Metabolic and Transcriptional Reprogramming in Developing Soybean (Glycine max) Embryos


Abstract
:1. Introduction
2. Results and Discussion
2.1. Metabolic Reprogramming in Developing Soybean Embryos
2.1.1. Lipid and Protein Accumulation in Developing Soybean Embryos
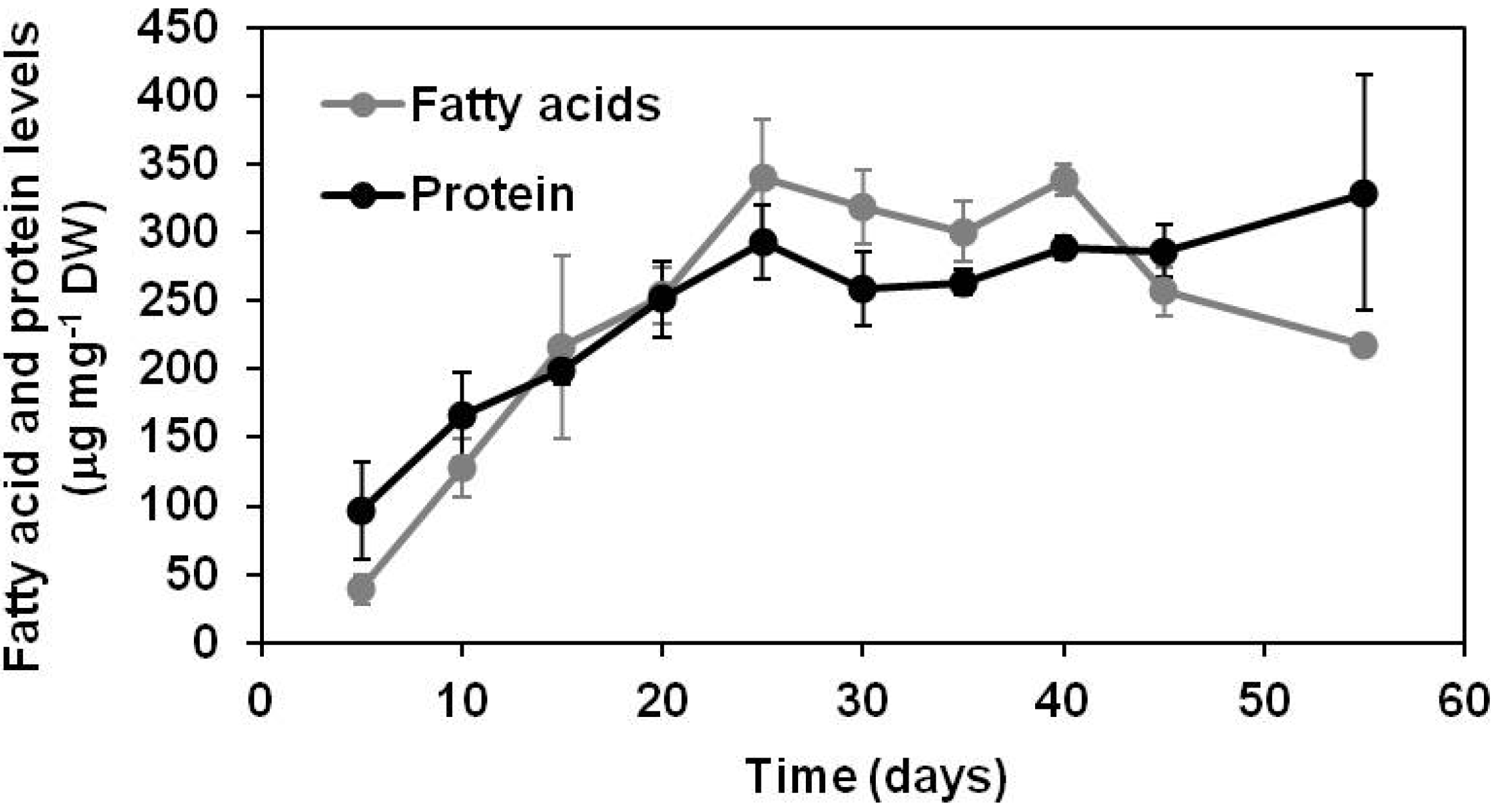
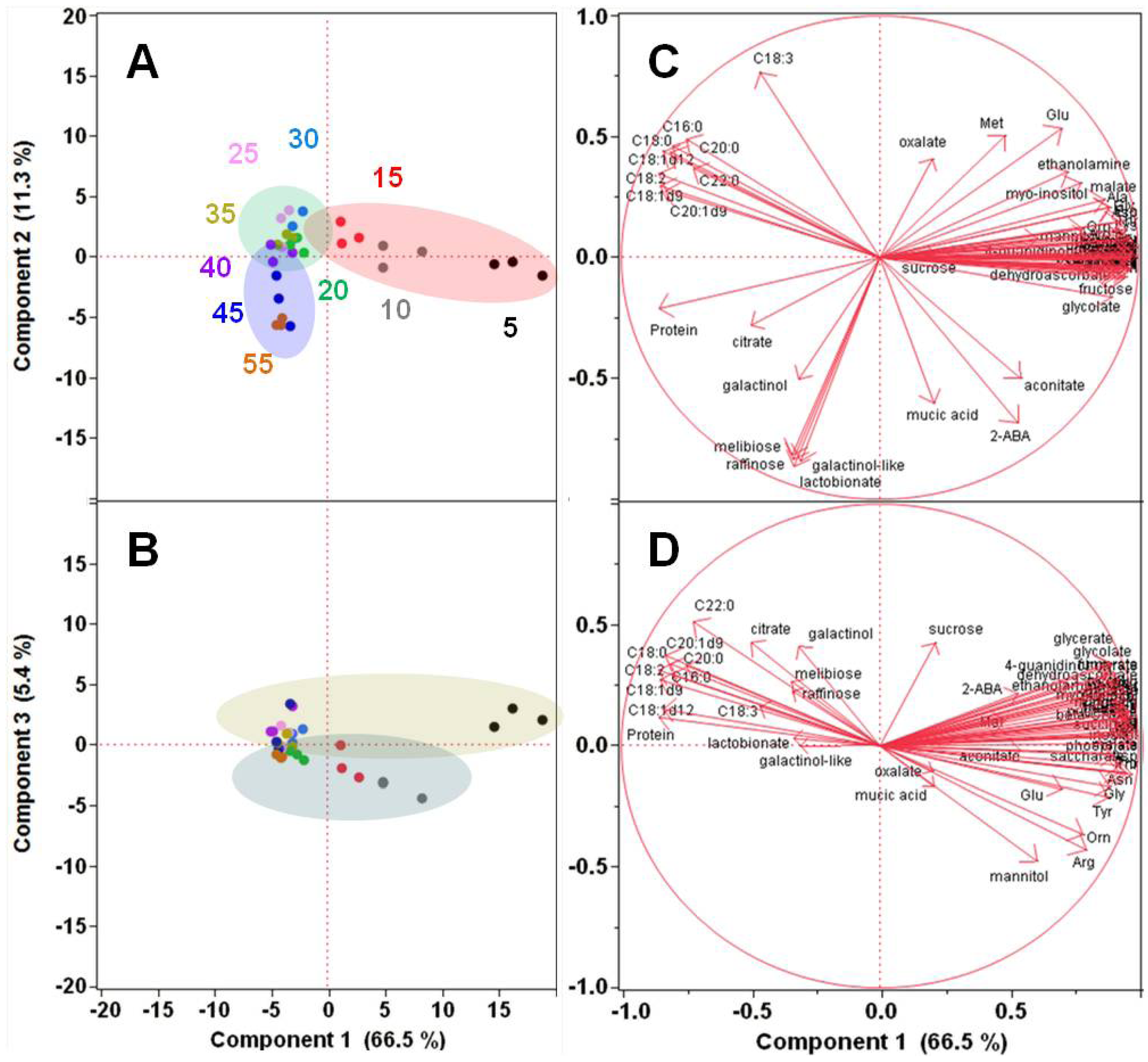
2.1.2. Polar Metabolomics in Developing Soybean Embryos

{kind=link}
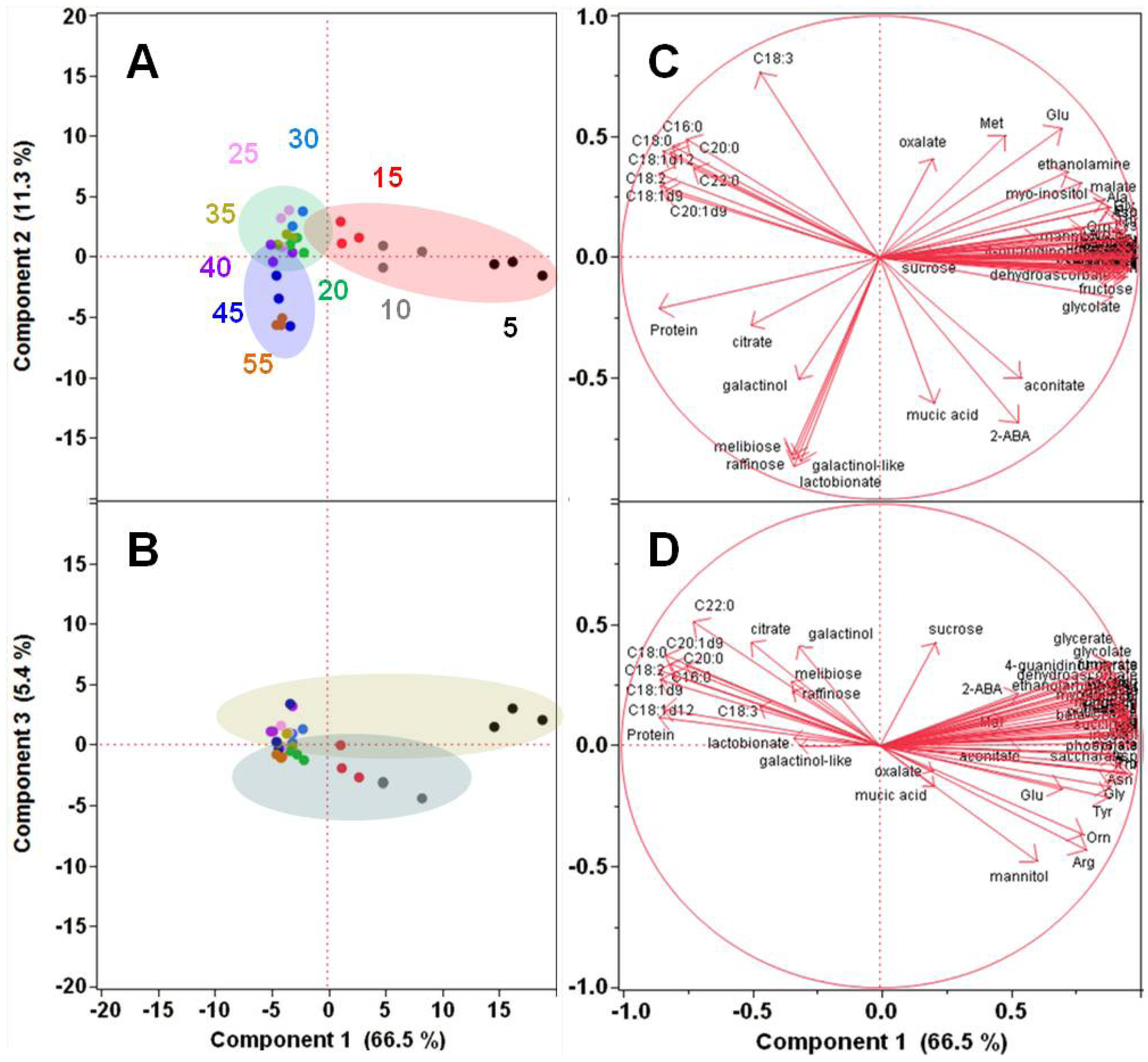{kind=link}
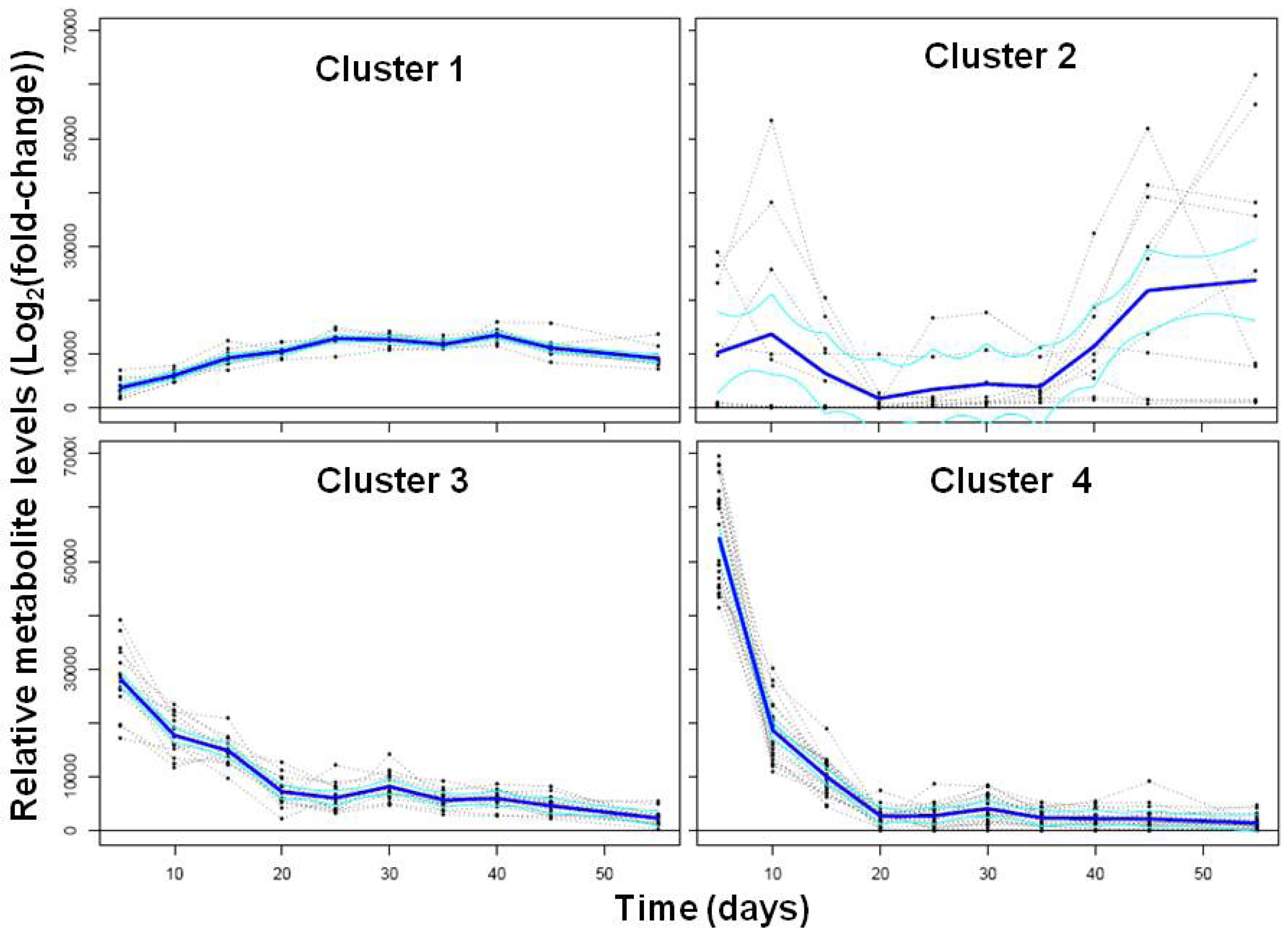{kind=link}
{kind=link}
{kind=link}
| Fatty Acids | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Day | 16:0 | 18:0 | 18:1 Δ9 | 18:1 Δ12 | 18:2 Δ9,12 | 18:3 Δ9,12,15 | 20:0 | 20:1 Δ11 | 22:0 |
| 5 | 8.0 ± 2.2 | 2.2 ± 0.4 | 6.1 ± 2.3 | 0.45 ± 0.19 | 14 ± 4 | 9.4 ± 2.5 | 0.40 ± 0.06 | 0.12 ± 0.04 | 0.34 ± 0.10 |
| 10 | 18 ± 2 | 6.1 ± 1.0 | 29 ± 9 | 1.9 ± 0.3 | 57 ± 8 | 16 ± 1 | 0.80 ± 0.14 | 0.31 ± 0.05 | 0.47 ± 0.07 |
| 15 | 29 ± 9 | 10 ± 3 | 52 ± 28 | 3.5 ± 0.9 | 98 ± 24 | 24 ± 2 | 1.1 ± 0.4 | 0.47 ± 0.23 | 0.77 ± 0.26 |
| 20 | 29 ± 2 | 11 ± 1 | 69 ± 7 | 3.8 ± 0.3 | 120 ± 9 | 20 ± 1 | 1.2 ± 0.1 | 0.56 ± 0.06 | 0.77 ± 0.08 |
| 25 | 38 ± 5 | 16 ± 2 | 73 ± 10 | 4.2 ± 0.6 | 183 ± 23 | 25 ± 4 | 1.5 ± 0.2 | 0.72 ± 0.09 | 1.2 ± 0.1 |
| 30 | 35 ± 2 | 14 ± 1 | 81 ± 11 | 4.2 ± 0.6 | 161 ± 16 | 22 ± 1 | 1.4 ± 0.0 | 0.76 ± 0.07 | 1.1 ± 0.1 |
| 35 | 33 ± 2 | 14 ± 0 | 66 ± 3 | 3.7 ± 0.4 | 162 ± 20 | 22 ± 2 | 1.3 ± 0.1 | 0.71 ± 0.05 | 1.0 ± 0.0 |
| 40 | 37 ± 0 | 15 ± 1 | 81 ± 10 | 4.2 ± 0.4 | 176 ± 10 | 22 ± 2 | 1.5 ± 0.1 | 0.83 ± 0.10 | 1.2 ± 0.0 |
| 45 | 29 ± 2 | 11 ± 0 | 65 ± 4 | 3.2 ± 0.4 | 132 ± 13 | 16 ± 1 | 1.2 ± 0.0 | 0.67 ± 0.06 | 0.93 ± 0.01 |
| 55 | 25 ± 1 | 9.0 ± 0.7 | 54 ± 6 | 2.8 ± 0.2 | 112 ± 10 | 14 ± 1 | 0.95 ± 0.10 | 0.52 ± 0.01 | 0.78 ± 0.07 |
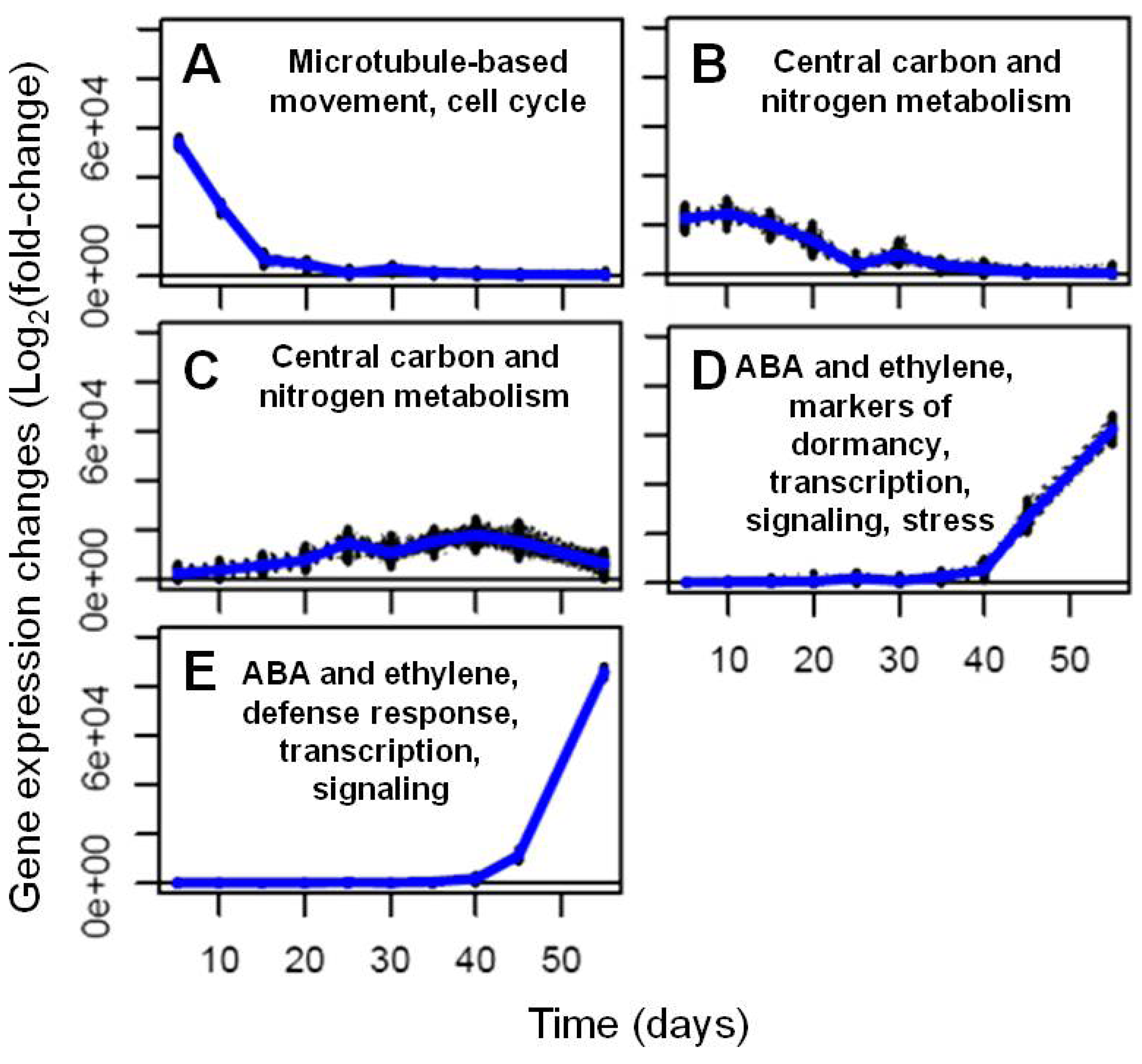
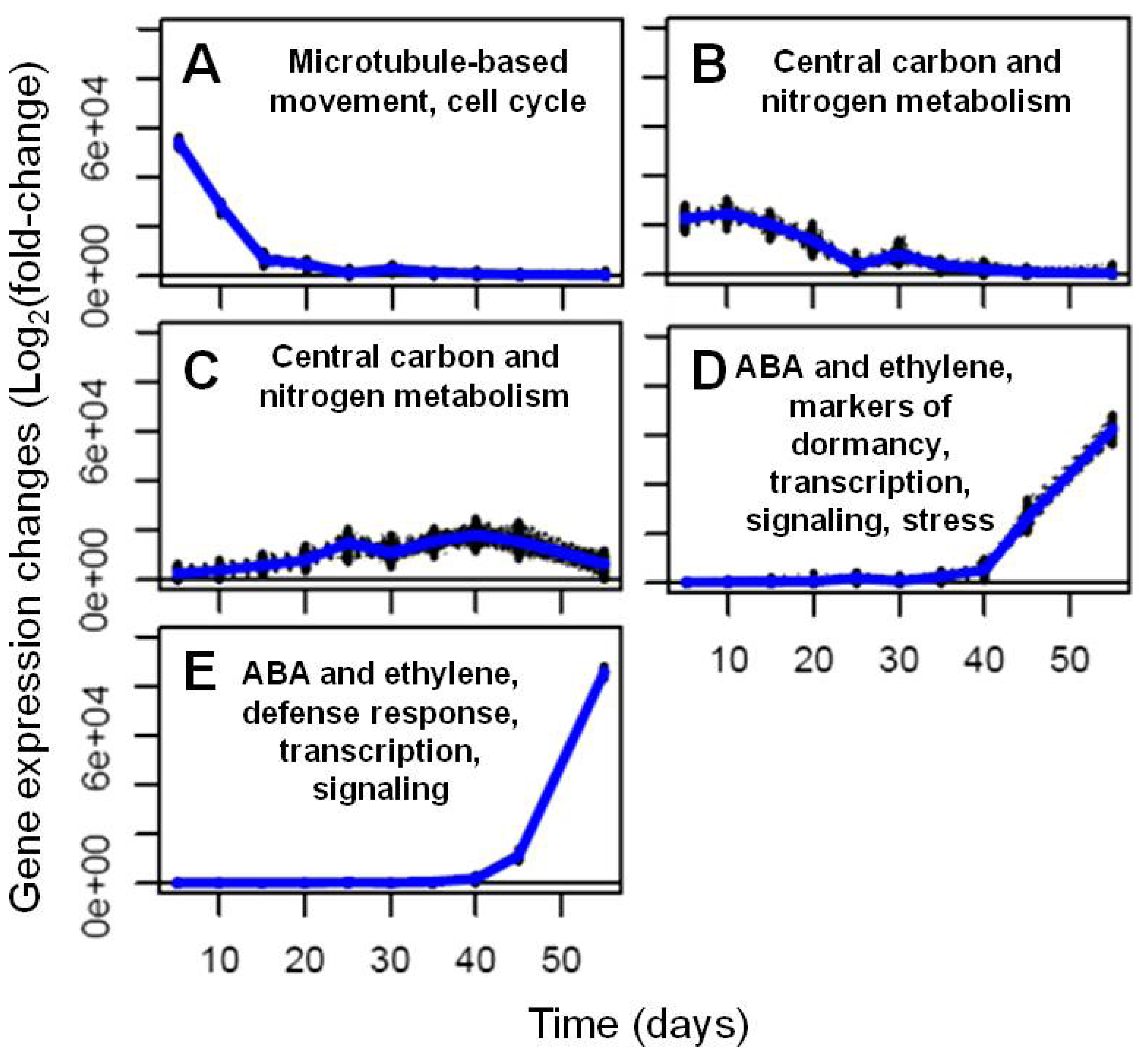
2.2. Transcriptional Reprogramming in Developing Soybean Embryos
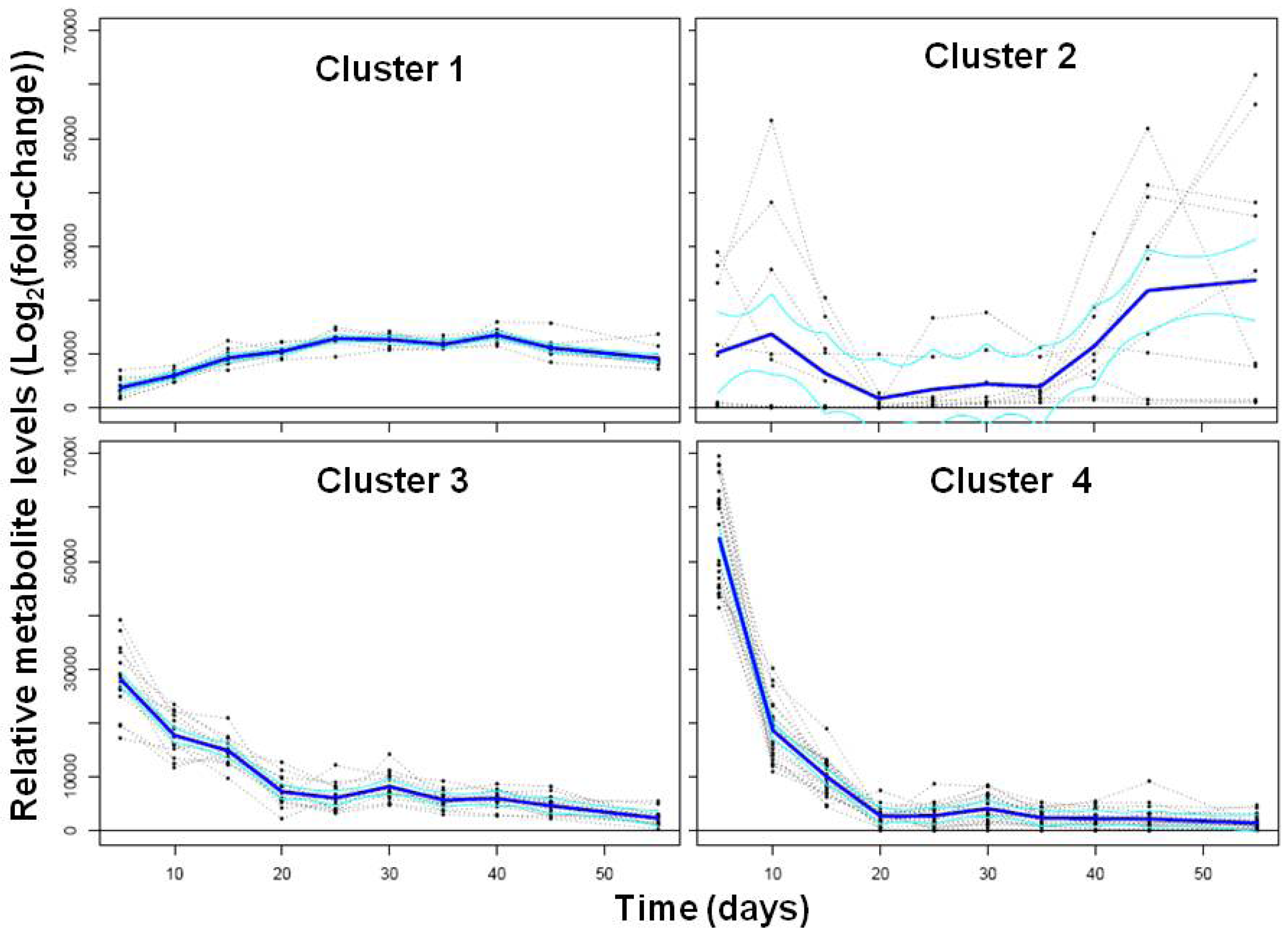
2.2.1. RNA Sequencing-Based Transcriptomics
2.2.2. Analysis and Visualization of Global Transcriptional Changes during Embryo Development
| GO Search Terms | Trend D | Trend E | Both Trends (D + E) | |||
|---|---|---|---|---|---|---|
| Number | % | Number | % | Number | % | |
| abscisic acid | 19 | 7.36 | 16 | 8.56 | 35 | 8.05 |
| ethylene | 9 | 3.49 | 11 | 5.88 | 20 | 4.60 |
| jasmonic acid | 16 | 6.20 | 13 | 6.95 | 29 | 6.67 |
| salicylic acid | 14 | 5.43 | 13 | 6.95 | 27 | 6.21 |
| chloroplast | 8 | 3.10 | 1 | 0.53 | 9 | 2.07 |
| redox | 4 | 1.55 | 3 | 1.60 | 7 | 1.61 |
| germination | 7 | 2.71 | 1 | 0.53 | 8 | 1.84 |
| flowering | 5 | 1.94 | 1 | 0.53 | 6 | 1.38 |
| dormancy | 9 | 3.49 | 6 | 3.21 | 15 | 3.45 |
| transcription | 37 | 14.34 | 28 | 14.97 | 65 | 14.94 |
| signaling | 28 | 10.85 | 21 | 11.23 | 49 | 11.26 |
| metal | 5 | 1.94 | 4 | 2.14 | 9 | 2.07 |
| iron | 8 | 3.10 | 2 | 1.07 | 10 | 2.30 |
| trehalose | 0 | 0.00 | 2 | 1.07 | 2 | 0.46 |
| stress | 36 | 13.95 | 24 | 12.83 | 60 | 13.79 |
| oxidative stress | 11 | 4.26 | 9 | 4.81 | 20 | 4.60 |
| salt stress | 20 | 7.75 | 9 | 4.81 | 29 | 6.67 |
| osmotic stress | 6 | 2.33 | 4 | 2.14 | 10 | 2.30 |
| biotic stimulus | 2 | 0.78 | 3 | 1.60 | 5 | 1.15 |
| defense response | 24 | 9.30 | 34 | 18.18 | 58 | 13.33 |
| water deprivation | 17 | 6.59 | 13 | 6.95 | 30 | 6.90 |
| Total number of genes | 258 | 187 | 435 | |||
2.3. Integrated Overview of Transcriptional and Metabolic Changes Representing Developmental and Metabolic Transitions during Soybean Embryo Development
3. Experimental Section
3.1. Plant Growth and Embryo Harvesting
3.2. Biomass Measurements
3.3. Metabolite Profiling
3.4. Transcriptomics
3.4.1. RNA Isolation, cDNA Library Preparation, and Illumina RNA Sequencing
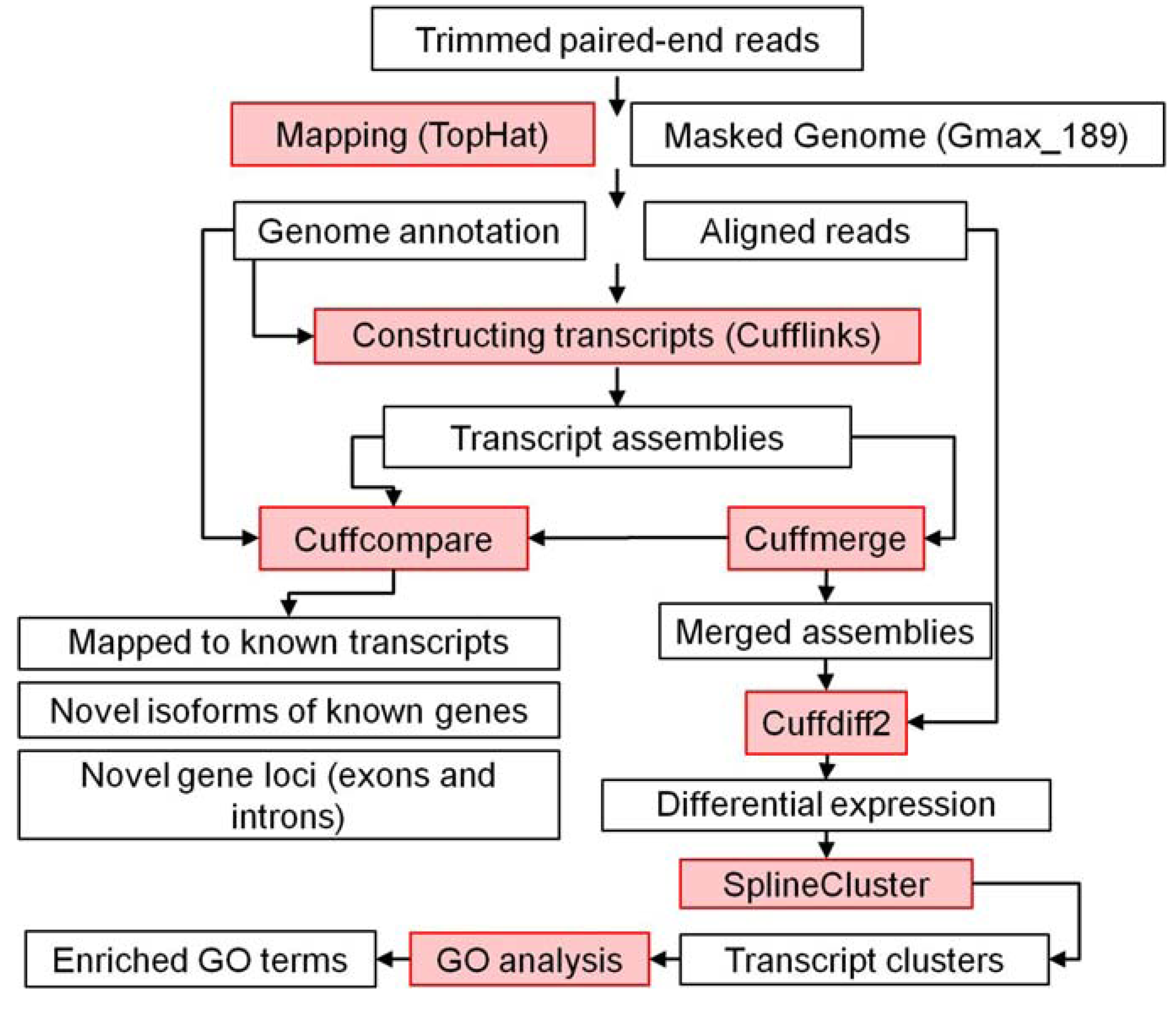
3.4.2. RNA Sequencing Data Processing, Differential Gene Expression, and Gene Coexpression Pipeline
| sample | raw | % | filtered | % | aligned | % |
|---|---|---|---|---|---|---|
| d5a | 50,277,367 | 100 | 48,847,168 | 97.2 | 45,036,342 | 89.6 |
| d5b | 62,438,141 | 100 | 60,618,219 | 97.1 | 55,916,016 | 89.6 |
| d5c | 42,789,850 | 100 | 41,586,412 | 97.2 | 38,479,330 | 89.9 |
| d10a | 72,420,225 | 100 | 69,364,427 | 95.8 | 61,431,613 | 84.8 |
| d10b | 46,641,758 | 100 | 44,850,498 | 96.2 | 39,812,742 | 85.4 |
| d10c | 85,142,664 | 100 | 81,774,580 | 96.0 | 71,198,971 | 83.6 |
| d15a | 42,701,829 | 100 | 39,342,420 | 92.1 | 33,742,148 | 79.0 |
| d15b | 55,919,488 | 100 | 51,665,677 | 92.4 | 44,507,297 | 79.6 |
| d15c | 98,613,720 | 100 | 91,253,129 | 92.5 | 78,873,912 | 80.0 |
| d20a | 71,059,794 | 100 | 65,828,827 | 92.6 | 52,933,436 | 74.5 |
| d20b | 44,455,535 | 100 | 41,248,905 | 92.8 | 33,195,196 | 74.7 |
| d20c | 54,423,534 | 100 | 50,618,077 | 93.0 | 40,743,929 | 74.9 |
| d25a | 61,500,744 | 100 | 56,010,032 | 91.1 | 46,786,403 | 76.1 |
| d25c | 83,670,143 | 100 | 76,387,754 | 91.3 | 63,785,353 | 76.2 |
| d30a | 74,112,923 | 100 | 68,885,120 | 92.9 | 59,572,944 | 80.4 |
| d30b | 77,985,515 | 100 | 72,428,008 | 92.9 | 61,711,555 | 79.1 |
| d30c | 100,671,683 | 100 | 93,523,791 | 92.9 | 81,026,533 | 80.5 |
| d35a | 67,498,080 | 100 | 62,861,610 | 93.1 | 54,691,875 | 81.0 |
| d35b | 87,943,727 | 100 | 82,007,125 | 93.2 | 71,352,895 | 81.1 |
| d35c | 99,568,258 | 100 | 92,761,463 | 93.2 | 80,856,912 | 81.2 |
| d40a | 135,660,811 | 100 | 124,626,322 | 91.9 | 107,084,232 | 78.9 |
| d40b | 54,135,432 | 100 | 49,929,189 | 92.2 | 43,065,040 | 79.6 |
| d40c | 55,241,634 | 100 | 51,064,159 | 92.4 | 44,214,195 | 80.0 |
| d45a | 67,554,585 | 100 | 63,069,990 | 93.4 | 53,773,223 | 79.6 |
| d45b | 62,187,743 | 100 | 58,293,335 | 93.7 | 50,756,256 | 81.6 |
| d45c | 74,873,986 | 100 | 70,329,261 | 93.9 | 60,877,029 | 81.3 |
| d55a | 74,383,353 | 100 | 68,110,555 | 91.6 | 56,475,935 | 75.9 |
| d55b | 86,213,964 | 100 | 79,106,644 | 91.8 | 65,108,144 | 75.5 |
| d55c | 47,160,359 | 100 | 43,338,248 | 91.9 | 35,914,805 | 76.2 |
3.4.3. MapMan
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Clemente, T.E.; Cahoon, E.B. Soybean oil: Genetic approaches for modification of functionality and total content. Plant Physiol. 2009, 151, 1030–1040. [Google Scholar] [CrossRef]
- Weselake, R.J.; Taylor, D.C.; Rahman, M.H.; Shah, S.; Laroche, A.; McVetty, P.B.E.; Harwood, J.L. Increasing the flow of carbon into seed oil. Biotechnol. Adv. 2009, 27, 866–878. [Google Scholar] [CrossRef]
- Eastmond, P.J.; Graham, I.A. Re-examining the role of the glyoxylate cycle in oilseeds. Trends Plant Sci. 2001, 6, 72–78. [Google Scholar] [CrossRef]
- Graham, I.A. Seed storage oil mobilization. Annu. Rev. Plant Biol. 2008, 59, 115–142. [Google Scholar] [CrossRef]
- Penfield, S.; Graham, S.; Graham, I.A. Storage reserve mobilization in germinating oilseeds: Arabidopsis as a model system. Biochem. Soc. Trans. 2005, 33, 380–383. [Google Scholar] [CrossRef]
- Meinke, D.W.; Chen, J.; Beachy, R.N. Expression of storage-protein genes during soybean seed development. Planta 1981, 153, 130–139. [Google Scholar] [CrossRef]
- Fehr, W.R.; Caviness, C.E.; Burmood, D.T.; Pennington, J.S. Stage of development descriptions for soybean, Glycine. max (L.) Merrill. Crop. Sci. 1971, 11, 929–931. [Google Scholar] [CrossRef]
- Le, B.H.; Wagmaister, J.A.; Kawashima, T.; Bui, A.Q.; Harada, J.J.; Goldberg, R.B. Using genomics to study legume seed development. Plant Physiol. 2007, 144, 562–574. [Google Scholar] [CrossRef]
- Hill, L.M.; Morley-Smith, E.R.; Rawsthorne, S. Metabolism of sugars in the endosperm of developing seeds of oilseed rape. Plant Physiol. 2003, 131, 228–236. [Google Scholar] [CrossRef]
- Hill, L.M.; Rawsthorne, S. Carbon supply for storage-product synthesis in developing seeds of oilseed rape. Biochem. Soc. Trans. 2000, 28, 667–669. [Google Scholar] [CrossRef]
- Allen, D.K.; Ohlrogge, J.B.; Shachar-Hill, Y. The role of light in soybean seed filling metabolism. Plant J. 2009, 58, 220–234. [Google Scholar] [CrossRef]
- Borisjuk, L.; Nguyen, T.H.; Neuberger, T.; Rutten, T.; Tschiersch, H.; Claus, B.; Feussner, I.; Webb, A.G.; Jakob, P.; Weber, H.; Wobus, U.; Rolletschek, H. Gradients of lipid storage, photosynthesis and plastid differentiation in developing soybean seeds. New Phytol. 2005, 167, 761–776. [Google Scholar] [CrossRef]
- Munier-Jolain, N.G.; Munier-Jolain, N.M.; Roche, R.; Ney, B.; Duthion, C. Seed growth rate in grain legumes - I. Effect of photoassimilate availability on seed growth rate. J. Exp. Bot. 1998, 49, 1963–1969. [Google Scholar]
- Rolletschek, H.; Radchuk, R.; Klukas, C.; Schreiber, F.; Wobus, U.; Borisjuk, L. Evidence of a key role for photosynthetic oxygen release in oil storage in developing soybean seeds. New Phytol. 2005, 167, 777–786. [Google Scholar] [CrossRef]
- Ruuska, S.A.; Schwender, J.; Ohlrogge, J.B. The capacity of green oilseeds to utilize photosynthesis to drive biosynthetic processes. Plant Physiol. 2004, 136, 2700–2709. [Google Scholar] [CrossRef]
- Allen, D.K.; Shachar-Hill, Y.; Ohlrogge, J.B. Compartment-specific labeling information in C-13 metabolic flux analysis of plants. Phytochemistry. 2007, 68, 2197–2210. [Google Scholar] [CrossRef]
- Bates, P.D.; Durrett, T.P.; Ohlrogge, J.B.; Pollard, M. Analysis of acyl fluxes through multiple pathways of triacylglycerol synthesis in developing soybean embryos. Plant Physiol. 2009, 150, 55–72. [Google Scholar] [CrossRef]
- Iyer, V.V.; Sriram, G.; Fulton, D.B.; Zhou, R.; Westgate, M.E.; Shanks, J.V. Metabolic flux maps comparing the effect of temperature on protein and oil biosynthesis in developing soybean cotyledons. Plant Cell Environ. 2008, 31, 506–517. [Google Scholar] [CrossRef]
- Sriram, G.; Fulton, D.B.; Iyer, V.V.; Peterson, J.M.; Zhou, R.; Westgate, M.E.; Spalding, M.H.; Shanks, J.V. Quantification of compartmented metabolic fluxes in developing soybean embryos by employing biosynthetically directed fractional 13C labeling, two-dimensional [13C, 1H] nuclear magnetic resonance, and comprehensive isotopomer balancing. Plant Physiol. 2004, 136, 3043–3057. [Google Scholar] [CrossRef]
- Angelovici, R.; Galili, G.; Fernie, A.R.; Fait, A. Seed desiccation: a bridge between maturation and germination. Trends Plant Sci. 2010, 15, 211–218. [Google Scholar] [CrossRef]
- Blochl, A.; Grenier-de March, G.; Sourdioux, M.; Peterbauer, T.; Richter, A. Induction of raffinose oligosaccharide biosynthesis by abscisic acid in somatic embryos of alfalfa (Medicago. sativa L.). Plant Sci. 2005, 168, 1075–1082. [Google Scholar] [CrossRef]
- Finkelstein, R.; Reeves, W.; Ariizumi, T.; Steber, C. Molecular aspects of seed dormancy. Annu. Rev. Plant Biol. 2008, 59, 387–415. [Google Scholar] [CrossRef]
- Gutierrez, L.; Van Wuytswinkel, O.; Castelain, M.; Bellini, C. Combined networks regulating seed maturation. Trends Plant Sci. 2007, 12, 294–300. [Google Scholar] [CrossRef]
- Baud, S.; Boutin, J.P.; Miquel, M.; Lepiniec, L.; Rochat, C. An integrated overview of seed development in Arabidopsis thaliana ecotype WS. Plant Physiol. Biochem. 2002, 40, 151–160. [Google Scholar] [CrossRef]
- Chia, T.Y.; Pike, M.J.; Rawsthorne, S. Storage oil breakdown during embryo development of Brassica. napus (L.). J. Exp. Bot. 2005, 56, 1285–1296. [Google Scholar] [CrossRef]
- Wilson, R.F.; Rinne, R.W. Lipid molecular species composition in developing soybean cotyledons. Plant Physiol. 1978, 61, 830–833. [Google Scholar] [CrossRef]
- Saha, R.; Suthers, P.F.; Maranas, C.D. Zea mays iRS1563: A comprehensive genome-scale metabolic reconstruction of maize metabolism. PLoS One 2011, 6, e21784. [Google Scholar]
- Heard, N.A.; Holmes, C.C.; Stephens, D.A.; Hand, D.J.; Dimopoulos, G. Bayesian coclustering of Anopheles gene expression time series: Study of immune defense response to multiple experimental challenges. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 16939–16944. [Google Scholar] [CrossRef]
- Kakumanu, A.; Ambavaram, M.M.; Klumas, C.; Krishnan, A.; Batlang, U.; Myers, E.; Grene, R.; Pereira, A. Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed by RNA-Seq. Plant Physiol. 2012, 160, 846–867. [Google Scholar]
- Thimm, O.; Blasing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Kruger, P.; Selbig, J.; Muller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Phytozome Homepage. Available online: www.phytozome.net/soybean/ (accessed on 16 April 2013).
- Liberman, L.M.; Sozzani, R.; Benfey, P.N. Integrative systems biology: an attempt to describe a simple weed. Curr. Opin. Plant Biol. 2012, 15, 162–167. [Google Scholar] [CrossRef]
- Wang, L.; Li, P.; Brutnell, T.P. Exploring plant transcriptomes using ultra high-throughput sequencing. Brief. Funct. Genomics 2010, 9, 118–128. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Collakova, E.; Yen, J.Y.; Senger, R.S. Are we ready for genome-scale modeling in plants? Plant Sci. 2012, 191–192, 53–70. [Google Scholar] [CrossRef]
- Dal'Molin, C.G.; Quek, L.E.; Palfreyman, R.W.; Brumbley, S.M.; Nielsen, L.K. AraGEM, a genome-scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiol. 2010, 152, 579–589. [Google Scholar] [CrossRef]
- Dal’Molin, C.G.; Quek, L.E.; Palfreyman, R.W.; Brumbley, S.M.; Nielsen, L.K. C4GEM, a genome-scale metabolic model to study C4 plant metabolism. Plant Physiol. 2010, 154, 1871–1885. [Google Scholar] [CrossRef]
- Gill, N.; Findley, S.; Walling, J.G.; Hans, C.; Ma, J.; Doyle, J.; Stacey, G.; Jackson, S.A. Molecular and chromosomal evidence for allopolyploidy in soybean. Plant Physiol. 2009, 151, 1167–1174. [Google Scholar] [CrossRef]
- Schlueter, J.A.; Lin, J.Y.; Schlueter, S.D.; Vasylenko-Sanders, I.F.; Deshpande, S.; Yi, J.; O'Bleness, M.; Roe, B.A.; Nelson, R.T.; Scheffler, B.E.; Jackson, S.A.; Shoemaker, R.C. Gene duplication and paleopolyploidy in soybean and the implications for whole genome sequencing. BMC Genomics 2007, 8, 330. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Roulin, A.; Auer, P.L.; Libault, M.; Schlueter, J.; Farmer, A.; May, G.; Stacey, G.; Doerge, R.W.; Jackson, S.A. The fate of duplicated genes in a polyploid plant genome. Plant J. 2013, 73, 143–153. [Google Scholar] [CrossRef]
- Goffard, N.; Weiller, G. Extending MapMan: application to legume genome arrays. Bioinformatics 2006, 22, 2958–2959. [Google Scholar] [CrossRef]
- Lonien, J.; Schwender, J. Analysis of metabolic flux phenotypes for two Arabidopsis mutants with severe impairment in seed storage lipid synthesis. Plant Physiol. 2009, 151, 1617–1634. [Google Scholar] [CrossRef]
- Schwender, J.; Goffman, F.; Ohlrogge, J.B.; Shachar-Hill, Y. Rubisco without the Calvin cycle improves the carbon efficiency of developing green seeds. Nature 2004, 432, 779–782. [Google Scholar] [CrossRef]
- Castillo, E.M.; Delumen, B.O.; Reyes, P.S.; Delumen, H.Z. Raffinose synthase and galactinol synthase in developing seeds and leaves of legumes. J. Agr. Food Chem. 1990, 38, 351–355. [Google Scholar] [CrossRef]
- Li, X.; Zhuo, J.J.; Jing, Y.; Liu, X.; Wang, X.F. Expression of a galactinol synthase gene is positively associated with desiccation tolerance of Brassica. napus seeds during development. J. Plant Physiol. 2012, 168, 1761–1770. [Google Scholar]
- Nishizawa, A.; Yabuta, Y.; Shigeoka, S. Galactinol and raffinose constitute a novel function to protect plants from oxidative damage. Plant Physiol. 2008, 147, 1251–1263. [Google Scholar] [CrossRef]
- Tan, J.L.; Wang, C.Y.; Xiang, B.; Han, R.H.; Guo, Z.F. Hydrogen peroxide and nitric oxide mediated cold- and dehydration-induced myo-inositol phosphate synthase that confers multiple resistances to abiotic stresses. Plant Cell Environ. 2013, 36, 288–299. [Google Scholar] [CrossRef]
- Falk, J.; Munne-Bosch, S. Tocochromanol functions in plants: antioxidation and beyond. J. Exp. Bot. 2010, 61, 1549–1566. [Google Scholar] [CrossRef]
- Maeda, H.; DellaPenna, D. Tocopherol functions in photosynthetic organisms. Curr. Opin. Plant Biol. 2007, 10, 260–265. [Google Scholar] [CrossRef]
- Mene-Saffrane, L.; Jones, A.D.; DellaPenna, D. Plastochromanol-8 and tocopherols are essential lipid-soluble antioxidants during seed desiccation and quiescence in Arabidopsis. Proc. Natl. Acad. Sci. USA 2010, 107, 17815–17820. [Google Scholar] [CrossRef]
- Sattler, S.E.; Gilliland, L.U.; Magallanes-Lundback, M.; Pollard, M.; DellaPenna, D. Vitamin E is essential for seed longevity and for preventing lipid peroxidation during germination. Plant Cell 2004, 16, 1419–1432. [Google Scholar] [CrossRef]
- Fisher, K.M. Bayesian reconstruction of ancestral expression of the LEA gene families reveals propagule-derived desiccation tolerance in resurrection plants. Am. J. Bot. 2008, 95, 506–515. [Google Scholar] [CrossRef]
- Gechev, T.S.; Dinakar, C.; Benina, M.; Toneva, V.; Bartels, D. Molecular mechanisms of desiccation tolerance in resurrection plants. Cell Mol. Life Sci. 2012, 69, 3175–3186. [Google Scholar] [CrossRef]
- Illing, N.; Denby, K.J.; Collett, H.; Shen, A.; Farrant, J.M. The signature of seeds in resurrection plants: a molecular and physiological comparison of desiccation tolerance in seeds and vegetative tissues. Integr. Comp. Biol. 2005, 45, 771–787. [Google Scholar] [CrossRef]
- Holdsworth, M.J.; Bentsink, L.; Soppe, W.J. Molecular networks regulating Arabidopsis seed maturation, after-ripening, dormancy and germination. New Phytol. 2008, 179, 33–54. [Google Scholar] [CrossRef]
- Sano, N.; Permana, H.; Kumada, R.; Shinozaki, Y.; Tanabata, T.; Yamada, T.; Hirasawa, T.; Kanekatsu, M. Proteomic analysis of embryonic proteins synthesized from long-lived mRNAs during germination of rice seeds. Plant Cell Physiol. 2012, 53, 687–698. [Google Scholar] [CrossRef]
- Fukushima, A.; Kusano, M.; Redestig, H.; Arita, M.; Saito, K. Integrated omics approaches in plant systems biology. Curr. Opin. Chem. Biol. 2009, 13, 532–538. [Google Scholar] [CrossRef]
- Urano, K.; Kurihara, Y.; Seki, M.; Shinozaki, K. “Omics” analyses of regulatory networks in plant abiotic stress responses. Curr. Opin. Plant Biol. 2010, 13, 132–138. [Google Scholar] [CrossRef]
- Fiehn, O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp. Funct. Genet. 2001, 2, 155–168. [Google Scholar] [CrossRef]
- Fiehn, O.; Kopka, J.; Dormann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nature Biotech. 2000, 18, 1157–1161. [Google Scholar] [CrossRef]
- Dai, S.; Chen, S. Single-cell-type proteomics: Toward a holistic understanding of plant function. Mol. Cell Proteomics 2012, 11, 1622–1630. [Google Scholar] [CrossRef]
- Kueger, S.; Steinhauser, D.; Willmitzer, L.; Giavalisco, P. High-resolution plant metabolomics: from mass spectral features to metabolites and from whole-cell analysis to subcellular metabolite distributions. Plant J. 2012, 70, 39–50. [Google Scholar] [CrossRef]
- Moco, S.; Schneider, B.; Vervoort, J. Plant micrometabolomics: The analysis of endogenous metabolites present in a plant cell or tissue. J. Proteome Res. 2009, 8, 1694–1703. [Google Scholar] [CrossRef]
- Klie, S.; Krueger, S.; Krall, L.; Giavalisco, P.; Flugge, U.I.; Willmitzer, L.; Steinhauser, D. Analysis of the compartmentalized metabolome - a validation of the non-aqueous fractionation technique. Front. Plant Sci. 2011, 2, 55. [Google Scholar]
- Rolletschek, H.; Melkus, G.; Grafahrend-Belau, E.; Fuchs, J.; Heinzel, N.; Schreiber, F.; Jakob, P.M.; Borisjuk, L. Combined noninvasive imaging and modeling approaches reveal metabolic compartmentation in the barley endosperm. Plant Cell 2011, 23, 3041–3054. [Google Scholar] [CrossRef]
- Braybrook, S.A.; Harada, J.J. LECs go crazy in embryo development. Trends Plant Sci. 2008, 13, 624–630. [Google Scholar] [CrossRef]
- Jeong, S.; Volny, M.; Lukowitz, W. Axis formation in Arabidopsis - transcription factors tell their side of the story. Curr. Opin. Plant Biol. 2012, 15, 4–9. [Google Scholar]
- Lau, S.; Slane, D.; Herud, O.; Kong, J.; Juergens, G. Early embryogenesis in flowering plants: Setting up the basic body pattern. Annu. Rev. Plant Biol. 2012, 63, 483–506. [Google Scholar] [CrossRef]
- Ueda, M.; Laux, T. The origin of the plant body axis. Curr. Opin. Plant Biol. 2012, 15, 578–584. [Google Scholar] [CrossRef]
- Alonso, A.P.; Piasecki, R.J.; Wang, Y.; LaClair, R.W.; Shachar-Hill, Y. Quantifying the labeling and the levels of plant cell wall precursors using ion chromatography tandem mass spectrometry. Plant Physiol. 2010, 153, 915–924. [Google Scholar] [CrossRef]
- Schwender, J.; Ohlrogge, J.B. Probing in vivo metabolism by stable isotope labeling of storage lipids and proteins in developing Brassica napus embryos. Plant Physiol. 2002, 130, 347–361. [Google Scholar] [CrossRef]
- Schwender, J.; Shachar-Hill, Y.; Ohlrogge, J.B. Mitochondrial metabolism in developing embryos of Brassica napus. J. Biol. Chem. 2006, 281, 34040–34047. [Google Scholar] [CrossRef]
- Lu, Y.; Savage, L.J.; Larson, M.D.; Wilkerson, C.G.; Last, R.L. Chloroplast 2010: A database for large-scale phenotypic screening of Arabidopsis mutants. Plant Physiol. 2011, 155, 1589–1600. [Google Scholar] [CrossRef]
- Collakova, E.; Goyer, A.; Naponelli, V.; Krassovskaya, I.; Gregory, J.F.; Hanson, A.D.; Shachar-Hill, Y. Arabidopsis 10-formyl tetrahydrofolate deformylases are essential for photorespiration. Plant Cell 2008, 20, 1818–1832. [Google Scholar] [CrossRef]
- Duran, A.L.; Yang, J.; Wang, L.J.; Sumner, L.W. Metabolomics spectral formatting, alignment and conversion tools (MSFACTs). Bioinformatics 2003, 19, 2283–2293. [Google Scholar] [CrossRef]
- Goyer, A.; Collakova, E.; de la Garza, R.D.; Quinlivan, E.P.; Williamson, J.; Gregory, J.F.; Shachar-Hill, Y.; Hanson, A.D. 5-Formyltetrahydrofolate is an inhibitory but well tolerated metabolite in Arabidopsis leaves. J. Biol. Chem. 2005, 280, 26137–26142. [Google Scholar] [CrossRef]
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10. [Google Scholar]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2012, 31, 46–53. [Google Scholar]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Collakova, E.; Aghamirzaie, D.; Fang, Y.; Klumas, C.; Tabataba, F.; Kakumanu, A.; Myers, E.; Heath, L.S.; Grene, R. Metabolic and Transcriptional Reprogramming in Developing Soybean (Glycine max) Embryos. Metabolites 2013, 3, 347-372. https://doi.org/10.3390/metabo3020347
Collakova E, Aghamirzaie D, Fang Y, Klumas C, Tabataba F, Kakumanu A, Myers E, Heath LS, Grene R. Metabolic and Transcriptional Reprogramming in Developing Soybean (Glycine max) Embryos. Metabolites. 2013; 3(2):347-372. https://doi.org/10.3390/metabo3020347
Chicago/Turabian StyleCollakova, Eva, Delasa Aghamirzaie, Yihui Fang, Curtis Klumas, Farzaneh Tabataba, Akshay Kakumanu, Elijah Myers, Lenwood S. Heath, and Ruth Grene. 2013. "Metabolic and Transcriptional Reprogramming in Developing Soybean (Glycine max) Embryos" Metabolites 3, no. 2: 347-372. https://doi.org/10.3390/metabo3020347
APA StyleCollakova, E., Aghamirzaie, D., Fang, Y., Klumas, C., Tabataba, F., Kakumanu, A., Myers, E., Heath, L. S., & Grene, R. (2013). Metabolic and Transcriptional Reprogramming in Developing Soybean (Glycine max) Embryos. Metabolites, 3(2), 347-372. https://doi.org/10.3390/metabo3020347