
Using Gene Essentiality and Synthetic Lethality Information to Correct Yeast and CHO Cell Genome-Scale Models

Abstract
:1. Introduction
2. Results and Discussion
2.1. S. Cerevisiae Model Yeast 7.11 Curation
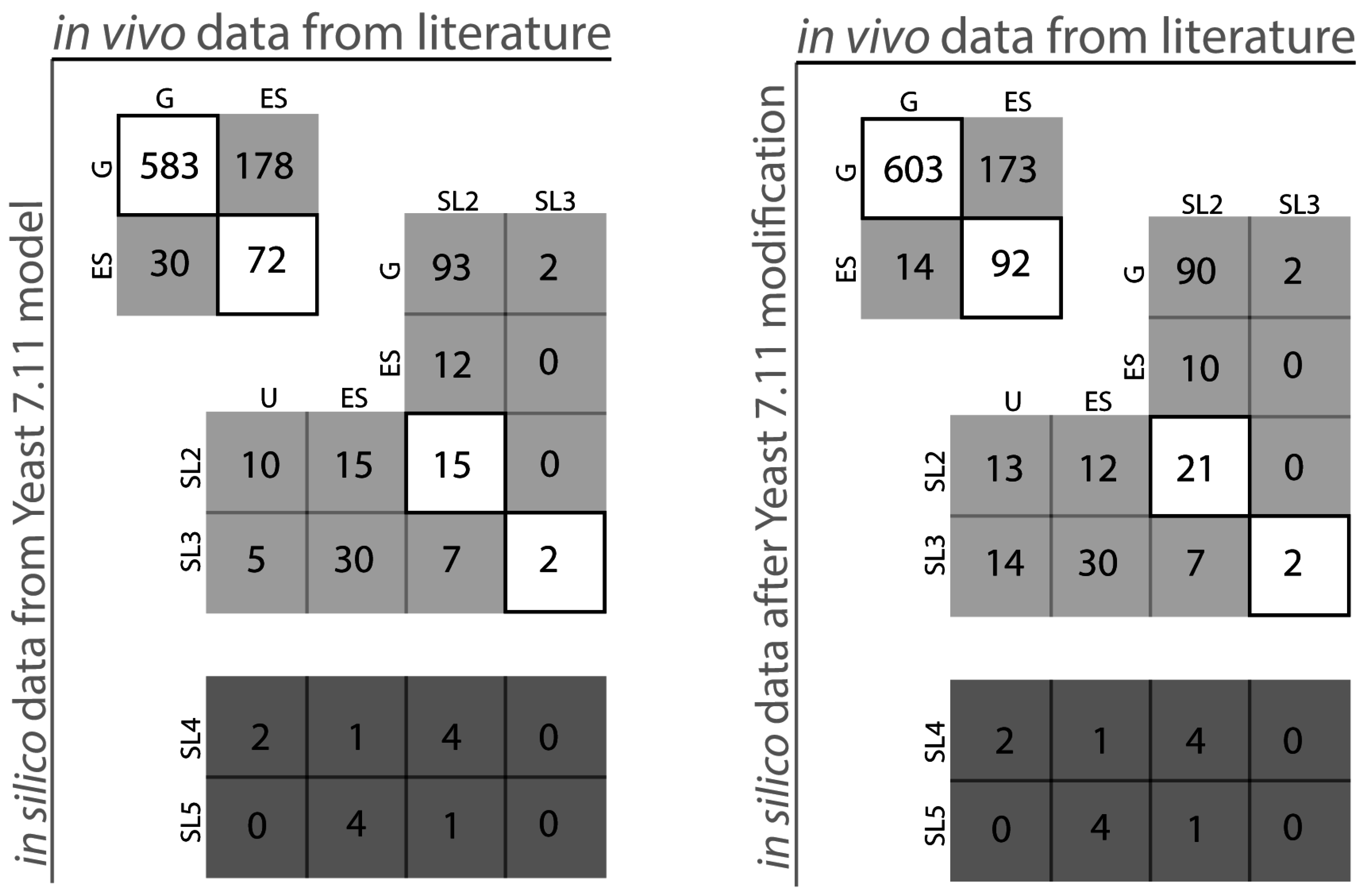

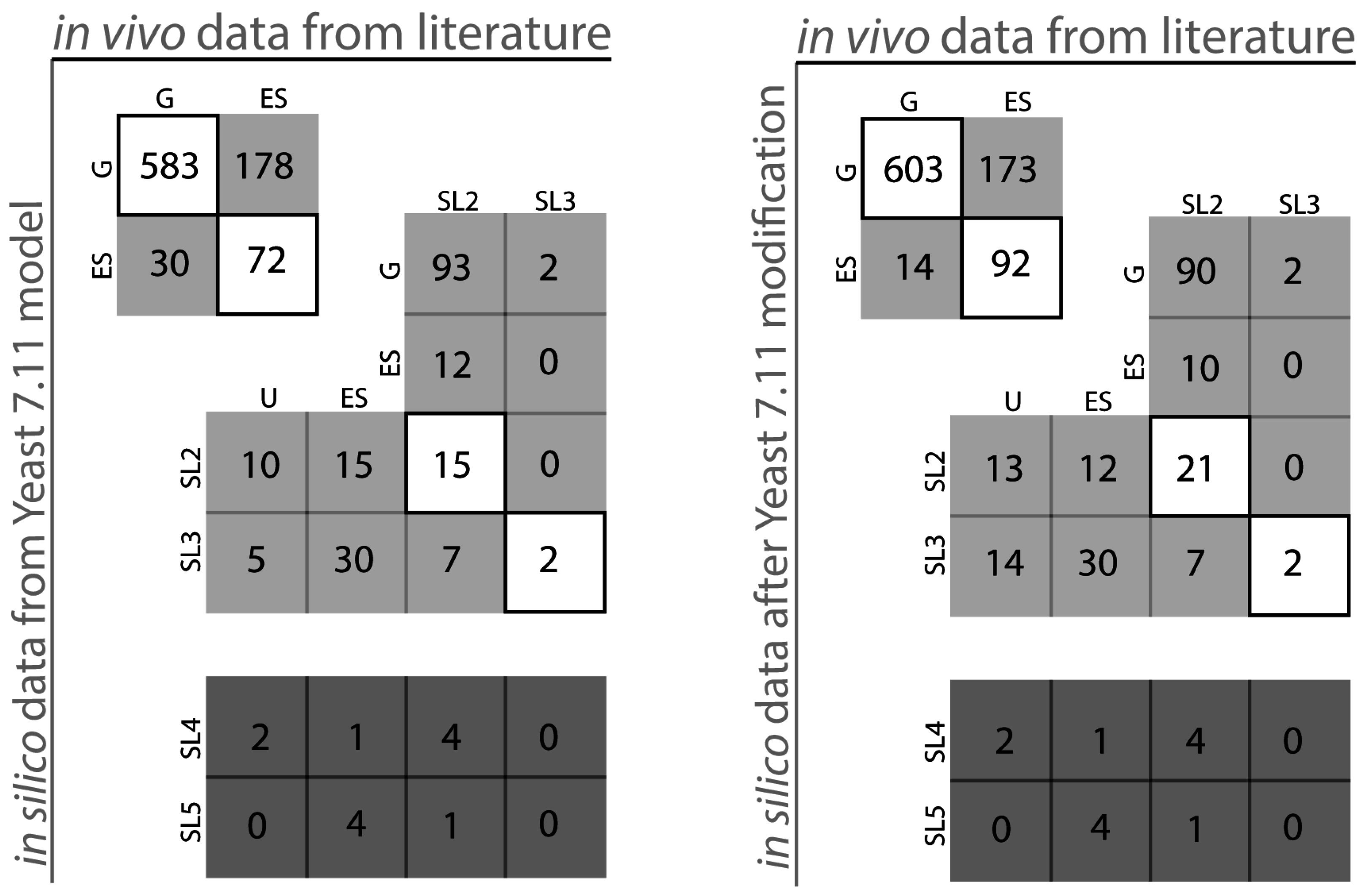{kind=link}
{kind=link}
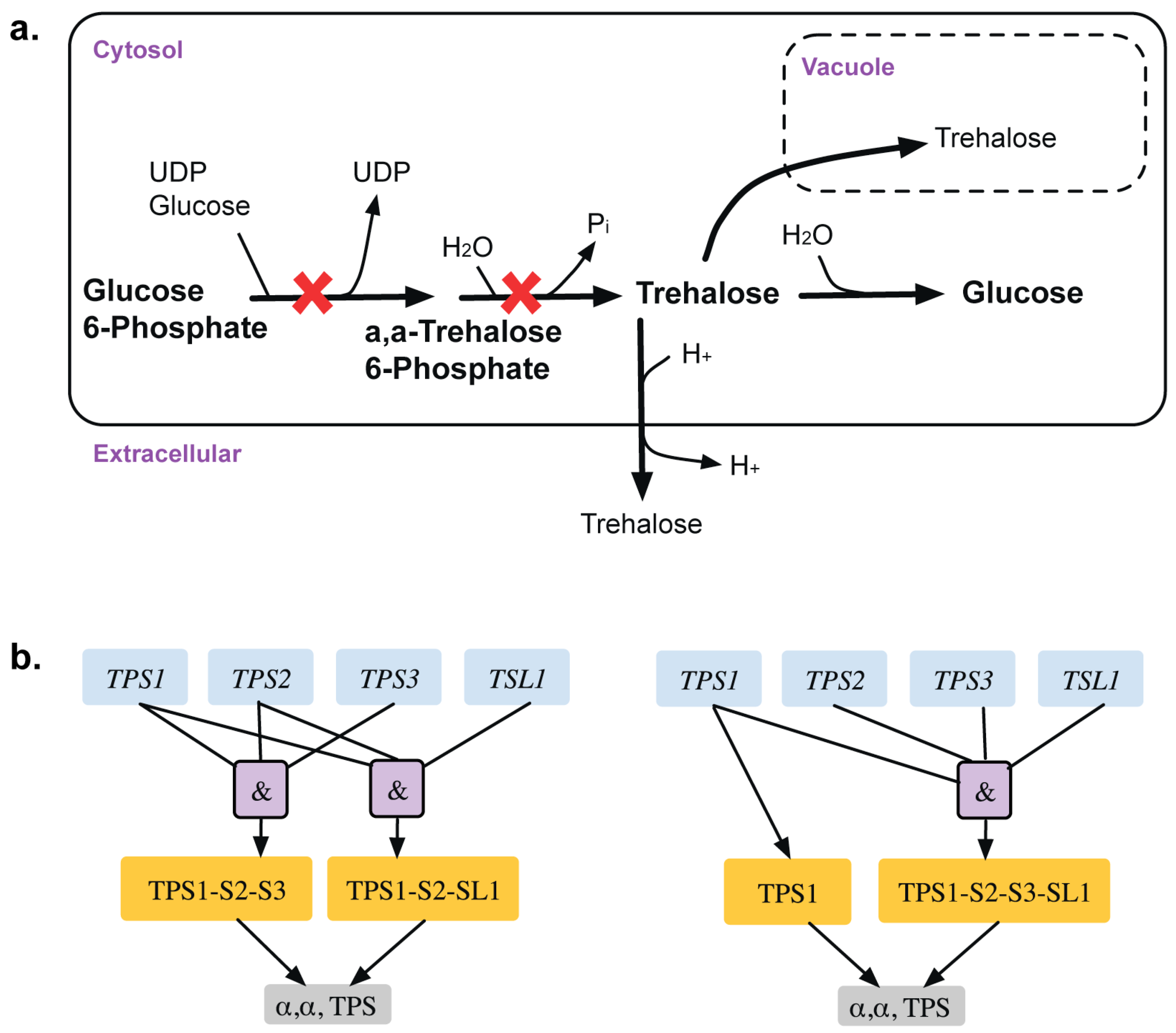{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
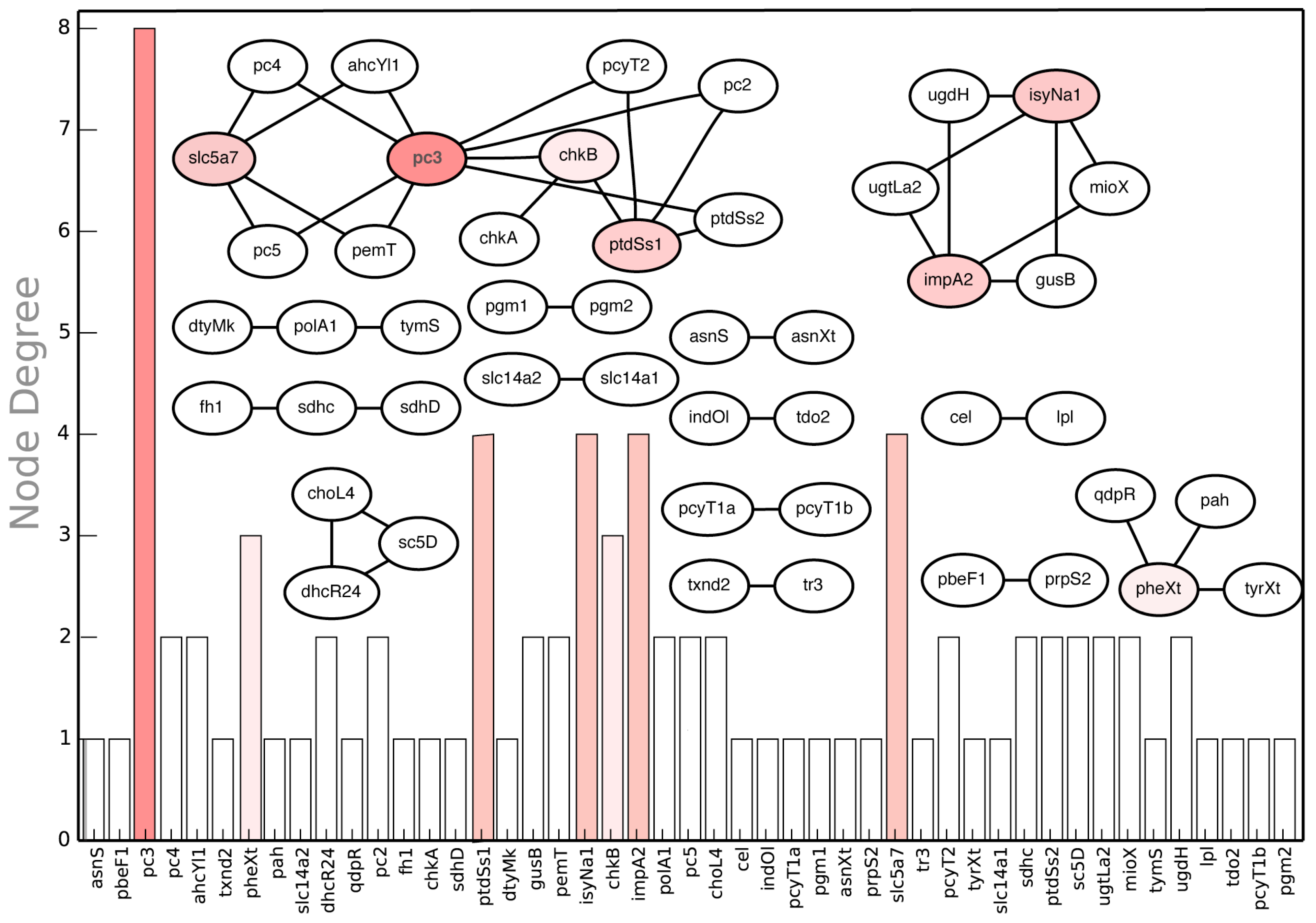{kind=link}
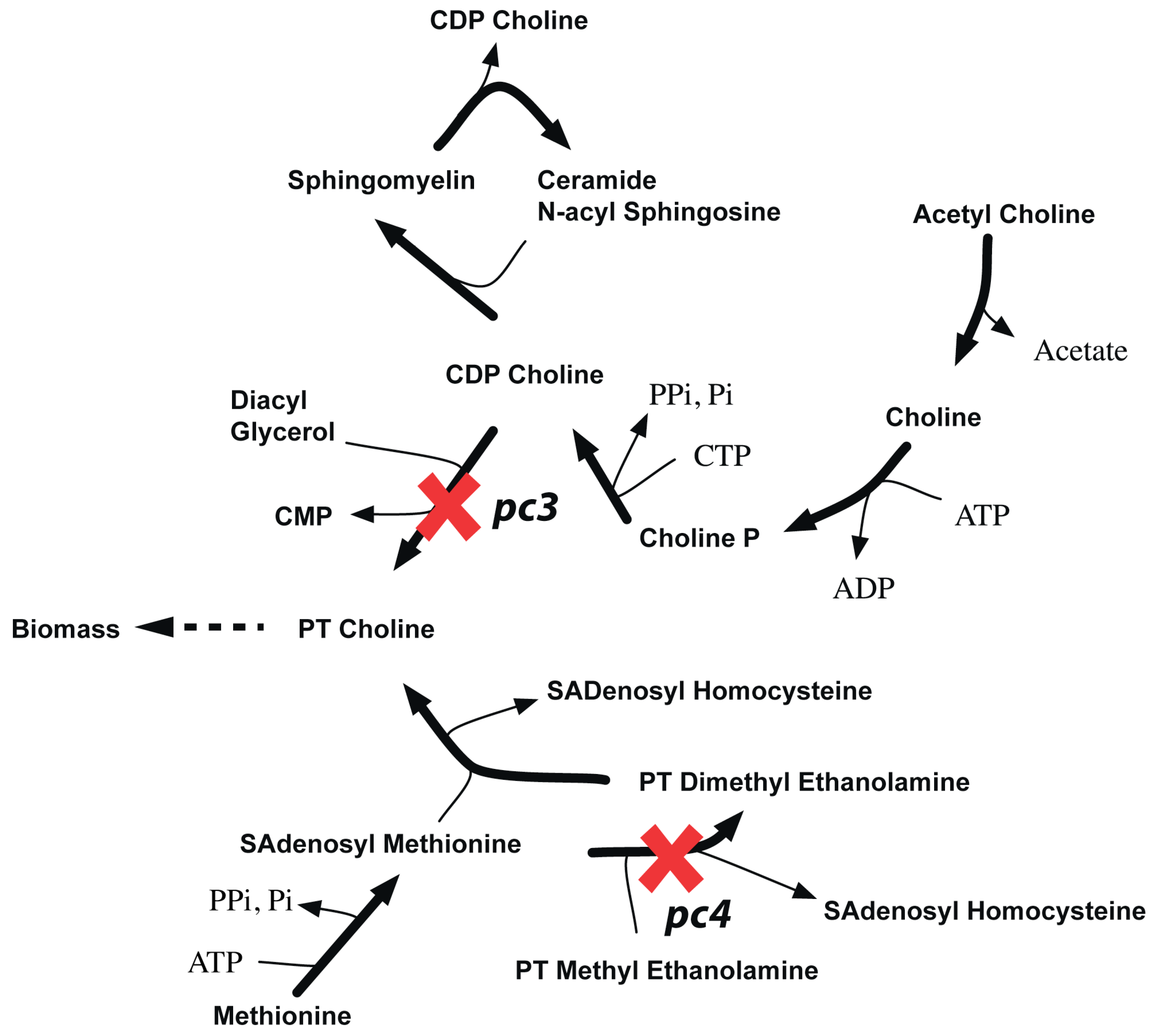{kind=link}
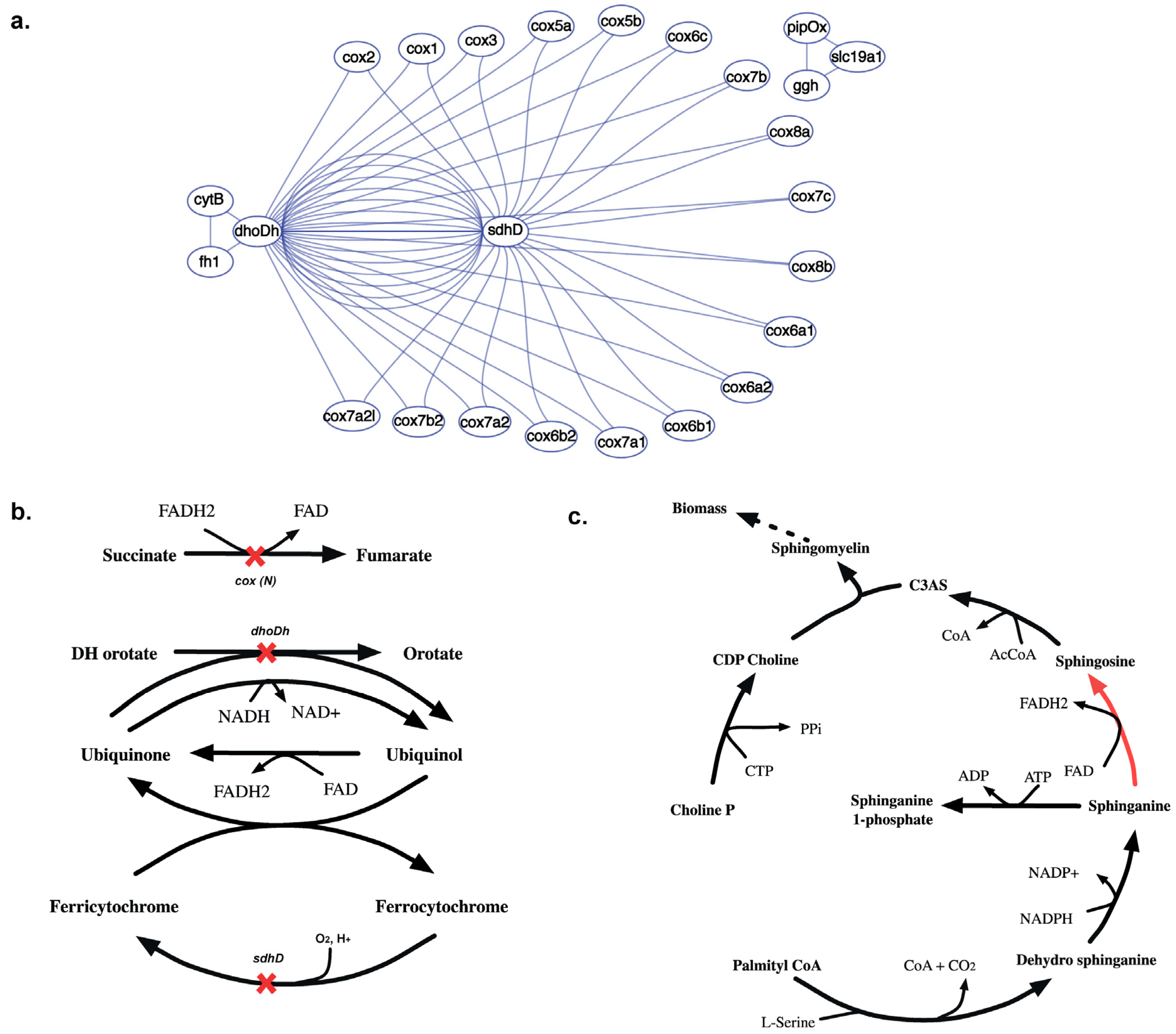{kind=link}
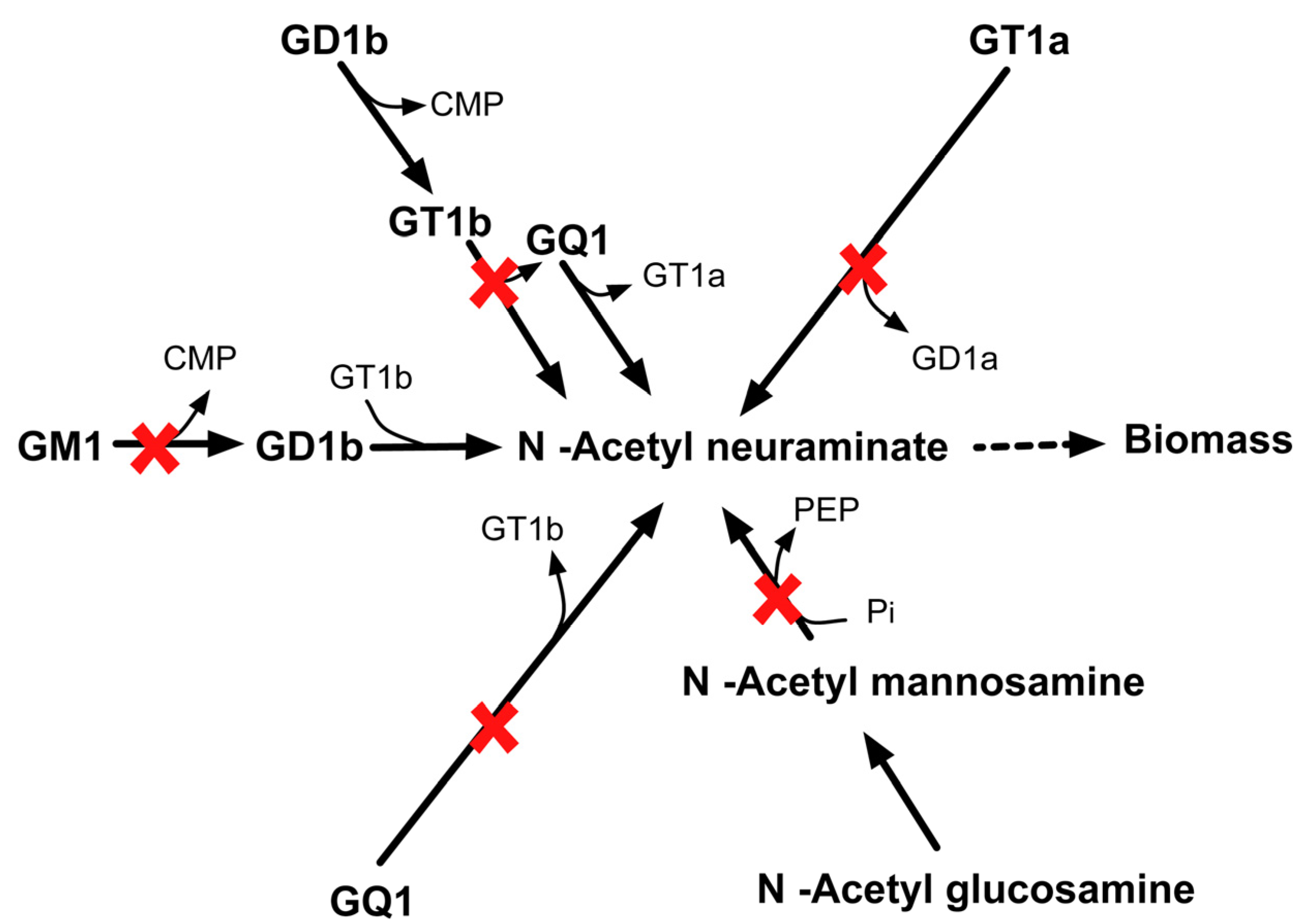{kind=link}
| Parameter | Count | |
|---|---|---|
| Essentiality information | Essential Reactions with GPRs | 195 |
| Essential Reactions without GPRs | 248 | |
| Essential genes | 151 | |
| Reaction level lethality | SL Pairs | 70 |
| SL Triplets | 21 | |
| SL Quadruplets | 11 | |
| SL Quintuplets | NP1 | |
| Gene level lethality | SL Pairs | 40 |
| SL Triplets | 44 | |
| SL Quadruplets | 7 | |
| SL Quintuplets | 5 |
| No. | Model Modification | Improvement on Yeast 7.11 | Remarks | Reference | |
|---|---|---|---|---|---|
| Addition of Reactions | 1 | Addition of alpha-keto isovalerate (KIV) transport 3-methyl 2-oxobutanoate [m] ⬄ 3-methyl 2-oxobutanoate [c] | BAT1 reconciled from ESG to GG BAT1-BAT2 reconciled from ESSL2 to SL2SL2 | The KIV transport provides an alternate path for cytosolic valine formation. | [25] [35] |
| 2 | Mitochondrial acetyl-transferase activity of glycine CoA [m] + L-2 amino 3-oxobutanoate [m] ⬄ acetyl-CoA [m] + L-glycine [m] GPR: YDL040C or YGR147C or YHR013C | Correctly adds NAT1, NAT2 and ARD1 as GG | This adds a missing reaction and identifies the associated genes correctly as non-essential. | [36] [37] | |
| GPR modifications | 3 | GPR modification for reaction r_0195 Old GPR: ((YBR126C and YDR074W and YMR261C) or (YML100W and YBR126C and YDR074W)) New GPR: ((YBR126C and YDR074W and YMR261C and YML100W) or YBR126C) | TPS1 gene is only essential in glucose media whereas both TPS1 and TPS2 genes are essential in galactose media reflected in old GPR TPS2 gene is restored as a GG from an ESG | This shows a media dependent gene essentiality. | [38] [39] [40] |
| 4 | GPR modification for reaction r_0995 Old GPR: YDR023W or YHR011W New GPR: YDR023W or (YDR023W and YHR011W) | SES1 gene is corrected from GES to ESES SES1-DIA1 is corrected from SL2ES and SL2G to ESES and GG cases respectively | The modification identifies SES1 as the major isoform consistent to in vivo information. | [41] [42] | |
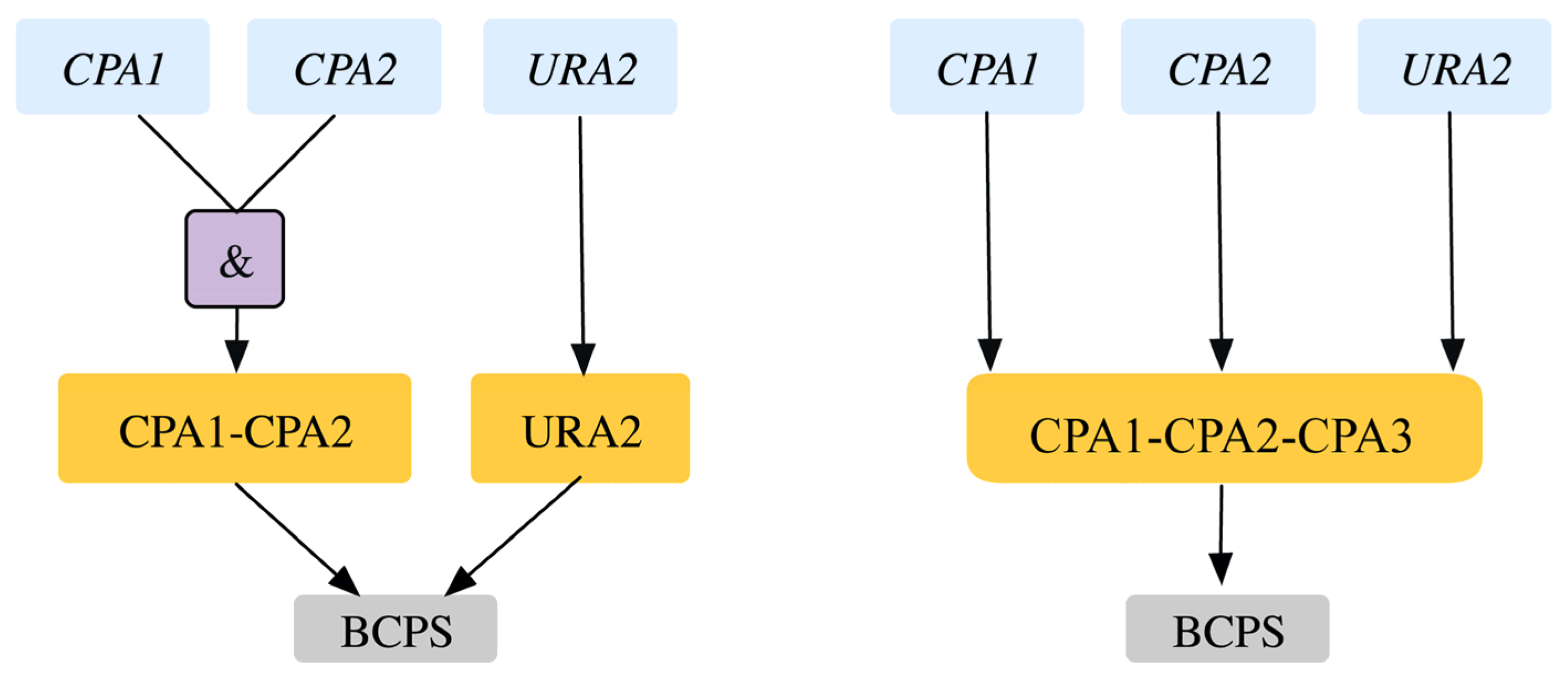
| 5 | GPR modification for reaction r_0250 Old GPR: ((YJR019C and YOR303W) or YJL130C) New GPR: YJR019C and YOR303W and YJL130C | SL2ES CPA2-URA2 is resolved correctly to 2 ESES for CPA2 and URA2 SL2ES CPA1-URA2 is resolved correctly to one more ESES case CPA1 At the same time it resolves 3 GES to ESES for the genes CPA1, CPA2, URA2 | This captures the essentiality of all three genes consistent with literature. | [43] [44] | |
| Removal of reactions | 6 | Remove orphan reaction r_2031 It was initially suggested in iAZ900 | Reconciles GSL2 of fur1-ura3 to SL2SL2 | This removes an orphan reaction that might have added extra alternate paths to uridine formation | [8] |
| Addition of GPR to orphan reactions | 7 | Add genes for reaction r_0094 L-alanine [c] +pimeloyl-CoA [c] ⬄ 8-amino-7 oxononanoate [c] + CO2 [c] + CoA [c] + 4H+ [c] | Adds GPR: YAR069W-A or YHR214W-F Adds genes BIO6 and BIO8 putative genes to the model and both are correctly predicted as GG. | This makes the model better in terms of correct identification of non-essential genes. | [45] |
| 8 | Add genes for reaction r_0475 H2O [c] + L-glutamine [c] ⬄ ammonium [c] + L-glutamate [c] | Adds GPR: YMR096W or (YMR095C and YMR096W) Adds genes SNZ1 and SNO1 to the model Correctly identifies SNZ1 and SNO1 genes as GG | This makes the model better in terms of correct identification of non-essential genes. | [46] |
| No. | Gene | Inconsistency | Remarks | Reference |
|---|---|---|---|---|
| 1 | SEC53 | ESG | SEC53 deletion is in silico and in vivo essential, but it was erroneously categorized as non-essential. | [24] [47] |
| 2 | HIS4 | ESG | HIS4 gene deletion is lysine auxotroph, which is in corroboration with in silico result. Yet the in vivo strain was categorized as viable hence causing ESG inconsistency. | [48] |
| 3 | ADK1 | ESG | ADK1 gene in vivo deletion is not inviable initially but over a period of 4 days, cells fail to survive. ADK1 in silico is adenine auxotroph as corroborated in vivo. | [49] |
| 4 | ERG20 | ESG | ERG20 deletion is in silico and in vivo essential, but it was erroneously categorized as non-essential. | [24] [50] |
| 6 | MET2 | ESG | MET2 gene deletion is methionine auxotroph and vegetative growth is reduced to less than 10%, which is in corroboration with in silico result. Yet, the in vivo strain was categorized as viable hence causing ESG inconsistency. | [51] [53] |
| 7 | LYS2 | ESG | LYS2 gene deletion is lysine auxotroph, which is in corroboration with in silico result. Yet, the in vivo strain was categorized as viable hence causing ESG inconsistency. | [54] [53] |
| 8 | DPS1 | ESG | DPS1 gene deletion is aspartate auxotroph, which is in corroboration with in silico result. Yet, the in vivo strain was categorized as viable hence causing ESG inconsistency. | [55] |
| 9 | FRS1 | ESG | FRS1 gene deletion is phenylalanine auxotroph, which is in corroboration with in silico result. Yet, the in vivo strain was categorized as viable hence causing ESG inconsistency. | [55] |
| 10 | ADE13 | ESG | ADE13 gene deletion is adenine auxotroph, which is in corroboration with in silico result. Yet, the in vivo strain was categorized as viable hence causing ESG inconsistency. | [54] [56] [57] [53] |
| 11 | ADE4 | ESG | ADE4 gene deletion is adenine auxotroph, which is in corroboration with in silico result. Yet, the in vivo strain was reported as viable hence causing ESG inconsistency. | [54] [56] [57] [53] |
| 12 | RIB4 | ESG | RIB4 gene deletion is riboflavin auxotroph, which is in corroboration with in silico result. Yet, the in vivo strain was categorized as viable hence causing ESG inconsistency. | [58] [53] |
| 13 | TPI1 | GES | TPI1 gene deletion is not in silico lethal. However, when PIT2m is suppressed, TPI1 is essential for viability. This could possibly be because of short-term Crabtree effect due to F16-bisphosphate accumulation under TPI1 deletion that suppresses mitochondrial respiratory enzymes. | [59] [60] |
| 14 | FBA1 | GES | FBA1 gene deletion is not in silico lethal. However, when PIT2m is suppressed, FBA1 is essential for viability. This could possibly be because of short-term Crabtree effect due to F16-bisphosphate accumulation under FBA1 deletion that suppresses mitochondrial respiratory enzymes. | [59] [60] |
| No. | in vivo Lethal Associations from Literature | Reason/Explanation | Reference |
|---|---|---|---|
| 1 | RIB7 gene forms 2 lethal pairs: RIB7-MAD1, RIB7-SGS1 | The candidate genes of lethal combination are non-metabolic and are involved in chromatid cohesion. | [57] |
| 2 | HIS7 gene forms 1 lethal pair: HIS7-RSP5 | RSP5 is involved in endocytosis signaling pathway, a non-metabolic function, hence unable to be captured in a metabolic model. | [61] |
| 3 | RIB5 gene forms 3 lethal pairs: RIB5-BUB1, RIB5-MAD1, RIB5-TAF1 | The candidate genes of lethal combination are non-metabolic and are involved in mitosis. | [57] |
| 4 | TSC10 gene forms 5 lethal pairs: TSC10-CDC74, TSC10-CHL1, TSC10-MAD1, TSC10-MRE11, TSC10-SGS1 | The candidate genes of lethal combination are non-metabolic and are involved in chromatid cohesion. | [57] |
| 5 | HEM13 gene forms 2 lethal pairs: HEM13-CDC73, HEM13-SMC3 | The candidate genes of lethal combination are non-metabolic and are involved in chromatid cohesion. | [57] |
| 6 | PRO3 gene forms 3 lethal pairs and 1 lethal triplet: PRO3-CDC73, PRO3-LRP1, PRO3-NIP7, PRO3-GAP1-PUT4 | The candidate genes are non-metabolic in function. | [54] [57] [62] |
| 7 | GNA1 forms 1 lethal pair: GNA1-CHL1 | The lethality is owing to chromosome loss which is a non-metabolic phenomenon. | [57] |
| 8 | FRS2 gene forms 5 lethal pairs: FRS2-CDC73, FRS2-ELG1, FRS2-RAD51, FRS2-SGS1, FRS2-SMC3 | The candidate genes of lethal combination are non-metabolic and are involved in chromatid cohesion. | [57] |
| 9 | TYS1 gene forms 2 lethal pairs: TYS1-BUB1, TYS1-SGS1 | The candidate genes of lethal combination are non-metabolic and are involved in mitosis. | [57] |
| 10 | ARG7 gene forms 1 lethal quadruplet: ARG7-ALP1-CAN1-GAP1 | The quadruplet association is not entirely metabolic hence cannot be captured by metabolic model. | [54] |
| 11 | OLE1 gene forms 3 lethal pairs: OLE1-BUB1, OLE1-ELO1, OLE1-RML2 | BUB1 gene is involved in mitosis. Δole1Δelo1 double mutant is inviable only in C:14 media RML2 is non-metabolic gene | [57] [63] [64] |
| 12 | YAH1 gene forms 1 lethal pair: YAH1-MRE11 | YAH1 has already been resolved as ESES MRE11-YAH1 double knockout strain will result in meiotic recombination disorder and will be lethal. This is a non-metabolic attribute of yeast. | [57] |
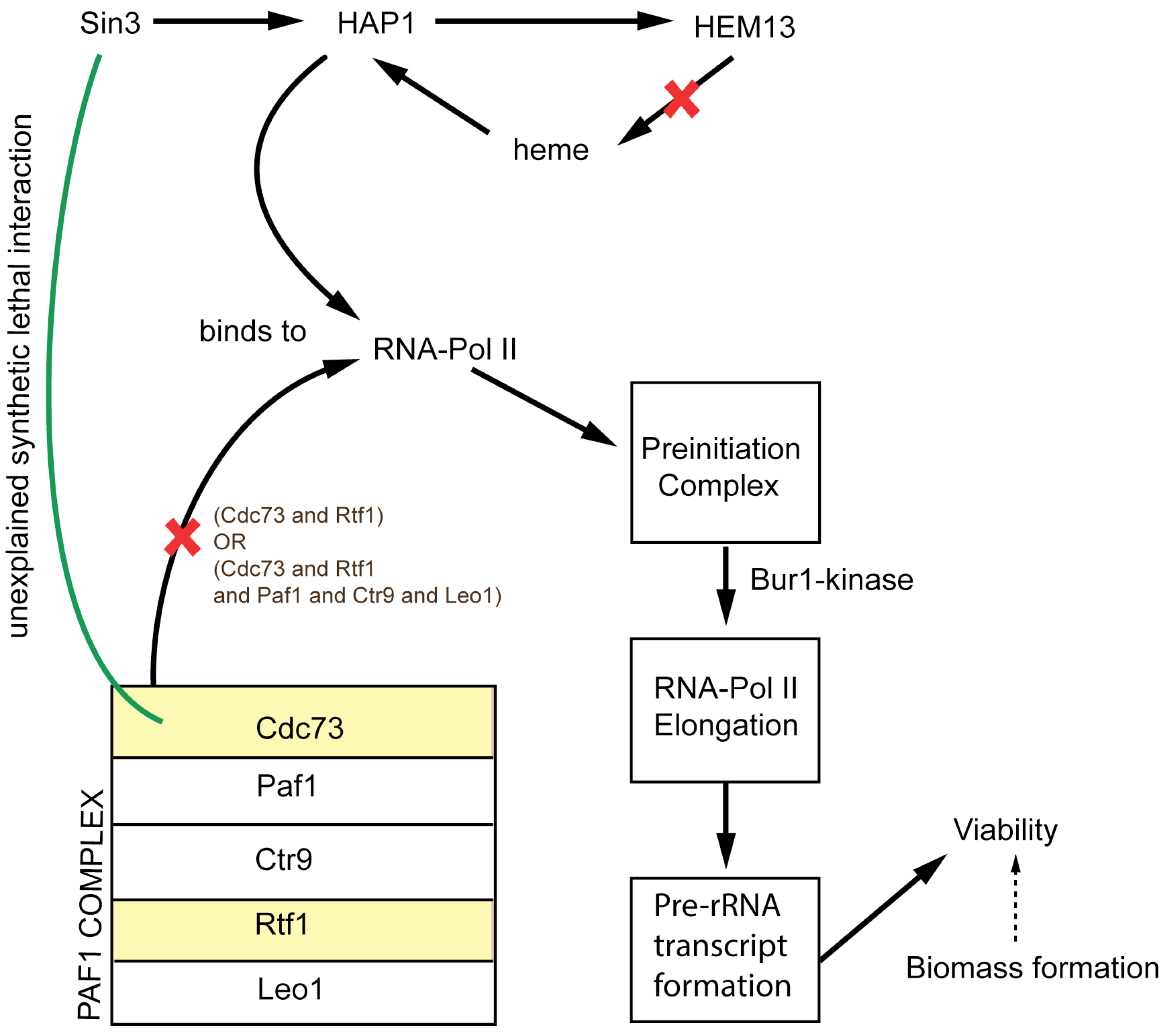
2.1.1. Addition of Reactions
2.1.2. Removal of Reactions
2.1.3. GPR Modifications
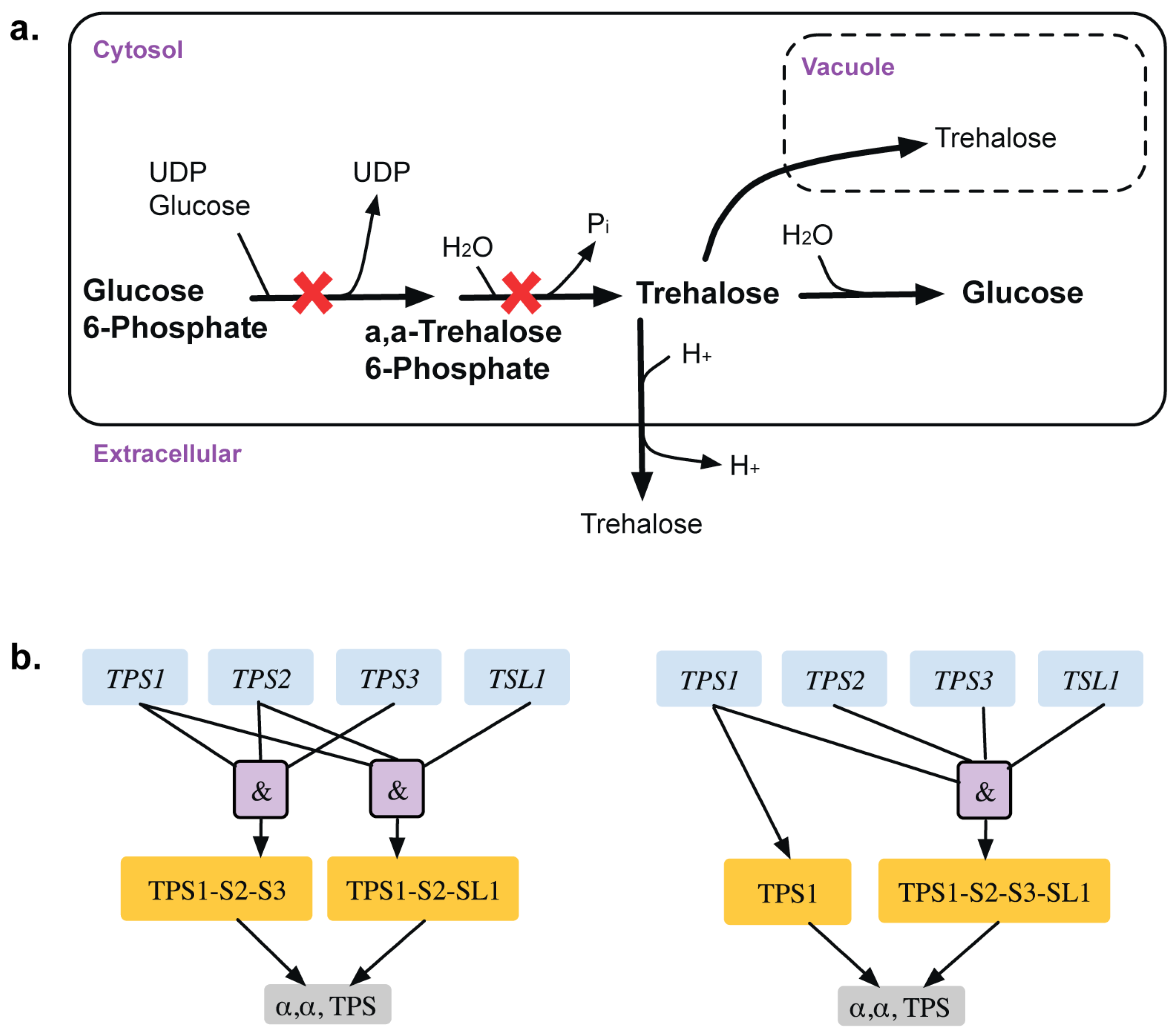
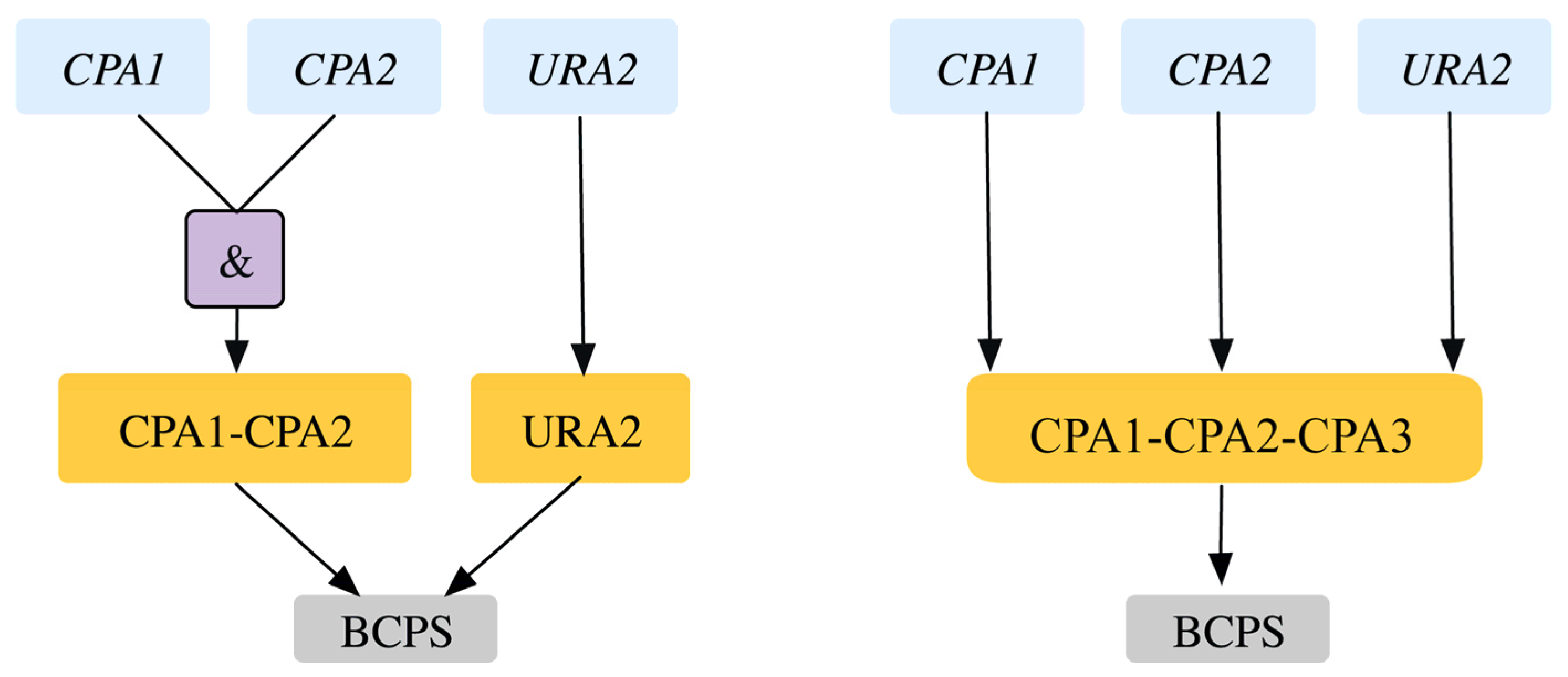
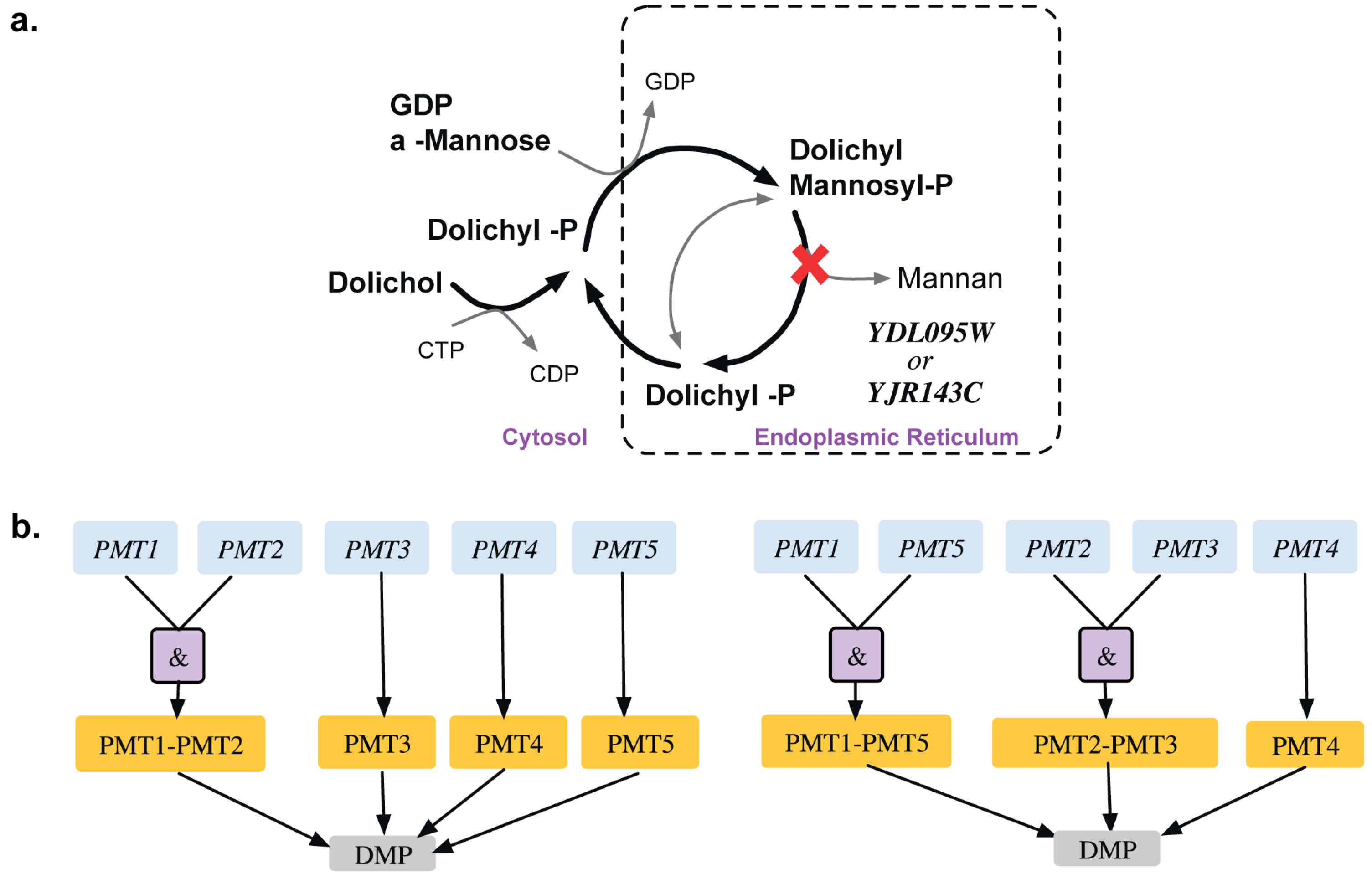
2.1.4. Addition of GPR to Orphan Reactions
2.1.5. MSL2 Gaps in the Model
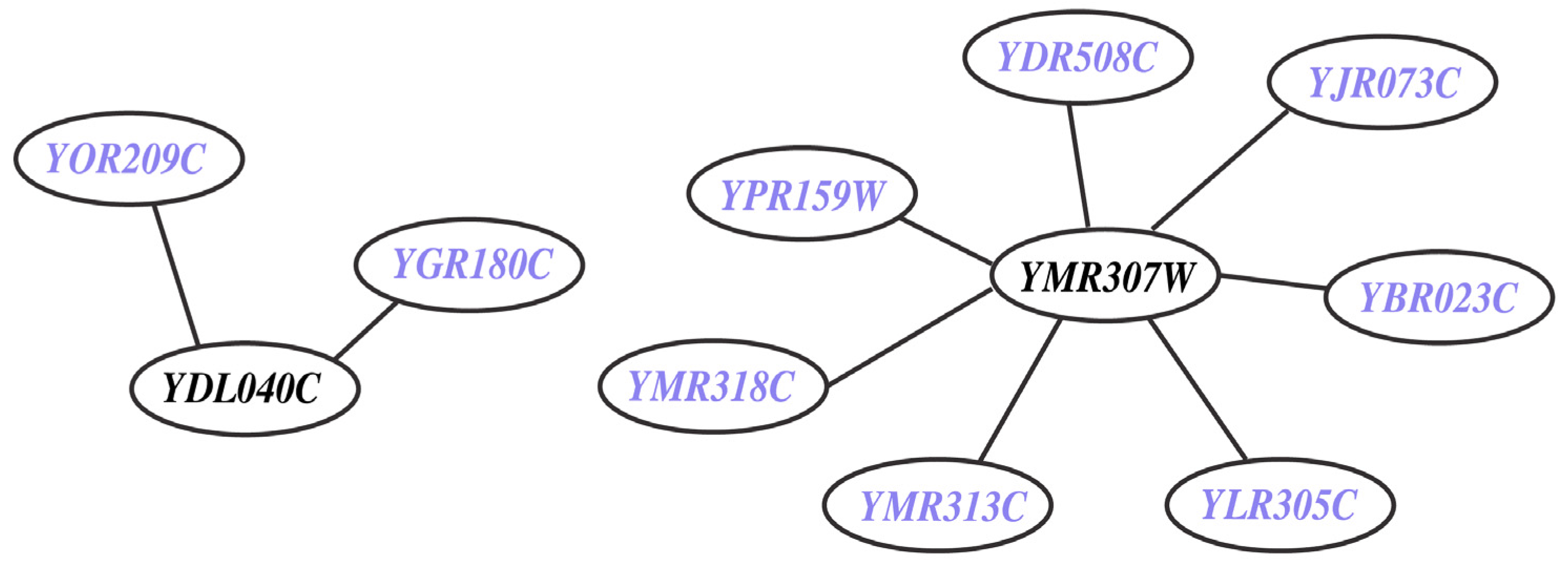
2.2. Model Predictions for Synthetic Lethals in S. Cerevisiae
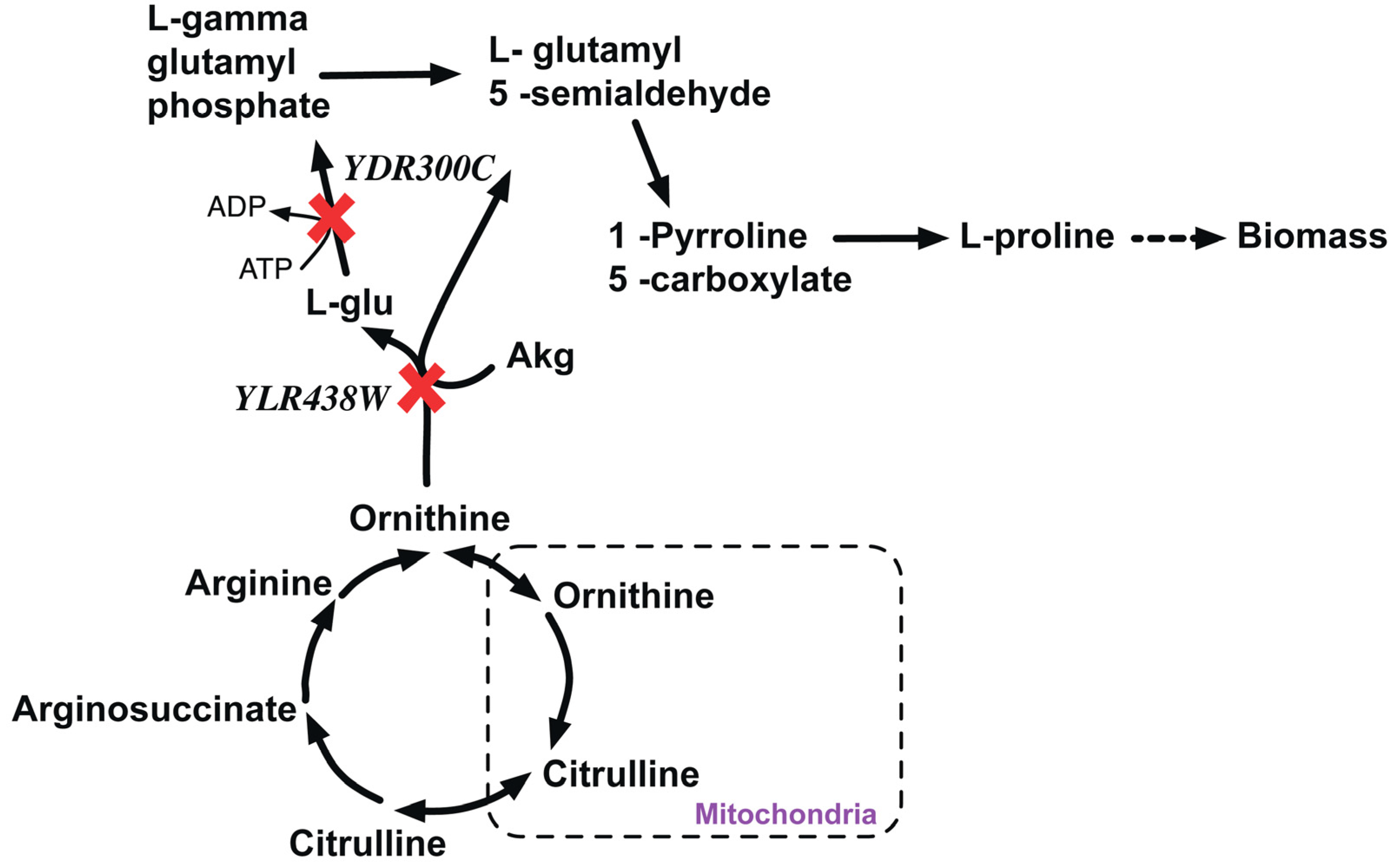
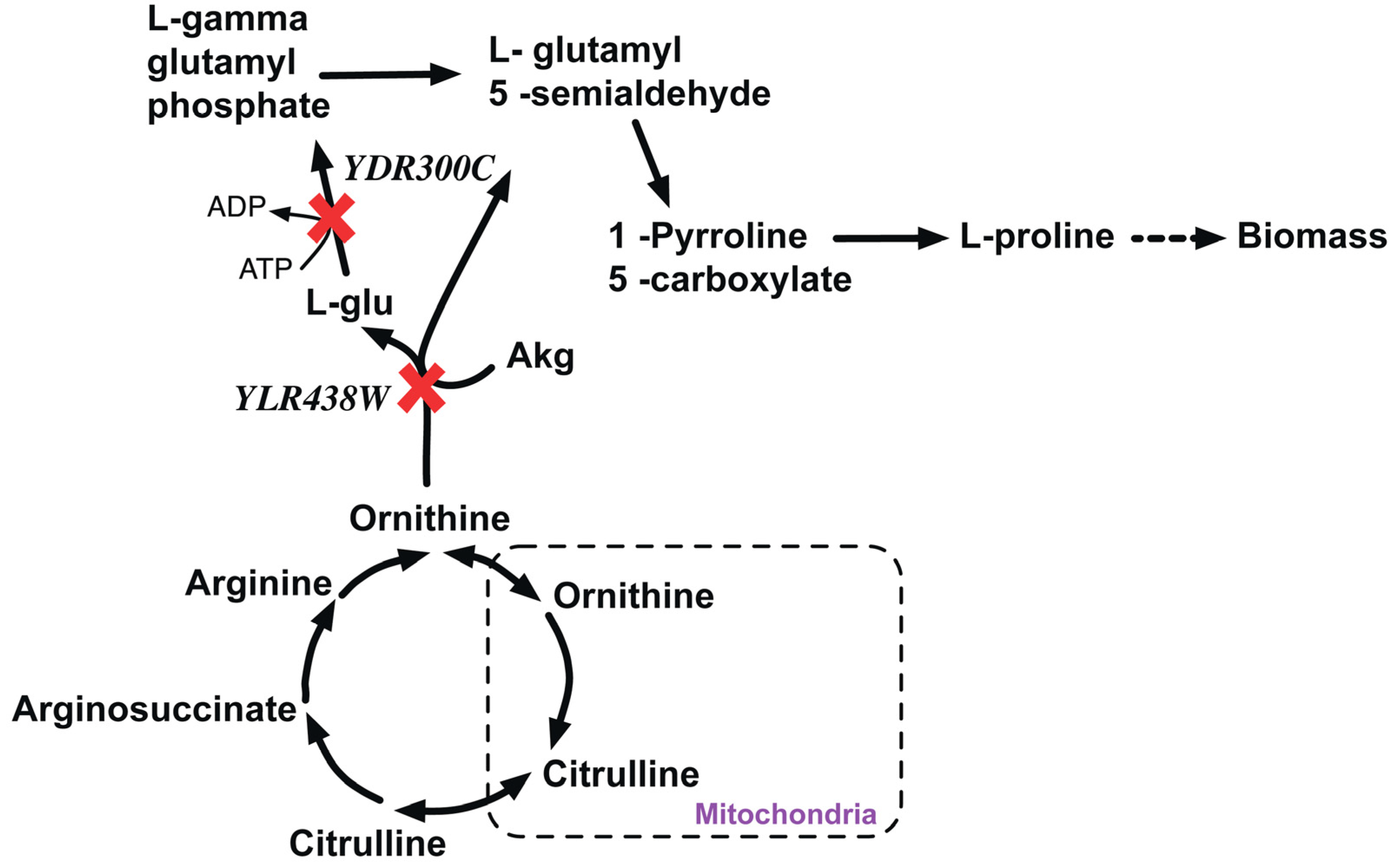
2.2.1. Proline Auxotrophy (Δpro1Δcar2 Double Mutant)
2.2.2. Leucine Auxotrophy (Δleu4Δleu9 Double Mutant)
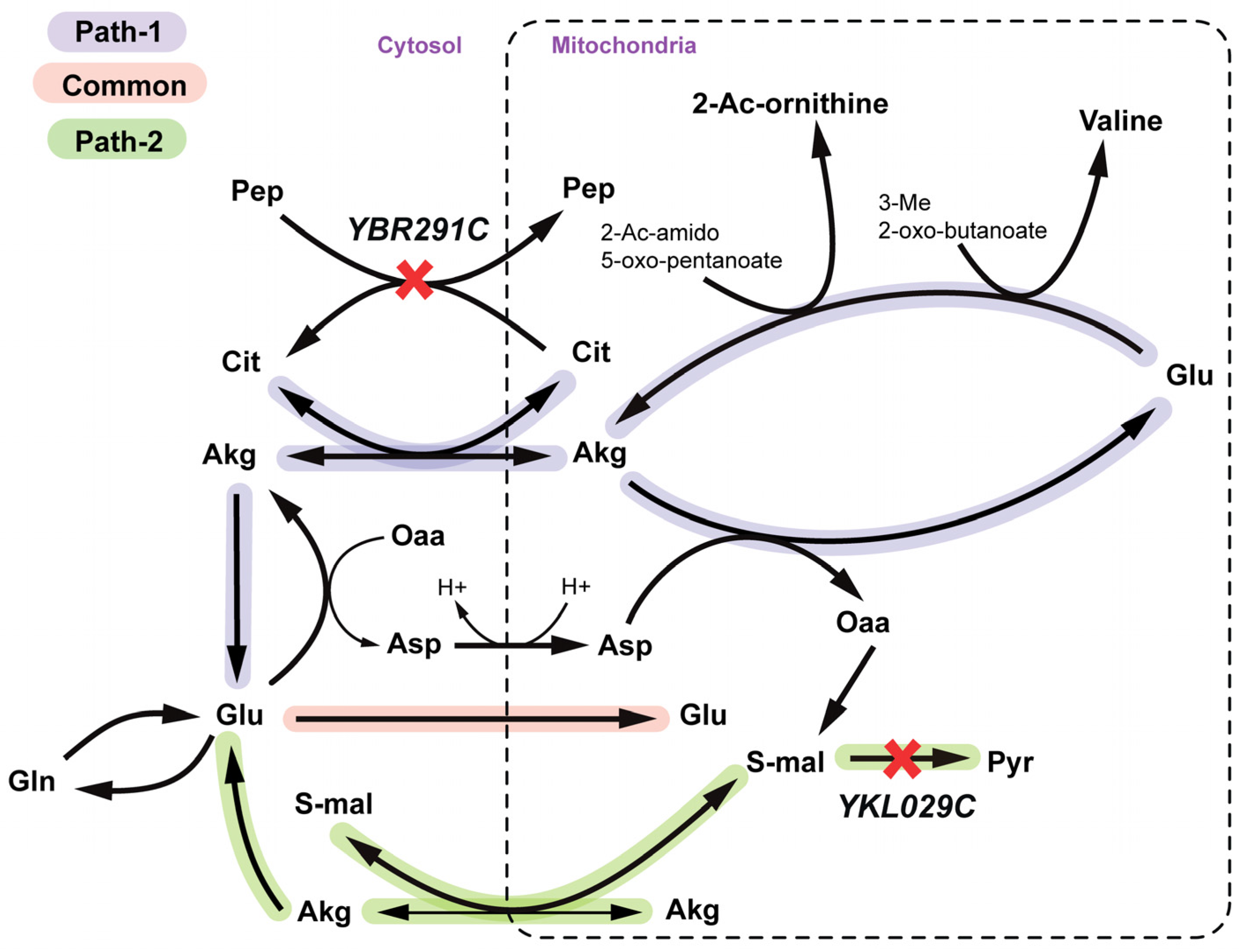
2.2.3. Arginine and Valine Auxotrophy (Δctp1Δmae1 Double Mutant)
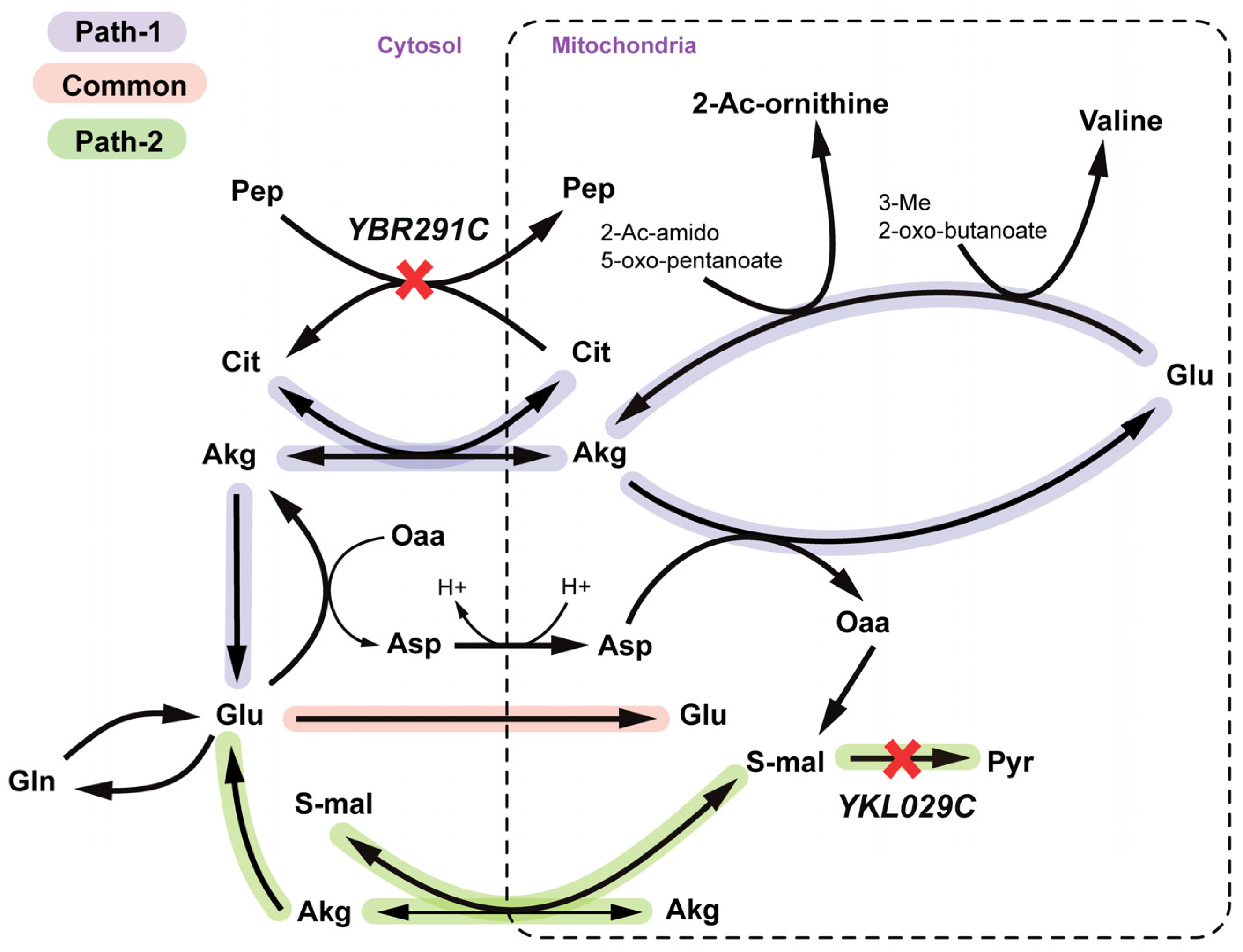
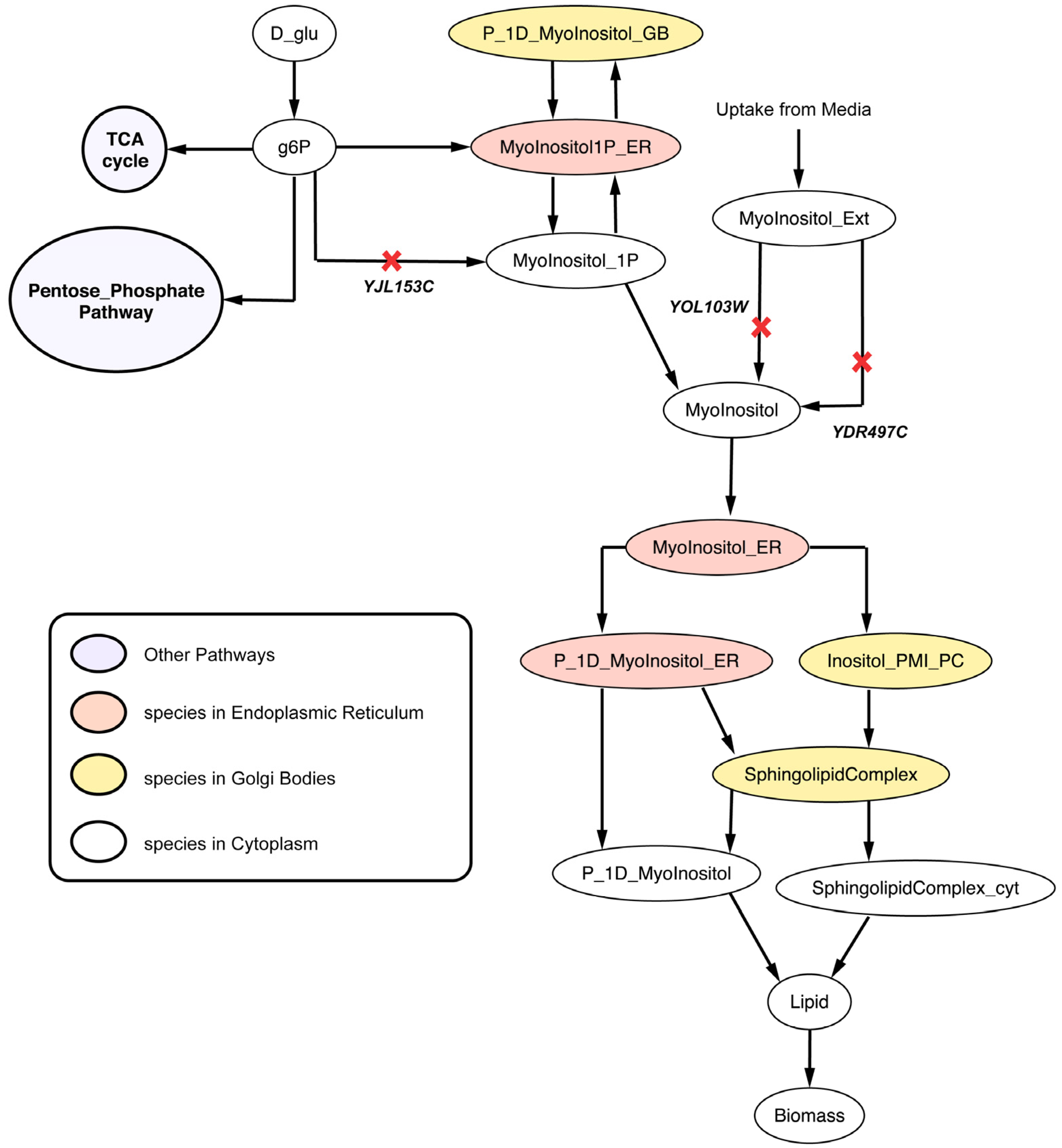
2.2.4. Disruption of Lipid Metabolism (Δitr1Δino1Δitr2 triple mutant)
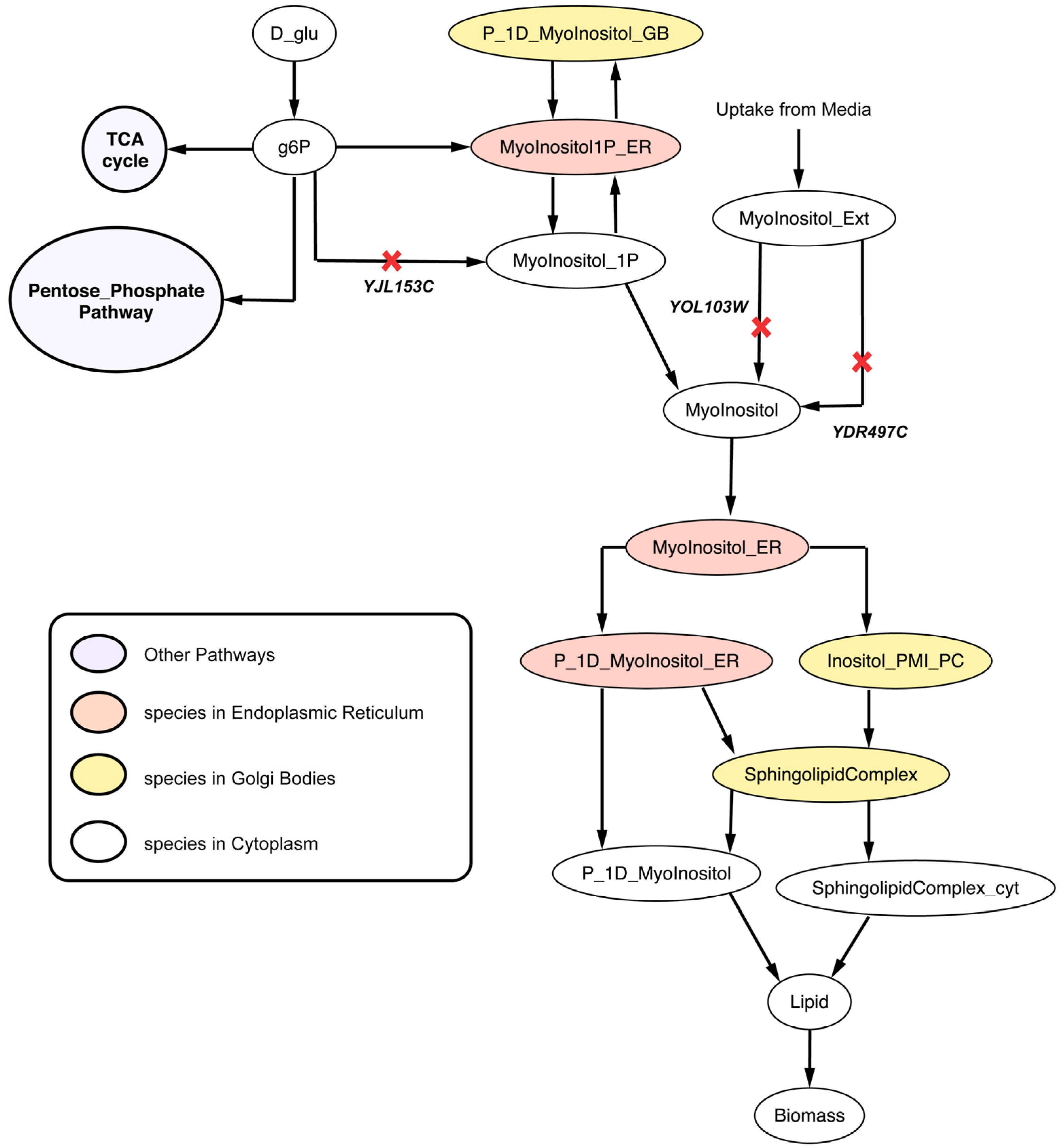
2.3. C. Griseus Model CHO 1.2 Curation and Suggestion of Gene Knockout Experiments
| Parameter | Count | |
|---|---|---|
| Essentiality information | Essential Reactions with GPRs | 82 |
| Essential Reactions without GPRs | 8 | |
| Essential genes | 57 | |
| Reaction level lethality | SL Pairs | 92 |
| SL Triplets | 57 | |
| SL Quadruplets | 3 | |
| Gene level lethality | SL Pairs | 43 |
| SL Triplets | 20 | |
| SL Quadruplets | 3 |
| Gene Name | Comments | Modifications | Reference | ||
|---|---|---|---|---|---|
| Single Gene Deletion | in silico matches in vivo | ggypS1 | ΔggypS1 mouse embyonic stem cells are embryonic lethal in vivo. In silico mutant strain cannot synthesize cholesterol, hence inviable. Thus in vivo result matches in silico predictions. | [82] [83] | |
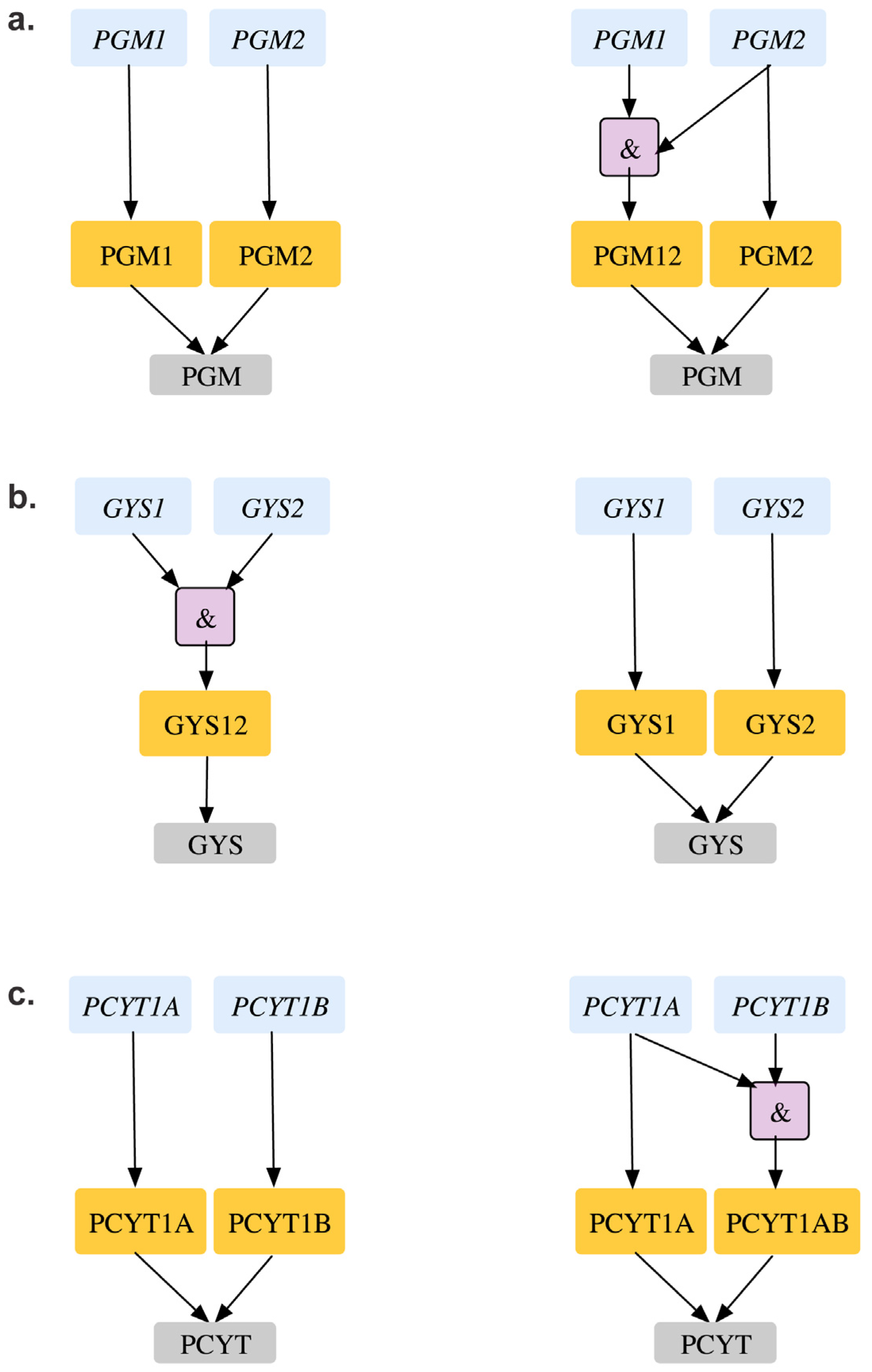
| GPR modifications to reconcile mismatch | gys1 | Δgys1 cannot produce glycogen in silico. However, in vivo studies show gys1- mutant is viable and forms SL2 with gys2. | GPR modified from: (gys1 and gys2) to (gys1 or gys2) gys1 and gys2 reconciled from ESG to GG gys1-gys2 reconciles from ESG to SL2SL2 | [84] | |
| acsL1, acsL3, acsL4 | ΔacsL4 in silico mutant is sphingomyelin auxotroph. However, in vivo data for mouse reveals that acsL4 deletion is viable. | r_0147 and r_0148 GPR was modified from acsL4 to (acsL1 or acsL3 or acsL4) r_0142 GPR was modified from acsL1 to (acsL1 or acsL3 or acsL4) r_0146 GPR was modified from acsL3 to (acsL1 or acsL3 or acsL4) acsL1, acsL3 and acsL4 were fixed from ESG to GG | [85] | ||
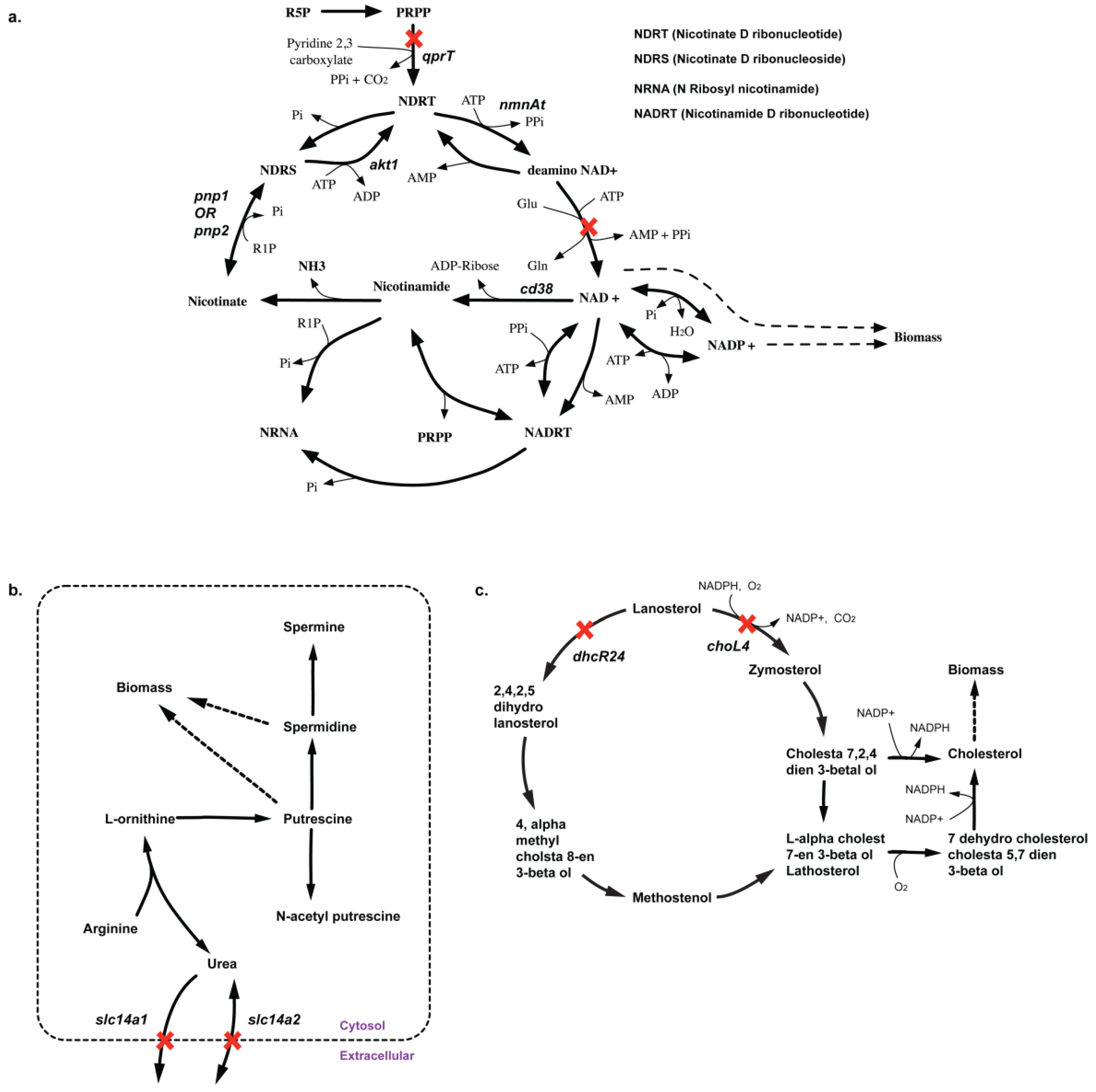
| Suggested experiment | qprT | ΔqprT mutant in silico causes auxotrophy of cofactors NAD+, NADH, NADP+ and NADPH. No experimental evidence of knockout data exists in CHO-K1 cell line. This serves as a potential non-intuitive essential gene. | NA | ||
| Double Gene Deletion | GPR modifications to reconcile mismatch | pgm1-pgm2 | Δpgm1Δpgm2 double mutant is lethal in silico causing glycogen auxotrophy. However, single gene mouse deletion shows Δpgm2 strain is inviable and there is more than 80% homology in mouse and CHO pgm2. Thierry-Mieg et al. shows that pgm2 is the major PGM isoform and is catalogued as MGI:97565. | GPR modification from pgm1 or pgm2 to pgm2 or (pgm1 and pgm2). pgm2 is fixed from GES to ESESSL2ES case is fixed to ESES | [86] [87] |
| pcyT1a-pcyT1b | ΔpcyT1aΔpcyT1b double mutant causes phosphatidylcholine and sphingomyelin auxotrophy in silico. However, in vivo studies reveal that pcyT1a deletion alone is seen to be lethal in mouse. | Changing GPR for phoshphate cytidyltransferase reaction (r_1023) from pcyT1a or pcyT1b to pcyT1a or (pcyT1a and pcyT1b) resolves SL2ES to ESES and GES to ESES with respect to pcyT1a. | [88] | ||
| Double Gene Deletion | GPR modifications to reconcile mismatch | chkA-cThkB | ΔchkA mouse strains have been shown to be embryonic lethal. However ΔchkB deletions have been non-lethal. | Changing GPR for choline-kinase reactions r_0359 and r_0360 from chkA or chkB to chkA or (chkA and chkB) resolved SL2ES to ESES and GES to ESES with respect to chkA | [89] |
| Suggested experiment | slc14a1-slc14a2 | Δslc14a1Δslc14a2 prevents spermidine and putrescine synthesis in silico. But there are no experimental evidence so it goes as a suggestion. | NA |
2.3.1. Suggested GPR Modifications to Reconcile Model Inconsistencies
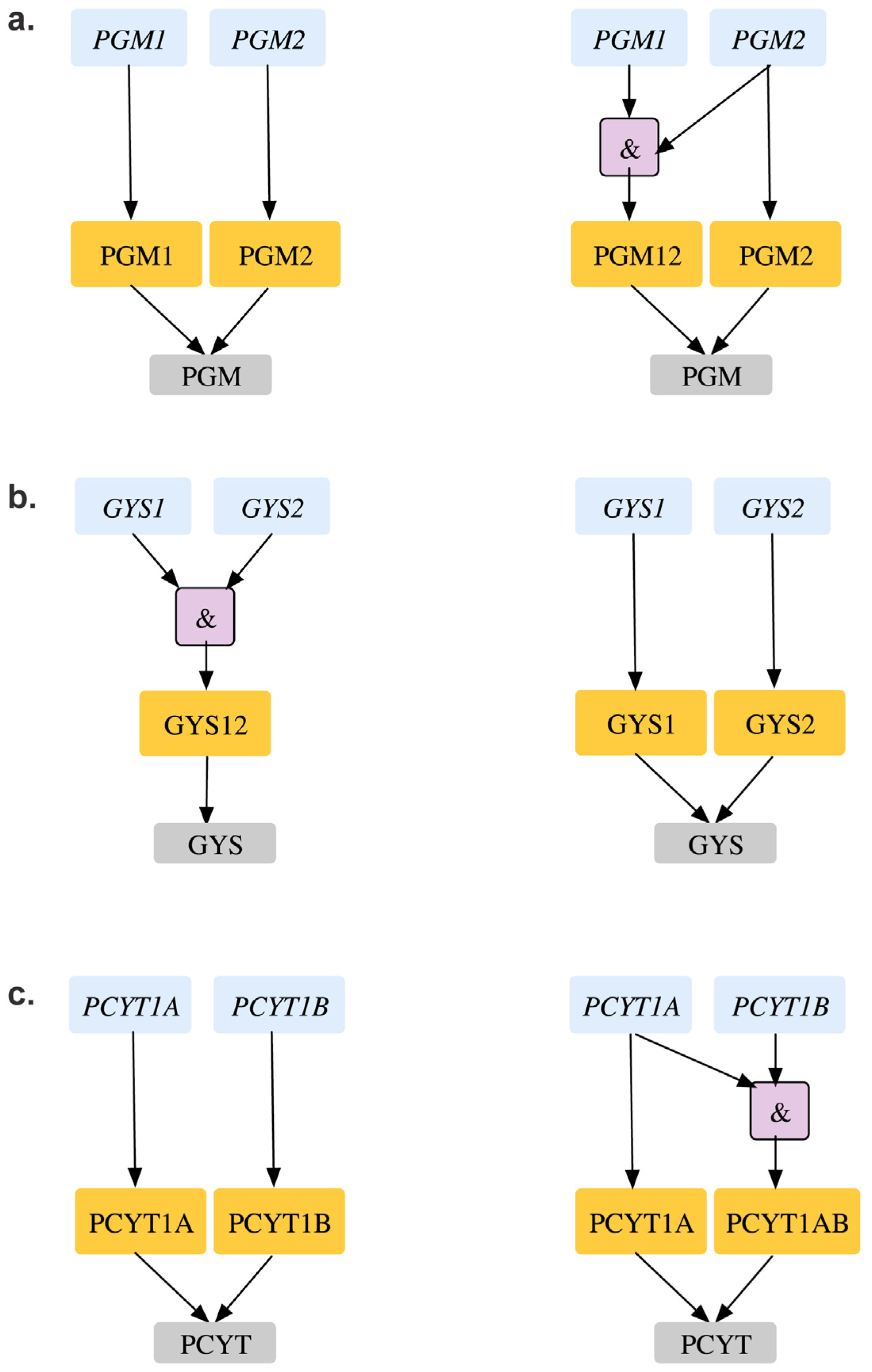
2.3.2. Suggested Single and Double Gene Deletion Experiments
2.3.3. SL2U Case ΔdhcR24ΔchoL4
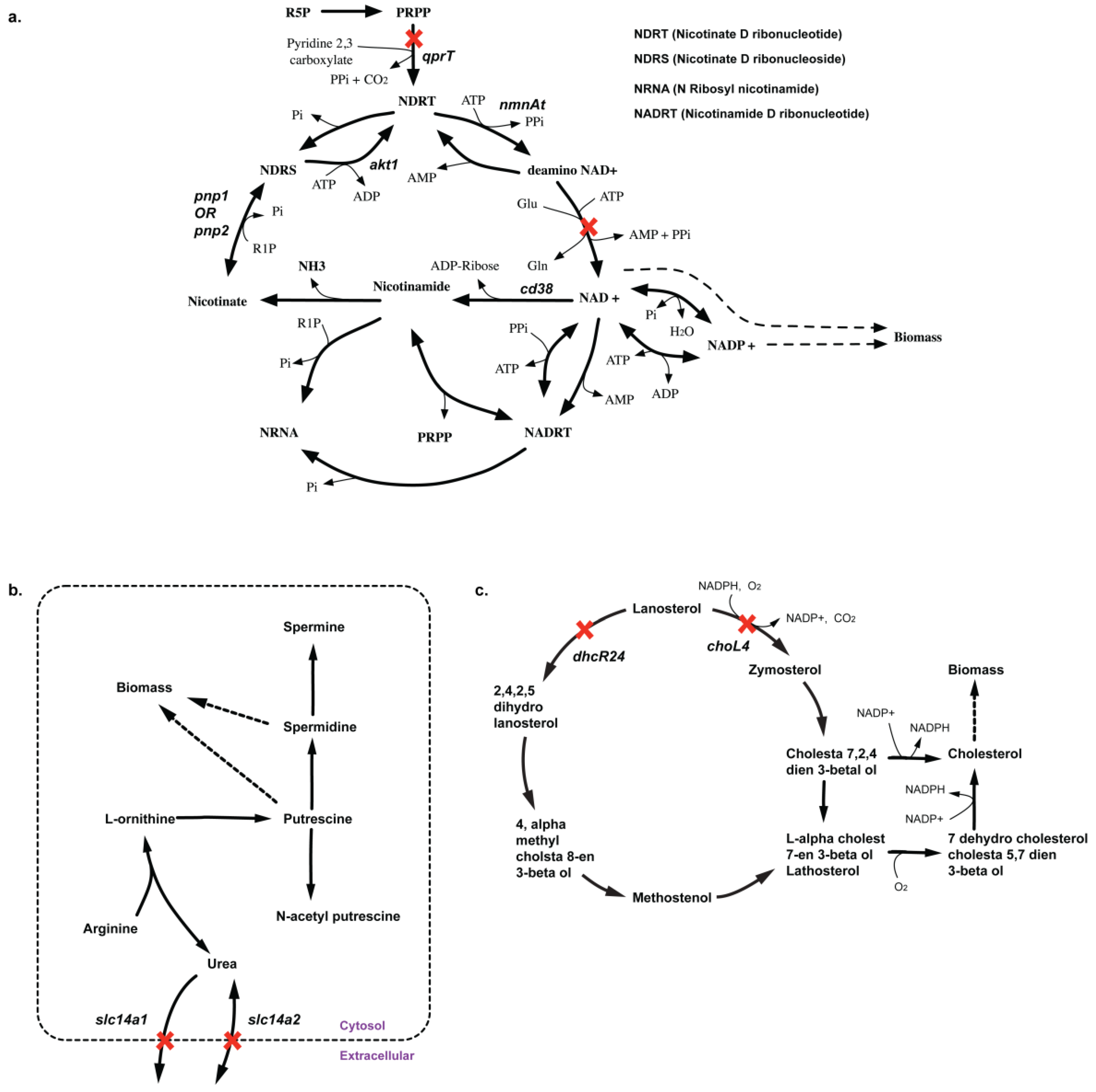
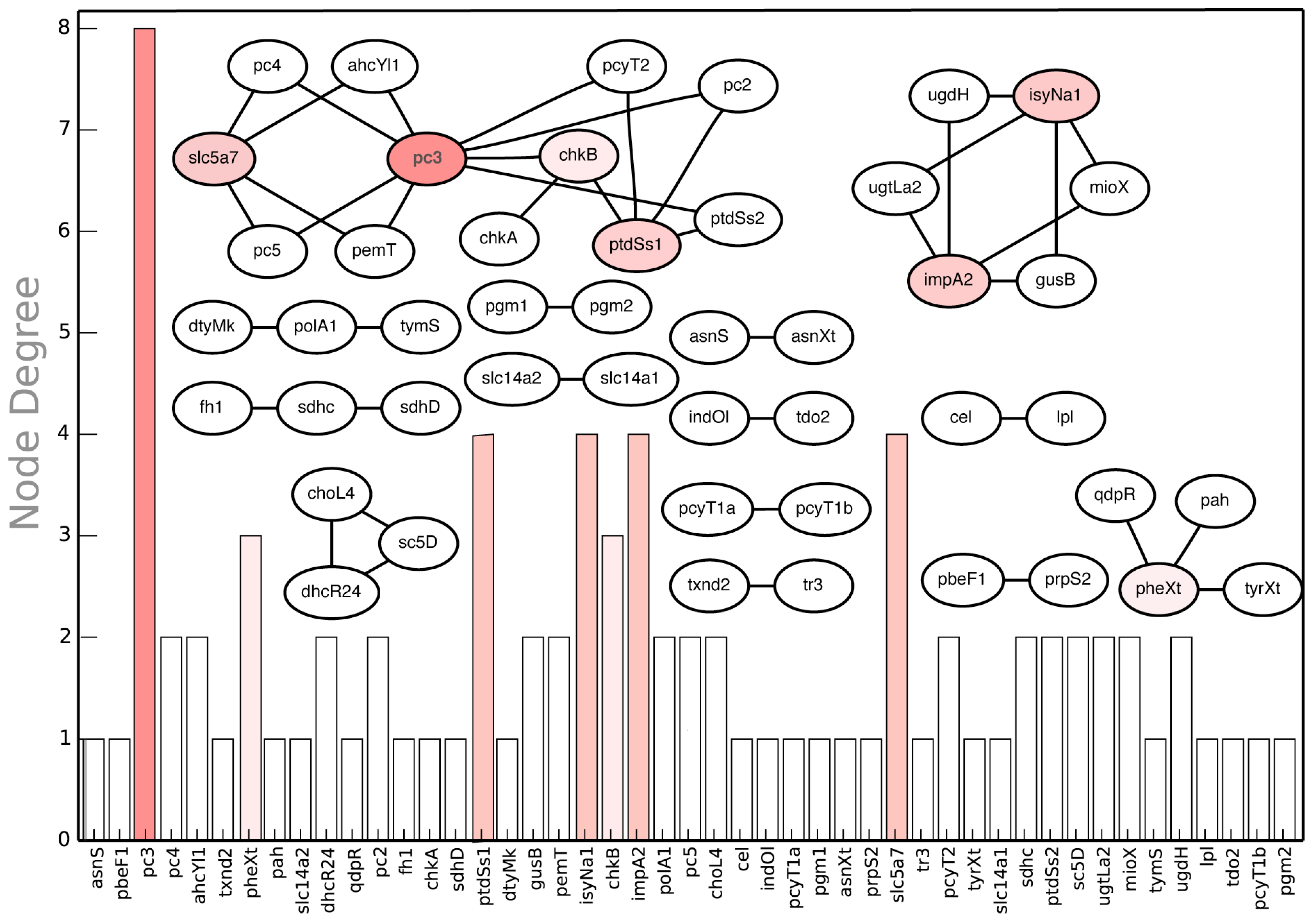
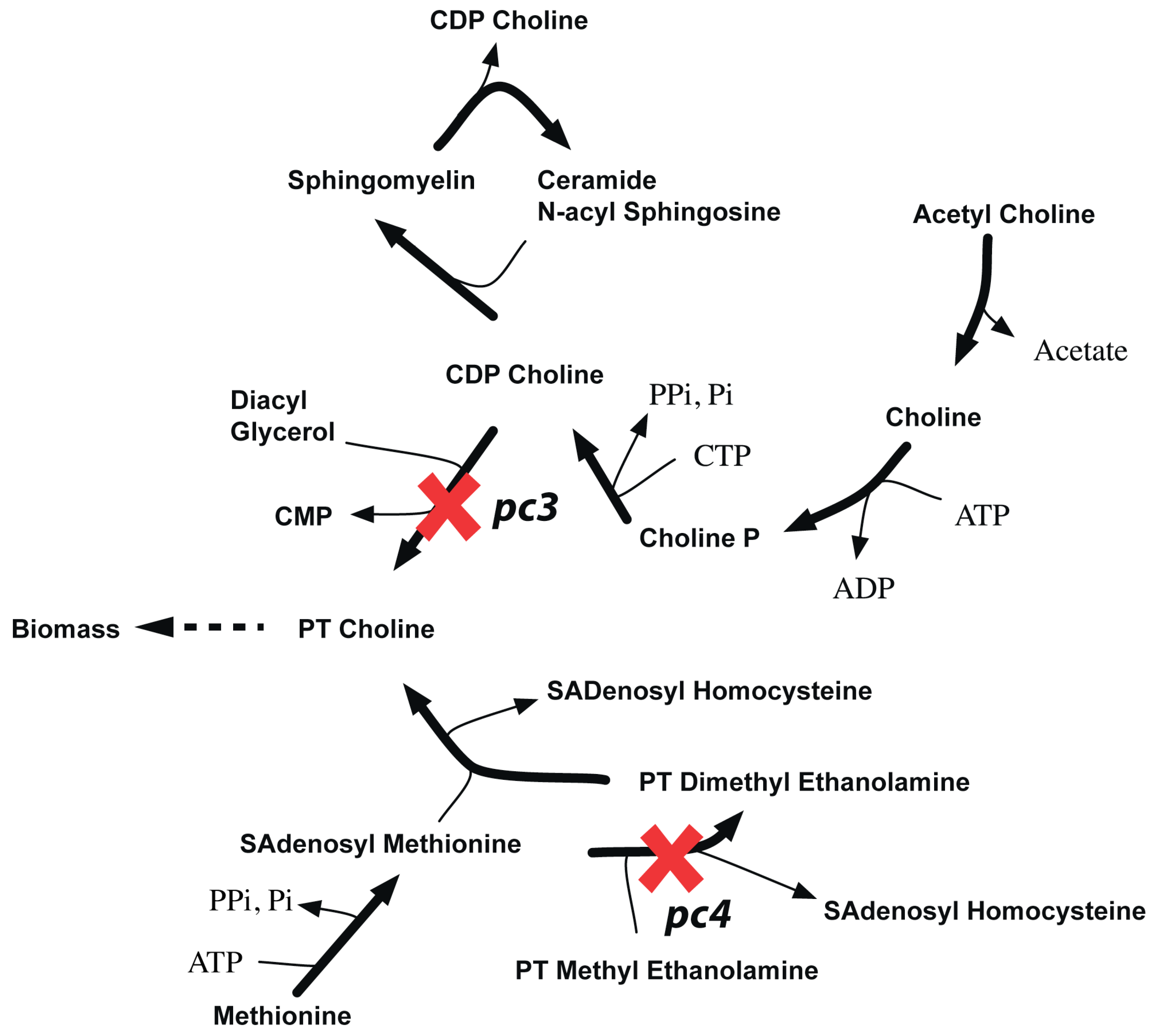
2.3.4. Suggested Experiments for Higher Order Gene Deletions
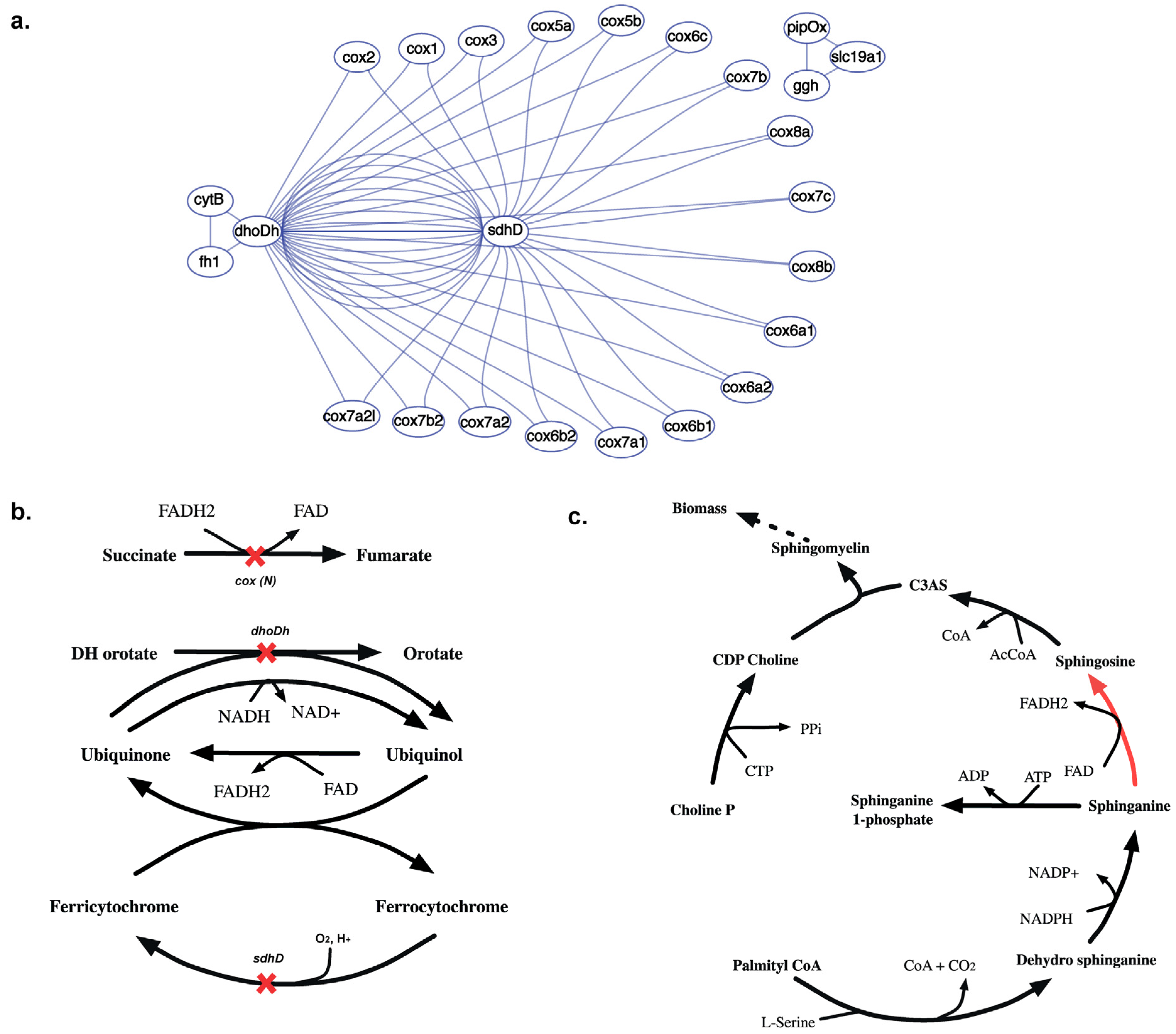
2.3.5. SL3U Case Δcox(N)ΔsdhDΔdhoDh
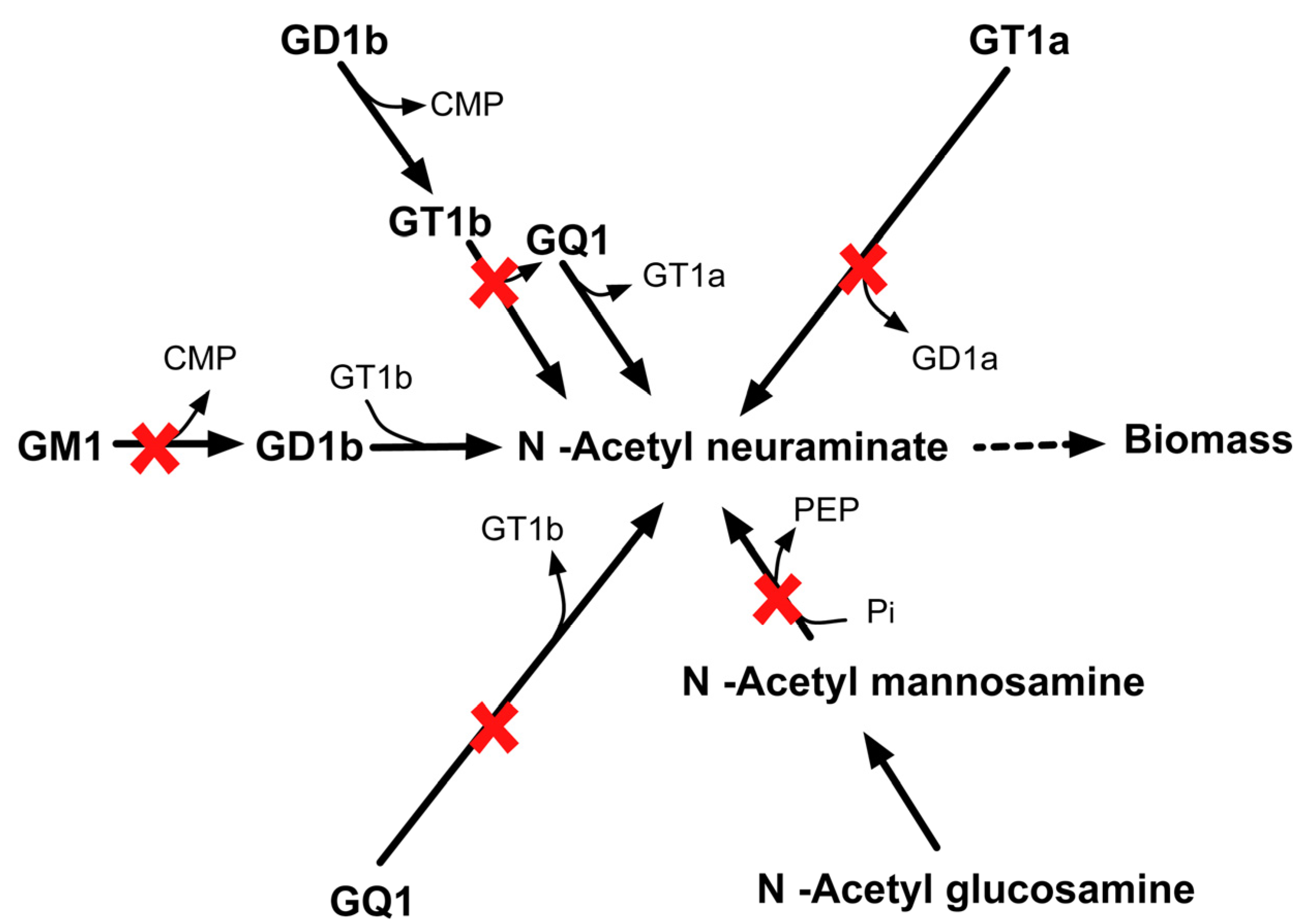
2.3.6. SL4U Case ΔnplΔnanSΔst8Sia1Δst8Sia5
3. Methods
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
Appendix
References
- Botstein, D.; Fink, G.R. Yeast: An experimental organism for 21st century biology. Genetics 2011, 189, 695–704. [Google Scholar] [CrossRef]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.M.; Ball, C.; Weng, S.; Juvik, G.; Schmidt, R.; Adler, C.; Dunn, B.; Dwight, S.; Riles, L.; Mortimer, R.K.; et al. Genetic and physical maps of saccharomyces cerevisiae. Nature 1997, 387, 67–73. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Orhan, I.E. Implications of some selected flavonoids towards alzheimer's disease with the emphasis on cholinesterase inhibition and their bioproduction by metabolic engineering. Curr. Pharm. Biotechno. 2014, 15, 352–361. [Google Scholar] [CrossRef]
- Forster, J.; Famili, I.; Fu, P.; Palsson, B.O.; Nielsen, J. Genome-scale reconstruction of the saccharomyces cerevisiae metabolic network. Genome Res. 2003, 13, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Feizi, A.; Osterlund, T.; Petranovic, D.; Bordel, S.; Nielsen, J. Genome-scale modeling of the protein secretory machinery in yeast. PloS one 2013, 8. [Google Scholar] [CrossRef]
- Aung, H.W.; Henry, S.A.; Walker, L.P. Revising the representation of fatty acid, glycerolipid, and glycerophospholipid metabolism in the consensus model of yeast metabolism. Ind. Biotechnol. 2013, 9, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Zomorrodi, A.R.; Maranas, C.D. Improving the imm904 S. Cerevisiae metabolic model using essentiality and synthetic lethality data. BMC Syst. Biol. 2010, 4. [Google Scholar] [CrossRef]
- Sanchez, B.J.; Nielsen, J. Genome scale models of yeast: Towards standardized evaluation and consistent omic integration. Integr. Biol. 2015, 7, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Lucas, B.K.; Giere, L.M.; DeMarco, R.A.; Shen, A.; Chisholm, V.; Crowley, C.W. High-level production of recombinant proteins in cho cells using a dicistronic dhfr intron expression vector. Nucleic Acids Res. 1996, 24, 1774–1779. [Google Scholar] [CrossRef]
- Li, F.; Vijayasankaran, N.; Shen, A.Y.; Kiss, R.; Amanullah, A. Cell culture processes for monoclonal antibody production. MAbs 2010, 2, 466–479. [Google Scholar] [CrossRef]
- Daramola, O.; Stevenson, J.; Dean, G.; Hatton, D.; Pettman, G.; Holmes, W.; Field, R. A high-yielding cho transient system: Coexpression of genes encoding ebna-1 and gs enhances transient protein expression. Biotechnol. Progr. 2014, 30, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Greenbaum, D.; Xin Lu, H.; Zhu, X.; Gerstein, M. Genomic analysis of essentiality within protein networks. Trends Genet. 2004, 20, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.L.; Pan, X.; Peyser, B.D.; Ye, P.; Meluh, P.B.; Yuan, D.S.; Irizarry, R.A.; Bader, J.S.; Spencer, F.A.; Boeke, J.D. Global synthetic-lethality analysis and yeast functional profiling. Trends Genet. 2006, 22, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of escherichia coli k-12 in-frame, single-gene knockout mutants: The keio collection. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [PubMed]
- Joyce, A.R.; Reed, J.L.; White, A.; Edwards, R.; Osterman, A.; Baba, T.; Mori, H.; Lesely, S.A.; Palsson, B.O.; Agarwalla, S. Experimental and computational assessment of conditionally essential genes in escherichia coli. J. Bacteriol. 2006, 188, 8259–8271. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.S.; Quek, L.E.; Nielsen, L.K. Network thermodynamic curation of human and yeast genome-scale metabolic models. Biophys. J. 2014, 107, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Stanford, N.J.; Lubitz, T.; Smallbone, K.; Klipp, E.; Mendes, P.; Liebermeister, W. Systematic construction of kinetic models from genome-scale metabolic networks. PloS one 2013, 8. [Google Scholar] [CrossRef]
- Soh, K.C.; Miskovic, L.; Hatzimanikatis, V. From network models to network responses: Integration of thermodynamic and kinetic properties of yeast genome-scale metabolic networks. FEMS Yeast Res. 2012, 12, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Selvarasu, S.; Karimi, I.A.; Ghim, G.H.; Lee, D.Y. Genome-scale modeling and in silico analysis of mouse cell metabolic network. Mol. Biosyst. 2010, 6, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.H.; Evangelista, M.; Parsons, A.B.; Xu, H.; Bader, G.D.; Page, N.; Robinson, M.; Raghibizadeh, S.; Hogue, C.W.; Bussey, H.; et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 2001, 294, 2364–2368. [Google Scholar] [CrossRef]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces genome database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef]
- Breslow, D.K.; Cameron, D.M.; Collins, S.R.; Schuldiner, M.; Stewart-Ornstein, J.; Newman, H.W.; Braun, S.; Madhani, H.D.; Krogan, N.J.; Weissman, J.S. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods 2008, 5, 711–718. [Google Scholar] [CrossRef]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Veronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; Andre, B.; et al. Functional profiling of the saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Baudin, A.; Ozier-Kalogeropoulos, O.; Denouel, A.; Lacroute, F.; Cullin, C. A simple and efficient method for direct gene deletion in saccharomyces cerevisiae. Nucleic Acids Res. 1993, 21, 3329–3330. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.C.; Polzin, K.; Labate, J.; Specht, J.; Brummer, E.C.; Olson, T.; Young, N.; Concibido, V.; Wilcox, J.; Tamulonis, J.P.; et al. Genome duplication in soybean (glycine subgenus soja). Genetics 1996, 144, 329–338. [Google Scholar] [PubMed]
- Wach, A.; Brachat, A.; Pohlmann, R.; Philippsen, P. New heterologous modules for classical or pcr-based gene disruptions in saccharomyces cerevisiae. Yeast 1994, 10, 1793–1808. [Google Scholar] [PubMed]
- Engelman, D.M.; Steitz, T.A.; Goldman, A. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys.Bio. 1986, 15, 321–353. [Google Scholar] [CrossRef] [PubMed]
- Mannhaupt, G.; Stucka, R.; Pilz, U.; Schwarzlose, C.; Feldmann, H. Characterization of the prephenate dehydrogenase-encoding gene, tyr1, from saccharomyces cerevisiae. Gene 1989, 85, 303–311. [Google Scholar] [CrossRef]
- Lesage, G.; Shapiro, J.; Specht, C.A.; Sdicu, A.M.; Menard, P.; Hussein, S.; Tong, A.H.; Boone, C.; Bussey, H. An interactional network of genes involved in chitin synthesis in saccharomyces cerevisiae. BMC Genet. 2005, 6. [Google Scholar] [CrossRef] [PubMed]
- Firon, A.; Lesage, G.; Bussey, H. Integrative studies put cell wall synthesis on the yeast functional map. Curr. Opin. Microbiol. 2004, 7, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, J.R. Pathways of leucine and valine catabolism in yeast. Methods Enzymol. 2000, 324, 80–92. [Google Scholar] [PubMed]
- Sentheshanmuganathan, S.; Elsden, S.R. The mechanism of the formation of tyrosol by saccharomyces cerevisiae. Biochem. J. 1958, 69, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Avalos, J.L.; Fink, G.R.; Stephanopoulos, G. Compartmentalization of metabolic pathways in yeast mitochondria improves the production of branched-chain alcohols. Nat. Biotechnol. 2013, 31, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.; Papp, B.; Pal, C.; Oliver, S.G.; Delneri, D. Plasticity of genetic interactions in metabolic networks of yeast. P. Natl. Acad. Sci. USA 2007, 104, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Gollub, E.G.; Liu, K.P.; Dayan, J.; Adlersberg, M.; Sprinson, D.B. Yeast mutants deficient in heme biosynthesis and a heme mutant additionally blocked in cyclization of 2,3-oxidosqualene. J. Biol. Chem. 1977, 252, 2846–2854. [Google Scholar]
- Oh-hama, T. Evolutionary consideration on 5-aminolevulinate synthase in nature. Origins life Evol. B. 1997, 27, 405–412. [Google Scholar] [CrossRef]
- De Silva-Udawatta, M.N.; Cannon, J.F. Roles of trehalose phosphate synthase in yeast glycogen metabolism and sporulation. Mol. Microbiol. 2001, 40, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- de Lichtenberg, U.; Jensen, L.J.; Brunak, S.; Bork, P. Dynamic complex formation during the yeast cell cycle. Science 2005, 307, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Bell, W.; Sun, W.; Hohmann, S.; Wera, S.; Reinders, A.; De Virgilio, C.; Wiemken, A.; Thevelein, J.M. Composition and functional analysis of the saccharomyces cerevisiae trehalose synthase complex. J. Biol. Chem. 1998, 273, 33311–33319. [Google Scholar] [CrossRef]
- Barros, M.H.; Nobrega, F.G. Yah1 of saccharomyces cerevisiae: A new essential gene that codes for a protein homologous to human adrenodoxin. Gene 1999, 233, 197–203. [Google Scholar] [PubMed]
- Barros, M.H.; Carlson, C.G.; Glerum, D.M.; Tzagoloff, A. Involvement of mitochondrial ferredoxin and cox15p in hydroxylation of heme o. Febs. Lett. 2001, 492, 133–138. [Google Scholar] [PubMed]
- Lim, A.L.; Powers-Lee, S.G. Requirement for the carboxyl-terminal domain of saccharomyces cerevisiae carbamoyl-phosphate synthetase. J. Biol. Chem. 1996, 271, 11400–11409. [Google Scholar] [PubMed]
- Inglis, D.O.; Arnaud, M.B.; Binkley, J.; Shah, P.; Skrzypek, M.S.; Wymore, F.; Binkley, G.; Miyasato, S.R.; Simison, M.; Sherlock, G. The candida genome database incorporates multiple candida species: Multispecies search and analysis tools with curated gene and protein information for candida albicans and candida glabrata. Nucleic Acids Res. 2012, 40, D667–D674. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Dietrich, F.S. The reacquisition of biotin prototrophy in saccharomyces cerevisiae involved horizontal gene transfer, gene duplication and gene clustering. Genetics 2007, 177, 2293–2307. [Google Scholar] [CrossRef] [PubMed]
- Tobias, S.; Rajic, I.; Vanyi, A. Effect of t-2 toxin on egg production and hatchability in laying hens. Acta Vet. Hung. 1992, 40, 47–54. [Google Scholar] [PubMed]
- Bernstein, M.; Hoffmann, W.; Ammerer, G.; Schekman, R. Characterization of a gene product (sec53p) required for protein assembly in the yeast endoplasmic reticulum. J. Cell Biol. 1985, 101, 2374–2382. [Google Scholar] [CrossRef] [PubMed]
- Hirschman, J.E.; Durbin, K.J.; Winston, F. Genetic evidence for promoter competition in saccharomyces cerevisiae. Mol. Cell. Biol. 1988, 8, 4608–4615. [Google Scholar] [PubMed]
- Dowell, R.D.; Ryan, O.; Jansen, A.; Cheung, D.; Agarwala, S.; Danford, T.; Bernstein, D.A.; Rolfe, P.A.; Heisler, L.E.; Chin, B.; et al. Genotype to phenotype: A complex problem. Science 2010, 328, 469. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.S.; Yarger, J.G.; Burck, C.L.; Poulter, C.D. Farnesyl diphosphate synthetase. Molecular cloning, sequence, and expression of an essential gene from saccharomyces cerevisiae. J. Biol. Chem. 1989, 264, 19176–19184. [Google Scholar]
- Lacroute, F. Regulation of pyrimidine biosynthesis in saccharomyces cerevisiae. J. Bacteriol. 1968, 95, 824–832. [Google Scholar] [PubMed]
- Petti, A.A.; Crutchfield, C.A.; Rabinowitz, J.D.; Botstein, D. Survival of starving yeast is correlated with oxidative stress response and nonrespiratory mitochondrial function. P. Natl. Acad. Sci. USA 2011, 108, E1089–E1098. [Google Scholar] [CrossRef]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting c metabolite labeling patterns from cells. Curr. Opin. Biotechn. 2015, 34, 189–201. [Google Scholar] [CrossRef]
- Deutscher, D.; Meilijson, I.; Kupiec, M.; Ruppin, E. Multiple knockout analysis of genetic robustness in the yeast metabolic network. Nat. Genet. 2006, 38, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Prevost, G.; Eriani, G.; Kern, D.; Dirheimer, G.; Gangloff, J. Study of the arrangement of the functional domains along the yeast cytoplasmic aspartyl-trna synthetase. Eur. J. Biochem. 1989, 180, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Rebora, K.; Laloo, B.; Daignan-Fornier, B. Revisiting purine-histidine cross-pathway regulation in saccharomyces cerevisiae: A central role for a small molecule. Genetics 2005, 170, 61–70. [Google Scholar] [CrossRef]
- van Pel, D.M.; Stirling, P.C.; Minaker, S.W.; Sipahimalani, P.; Hieter, P. Saccharomyces cerevisiae genetics predicts candidate therapeutic genetic interactions at the mammalian replication fork. G3 2013, 3, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Berthelet, S.; Usher, J.; Shulist, K.; Hamza, A.; Maltez, N.; Johnston, A.; Fong, Y.; Harris, L.J.; Baetz, K. Functional genomics analysis of the saccharomyces cerevisiae iron responsive transcription factor aft1 reveals iron-independent functions. Genetics 2010, 185, 1111–1128. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ruiz, R.; Averet, N.; Araiza, D.; Pinson, B.; Uribe-Carvajal, S.; Devin, A.; Rigoulet, M. Mitochondrial oxidative phosphorylation is regulated by fructose 1,6-bisphosphate. A possible role in crabtree effect induction? J. Biol. chem. 2008, 283, 26948–26955. [Google Scholar] [PubMed]
- Guaragnella, N.; Zdralevic, M.; Antonacci, L.; Passarella, S.; Marra, E.; Giannattasio, S. The role of mitochondria in yeast programmed cell death. Front. Oncol. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Kus, B.; Gajadhar, A.; Stanger, K.; Cho, R.; Sun, W.; Rouleau, N.; Lee, T.; Chan, D.; Wolting, C.; Edwards, A.; et al. A high throughput screen to identify substrates for the ubiquitin ligase rsp5. J. Biol. Chem. 2005, 280, 29470–29478. [Google Scholar] [CrossRef] [PubMed]
- Davierwala, A.P.; Haynes, J.; Li, Z.; Brost, R.L.; Robinson, M.D.; Yu, L.; Mnaimneh, S.; Ding, H.; Zhu, H.; Chen, Y.; et al. The synthetic genetic interaction spectrum of essential genes. Nat. Genet. 2005, 37, 1147–1152. [Google Scholar] [CrossRef]
- Schneiter, R.; Tatzer, V.; Gogg, G.; Leitner, E.; Kohlwein, S.D. Elo1p-dependent carboxy-terminal elongation of c14:1delta(9) to c16:1delta(11) fatty acids in saccharomyces cerevisiae. J. Bacteriol. 2000, 182, 3655–3660. [Google Scholar] [CrossRef] [PubMed]
- Trotter, P.J.; Hagerman, R.A.; Voelker, D.R. A yeast strain defective in oleic acid utilization has a mutation in the rml2 gene. Biochim. Biophys. Acta 1999, 1438, 223–238. [Google Scholar] [CrossRef]
- Keng, T. Hap1 and rox1 form a regulatory pathway in the repression of hem13 transcription in saccharomyces cerevisiae. Mol. Cell. Biol. 1992, 12, 2616–2623. [Google Scholar]
- Chu, X.; Qin, X.; Xu, H.; Li, L.; Wang, Z.; Li, F.; Xie, X.; Zhou, H.; Shen, Y.; Long, J. Structural insights into paf1 complex assembly and histone binding. Nucleic Acids Res. 2013, 41, 10619–10629. [Google Scholar] [CrossRef]
- Keogh, M.C.; Podolny, V.; Buratowski, S. Bur1 kinase is required for efficient transcription elongation by rna polymerase ii. Mol. Cell. Biol. 2003, 23, 7005–7018. [Google Scholar] [CrossRef] [PubMed]
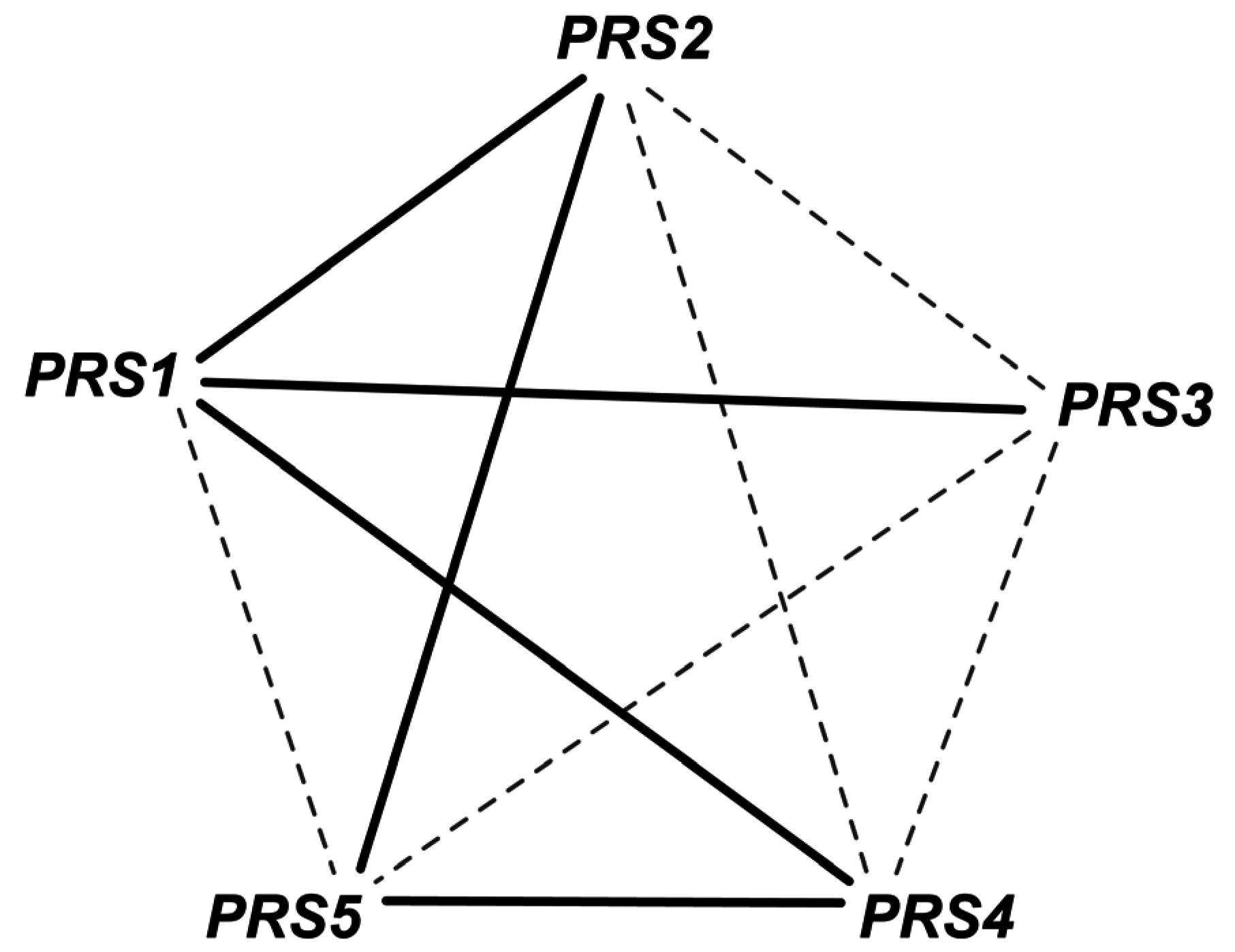
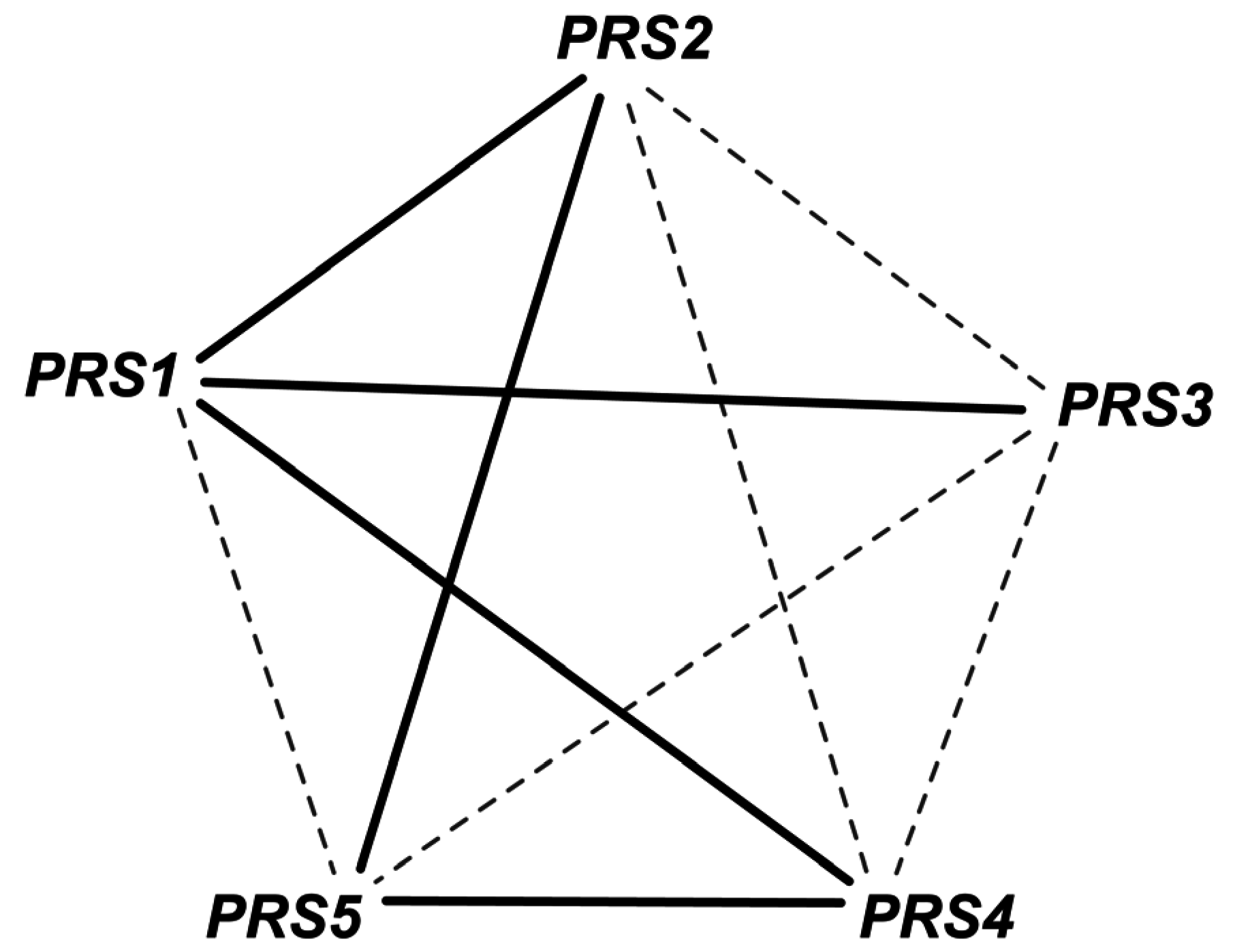
- Hove-Jensen, B. Heterooligomeric phosphoribosyl diphosphate synthase of saccharomyces cerevisiae: Combinatorial expression of the five prs genes in escherichia coli. J. Biol. Chem. 2004, 279, 40345–40350. [Google Scholar] [CrossRef] [PubMed]
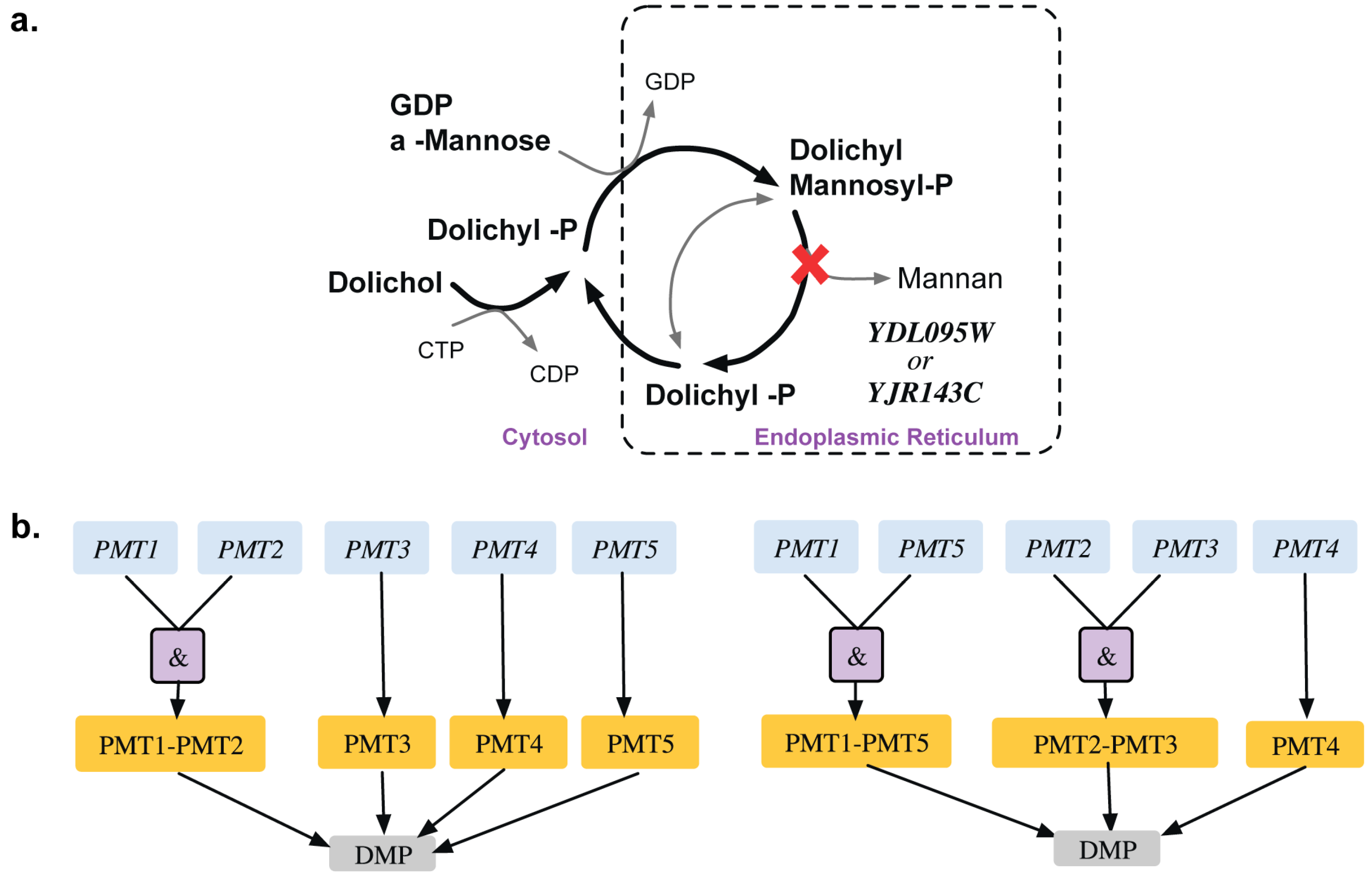
- Girrbach, V.; Strahl, S. Members of the evolutionarily conserved pmt family of protein o-mannosyltransferases form distinct protein complexes among themselves. J. Biol. Chem. 2003, 278, 12554–12562. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.F.; Prill, S.K. O-glycosylation. Med. Mycol. 2001, 39 (Suppl 1), 67–74. [Google Scholar]
- Nuoffer, C.; Jeno, P.; Conzelmann, A.; Riezman, H. Determinants for glycophospholipid anchoring of the saccharomyces cerevisiae gas1 protein to the plasma membrane. Mol. Cell. Biol. 1991, 11, 27–37. [Google Scholar]
- Hirayama, H.; Fujita, M.; Yoko-o, T.; Jigami, Y. O-mannosylation is required for degradation of the endoplasmic reticulum-associated degradation substrate gas1*p via the ubiquitin/proteasome pathway in saccharomyces cerevisiae. J. Biochem. 2008, 143, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.R.; Kayne, P.S.; Moerschell, R.P.; Tsunasawa, S.; Gribskov, M.; Colavito-Shepanski, M.; Grunstein, M.; Sherman, F.; Sternglanz, R. Identification and characterization of genes and mutants for an n-terminal acetyltransferase from yeast. EMBO J. 1989, 8, 2067–2075. [Google Scholar] [PubMed]
- Levin, D.E. Cell wall integrity signaling in saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2005, 69, 262–291. [Google Scholar] [CrossRef] [PubMed]
- Arias, P.; Diez-Muniz, S.; Garcia, R.; Nombela, C.; Rodriguez-Pena, J.M.; Arroyo, J. Genome-wide survey of yeast mutations leading to activation of the yeast cell integrity mapk pathway: Novel insights into diverse mapk outcomes. BMC Genomics 2011, 12. [Google Scholar] [CrossRef]
- Prohl, C.; Pelzer, W.; Diekert, K.; Kmita, H.; Bedekovics, T.; Kispal, G.; Lill, R. The yeast mitochondrial carrier leu5p and its human homologue graves' disease protein are required for accumulation of coenzyme a in the matrix. Mol. Cell. Biol. 2001, 21, 1089–1097. [Google Scholar] [PubMed]
- Nikawa, J.; Tsukagoshi, Y.; Yamashita, S. Isolation and characterization of two distinct myo-inositol transporter genes of saccharomyces cerevisiae. J. Biol. Chem. 1991, 266, 11184–11191. [Google Scholar] [PubMed]
- Culbertson, M.R.; Henry, S.A. Inositol-requiring mutants of saccharomyces cerevisiae. Genetics 1975, 80, 23–40. [Google Scholar] [PubMed]
- Dean-Johnson, M.; Henry, S.A. Biosynthesis of inositol in yeast. Primary structure of myo-inositol-1-phosphate synthase (ec 5.5.1.4) and functional analysis of its structural gene, the ino1 locus. J. Biol. Chem. 1989, 264, 1274–1283. [Google Scholar]
- Selvarasu, S.; Ho, Y.S.; Chong, W.P.; Wong, N.S.; Yusufi, F.N.; Lee, Y.Y.; Yap, M.G.; Lee, D.Y. Combined in silico modeling and metabolomics analysis to characterize fed-batch cho cell culture. Biotechnol. Bioeng. 2012, 109, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. Omim.Org: Online mendelian inheritance in man (omim(r)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43. [Google Scholar] [CrossRef] [PubMed]
- Ruppel, N.J.; Kropp, K.N.; Davis, P.A.; Martin, A.E.; Luesse, D.R.; Hangarter, R.P. Mutations in geranylgeranyl diphosphate synthase 1 affect chloroplast development in arabidopsis thaliana (brassicaceae). Am. J. Bot. 2013, 100, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Kainou, T.; Kawamura, K.; Tanaka, K.; Matsuda, H.; Kawamukai, M. Identification of the ggps1 genes encoding geranylgeranyl diphosphate synthases from mouse and human. Biochim. Biophys. Acta 1999, 1437, 333–340. [Google Scholar] [CrossRef]
- Douillard-Guilloux, G.; Raben, N.; Takikita, S.; Ferry, A.; Vignaud, A.; Guillet-Deniau, I.; Favier, M.; Thurberg, B.L.; Roach, P.J.; Caillaud, C.; et al. Restoration of muscle functionality by genetic suppression of glycogen synthesis in a murine model of pompe disease. Hum. Mol. Genet. 2010, 19, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.Y.; Kang, M.J.; Sone, H.; Suzuki, T.; Abe, M.; Igarashi, M.; Tokunaga, T.; Ogawa, S.; Takei, Y.A.; Miyazawa, T.; et al. Abnormal uterus with polycysts, accumulation of uterine prostaglandins, and reduced fertility in mice heterozygous for acyl-coa synthetase 4 deficiency. Biochem. Bioph. Res. Co. 2001, 284, 993–997. [Google Scholar]
- Nadeau, J.H.; Kompf, J.; Siebert, G.; Taylor, B.A. Linkage of pgm-3 in the house mouse and homologies of three phosphoglucomutase loci in mouse and man. Biochem. Genet. 1981, 19, 465–474. [Google Scholar] [PubMed]
- Thierry-Mieg, D.; Thierry-Mieg, J. Aceview: A comprehensive cdna-supported gene and transcripts annotation. Genome Biol. 2006, 7 (Suppl 1), 11–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Magdaleno, S.; Tabas, I.; Jackowski, S. Early embryonic lethality in mice with targeted deletion of the ctp:Phosphocholine cytidylyltransferase alpha gene (pcyt1a). Mol. Cell. Biol. 2005, 25, 3357–3363. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Aoyama, C.; Young, S.G.; Vance, D.E. Early embryonic lethality caused by disruption of the gene for choline kinase alpha, the first enzyme in phosphatidylcholine biosynthesis. J. Biol. Chem. 2008, 283, 1456–1462. [Google Scholar] [CrossRef]
- Fan, L.; Kadura, I.; Krebs, L.E.; Hatfield, C.C.; Shaw, M.M.; Frye, C.C. Improving the efficiency of cho cell line generation using glutamine synthetase gene knockout cells. Biotechn. Bioeng. 2012, 109, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Greig, K.T.; Antonchuk, J.; Metcalf, D.; Morgan, P.O.; Krebs, D.L.; Zhang, J.G.; Hacking, D.F.; Bode, L.; Robb, L.; Kranz, C.; et al. Agm1/pgm3-mediated sugar nucleotide synthesis is essential for hematopoiesis and development. Mol. Cell. Biol. 2007, 27, 5849–5859. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.W.; Zeng, A.P. The connectivity structure, giant strong component and centrality of metabolic networks. Bioinformatics 2003, 19, 1423–1430. [Google Scholar] [CrossRef]
- Suthers, P.F.; Zomorrodi, A.; Maranas, C.D. Genome-scale gene/reaction essentiality and synthetic lethality analysis. Mol. Syst. Biol. 2009, 5. [Google Scholar] [CrossRef]
- Hsiang, Y.H.; Liu, L.F. Identification of mammalian DNA topoisomerase i as an intracellular target of the anticancer drug camptothecin. Cancer Res. 1988, 48, 1722–1726. [Google Scholar] [PubMed]
- Mo, M.L.; Palsson, B.O.; Herrgard, M.J. Connecting extracellular metabolomic measurements to intracellular flux states in yeast. BMC Syst. Biol. 2009, 3. [Google Scholar] [CrossRef] [PubMed]
- Otterstedt, K.; Larsson, C.; Bill, R.M.; Stahlberg, A.; Boles, E.; Hohmann, S.; Gustafsson, L. Switching the mode of metabolism in the yeast saccharomyces cerevisiae. EMBO Rep. 2004, 5, 532–537. [Google Scholar] [CrossRef]
- Riordan, J.R.; Ling, V. Purification of p-glycoprotein from plasma membrane vesicles of chinese hamster ovary cell mutants with reduced colchicine permeability. J. Biol. Chem. 1979, 254, 12701–12705. [Google Scholar] [PubMed]
- Stanners, C.P.; Eliceiri, G.L.; Green, H. Two types of ribosome in mouse-hamster hybrid cells. Nature 1971, 230, 52–54. [Google Scholar] [CrossRef]
- Chowdhury, A.; Zomorrodi, A.R.; Maranas, C.D. K-optforce: Integrating kinetics with flux balance analysis for strain design. PLoS Comput. Biol. 2014, 10. [Google Scholar] [CrossRef]
- Schellenberger, J.; Que, R.; Fleming, R.M.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The cobra toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef]
- O'Brien, E.J.; Lerman, J.A.; Chang, R.L.; Hyduke, D.R.; Palsson, B.O. Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 2013, 9. [Google Scholar] [CrossRef]
- Lerman, J.A.; Hyduke, D.R.; Latif, H.; Portnoy, V.A.; Lewis, N.E.; Orth, J.D.; Schrimpe-Rutledge, A.C.; Smith, R.D.; Adkins, J.N.; Zengler, K.; et al. In silico method for modelling metabolism and gene product expression at genome scale. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chowdhury, R.; Chowdhury, A.; Maranas, C.D. Using Gene Essentiality and Synthetic Lethality Information to Correct Yeast and CHO Cell Genome-Scale Models. Metabolites 2015, 5, 536-570. https://doi.org/10.3390/metabo5040536
Chowdhury R, Chowdhury A, Maranas CD. Using Gene Essentiality and Synthetic Lethality Information to Correct Yeast and CHO Cell Genome-Scale Models. Metabolites. 2015; 5(4):536-570. https://doi.org/10.3390/metabo5040536
Chicago/Turabian StyleChowdhury, Ratul, Anupam Chowdhury, and Costas D. Maranas. 2015. "Using Gene Essentiality and Synthetic Lethality Information to Correct Yeast and CHO Cell Genome-Scale Models" Metabolites 5, no. 4: 536-570. https://doi.org/10.3390/metabo5040536
APA StyleChowdhury, R., Chowdhury, A., & Maranas, C. D. (2015). Using Gene Essentiality and Synthetic Lethality Information to Correct Yeast and CHO Cell Genome-Scale Models. Metabolites, 5(4), 536-570. https://doi.org/10.3390/metabo5040536