.org/10.3390/metabo5040571
Chicago/Turabian StyleRahman, Mahbuba, and Mohammad Rubayet Hasan. 2015. "Cancer Metabolism and Drug Resistance" Metabolites 5, no. 4: 571-600. https://doi.org/10.3390/metabo5040571
APA StyleRahman, M., & Hasan, M. R. (2015). Cancer Metabolism and Drug Resistance. Metabolites, 5(4), 571-600. https://doi.org/10.3390/metabo5040571