Comparative Analysis of End Point Enzymatic Digests of Arabino-Xylan Isolated from Switchgrass (Panicum virgatum L) of Varying Maturities using LC-MSn †

Abstract
:1. Introduction
2. Results and Discussion
2.1. Xylan Isolation
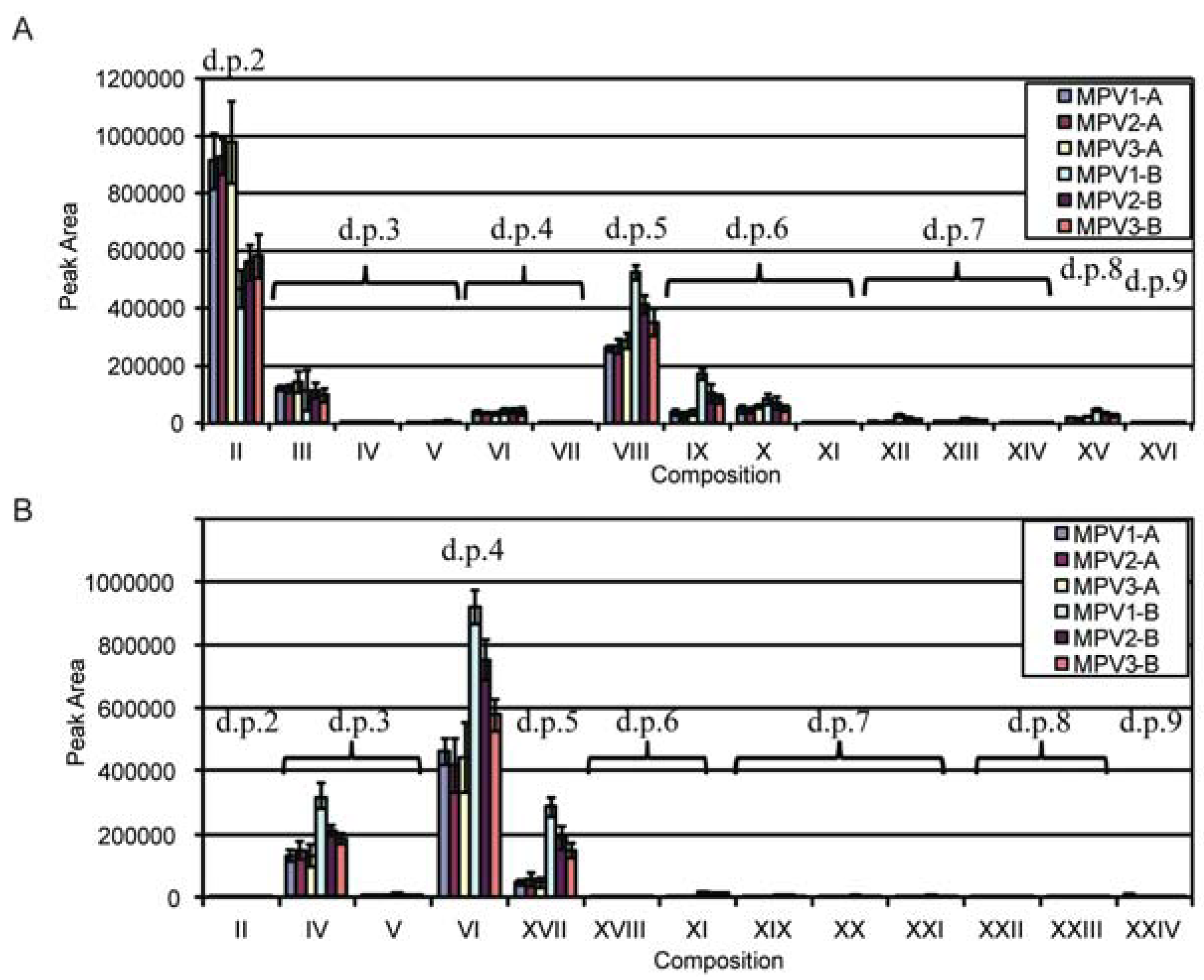
2.2. Enzymatic Depolymerization
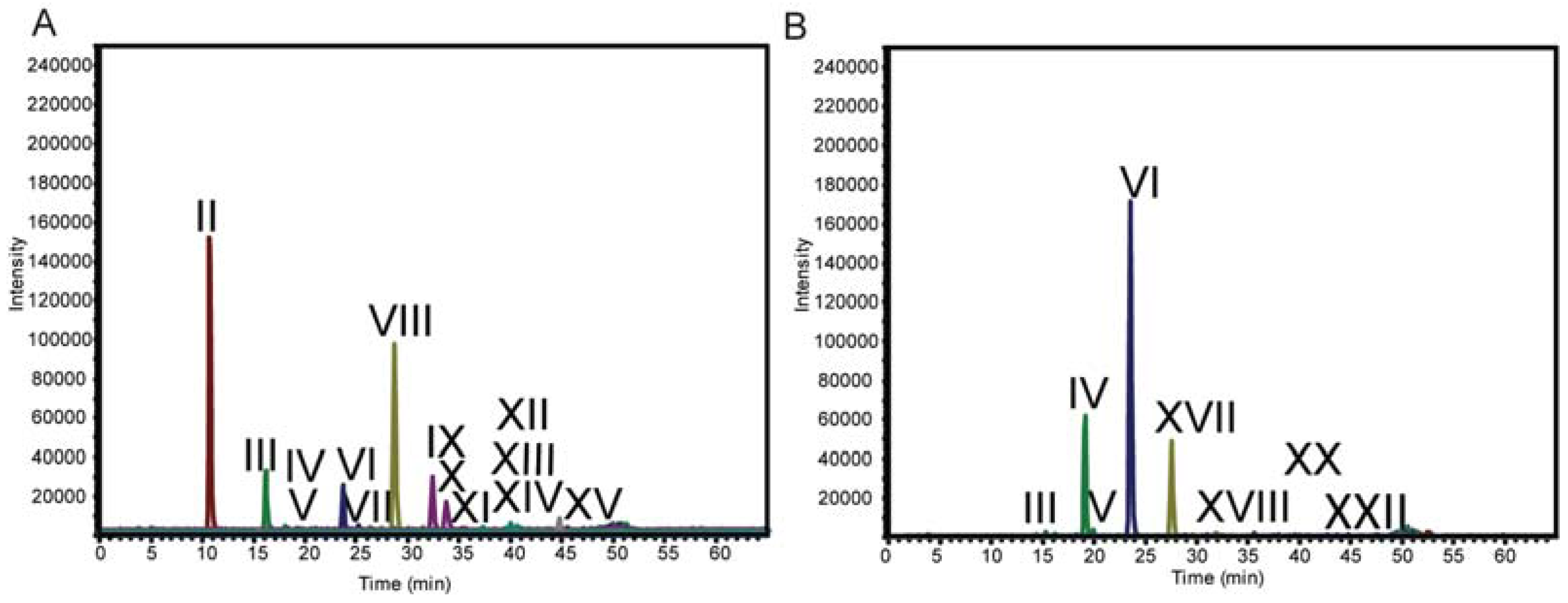
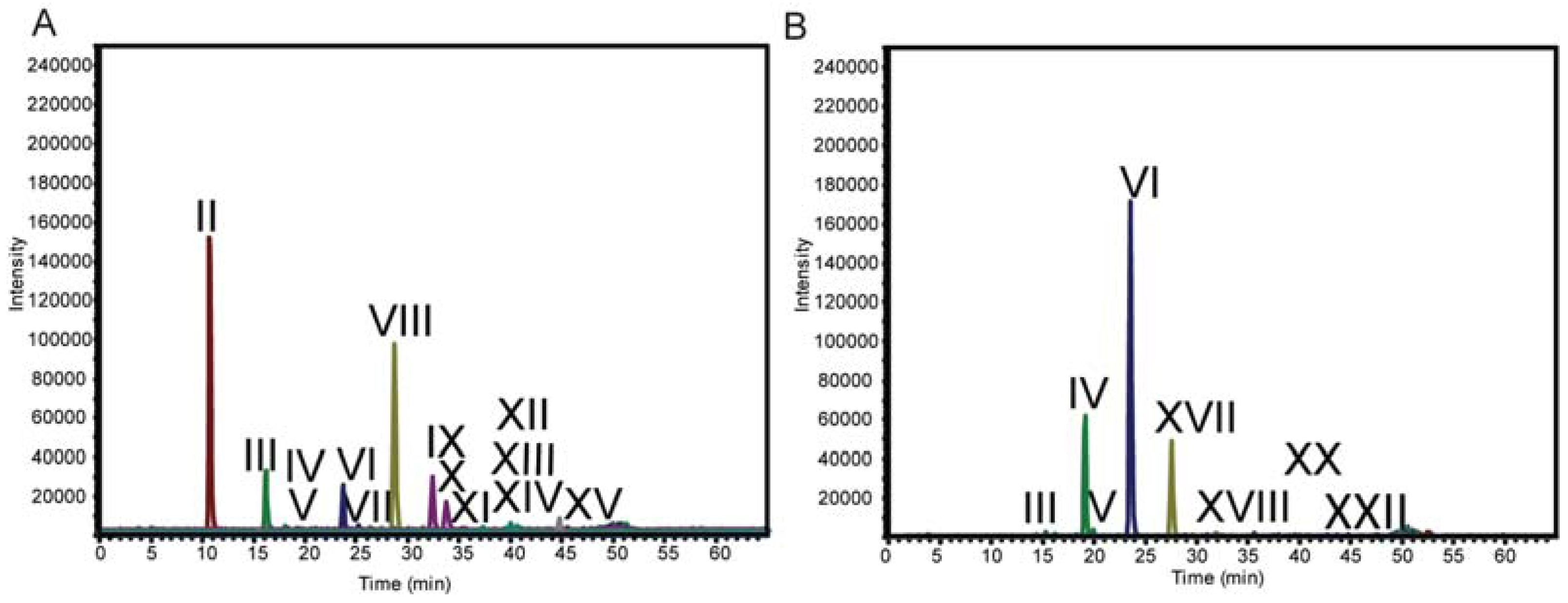
2.3. LC-MS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
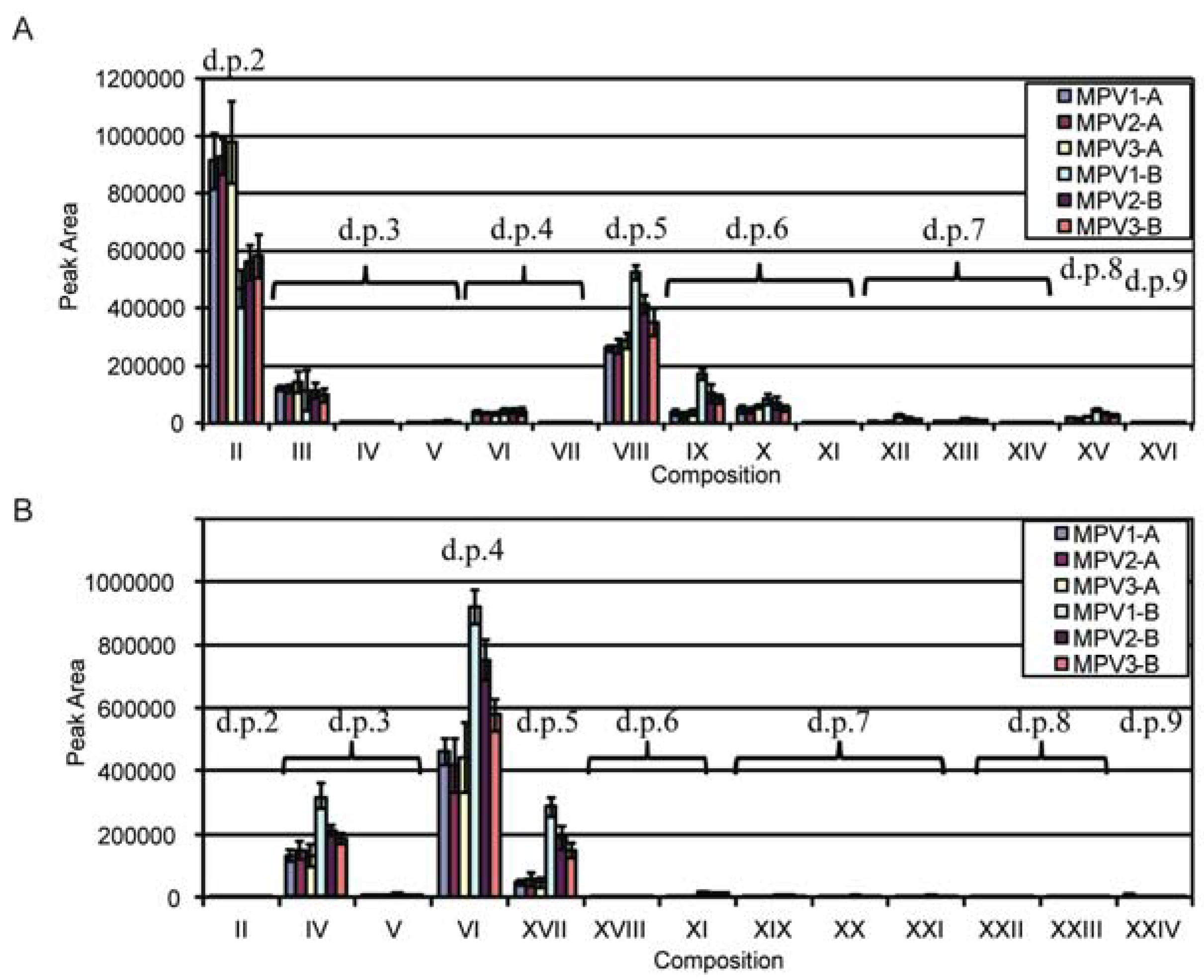
| Structure | d.p.a (RT, min) | Underivatized Molecular Formula | Reduced, Permethylated m/z (M+Na)+ | Assignment by | Present in | Additional Information/References |
|---|---|---|---|---|---|---|
| I | 1 | C5H12O5 | 245.1 | Standards | T. reesei digestion | |
| II | 2 (10.9) | C10H20O9 | 405.2 | Standard | T. viride digestion | |
| III | 3 (16.3) | C15H28O13 | 565.3 | Standard | T. viride digestion | |
| IV | 3 (19.2) | C15H28O13 | 565.3 | MS n Fragmentation | T. viride and T. reesei digestion | Linear sequence that does not correlate with Xyl3 standard |
| V | 3 (19.8) | C15H28O13 | 565.3 | MS n Fragmentation | T. viride and T. reesei digestion | |
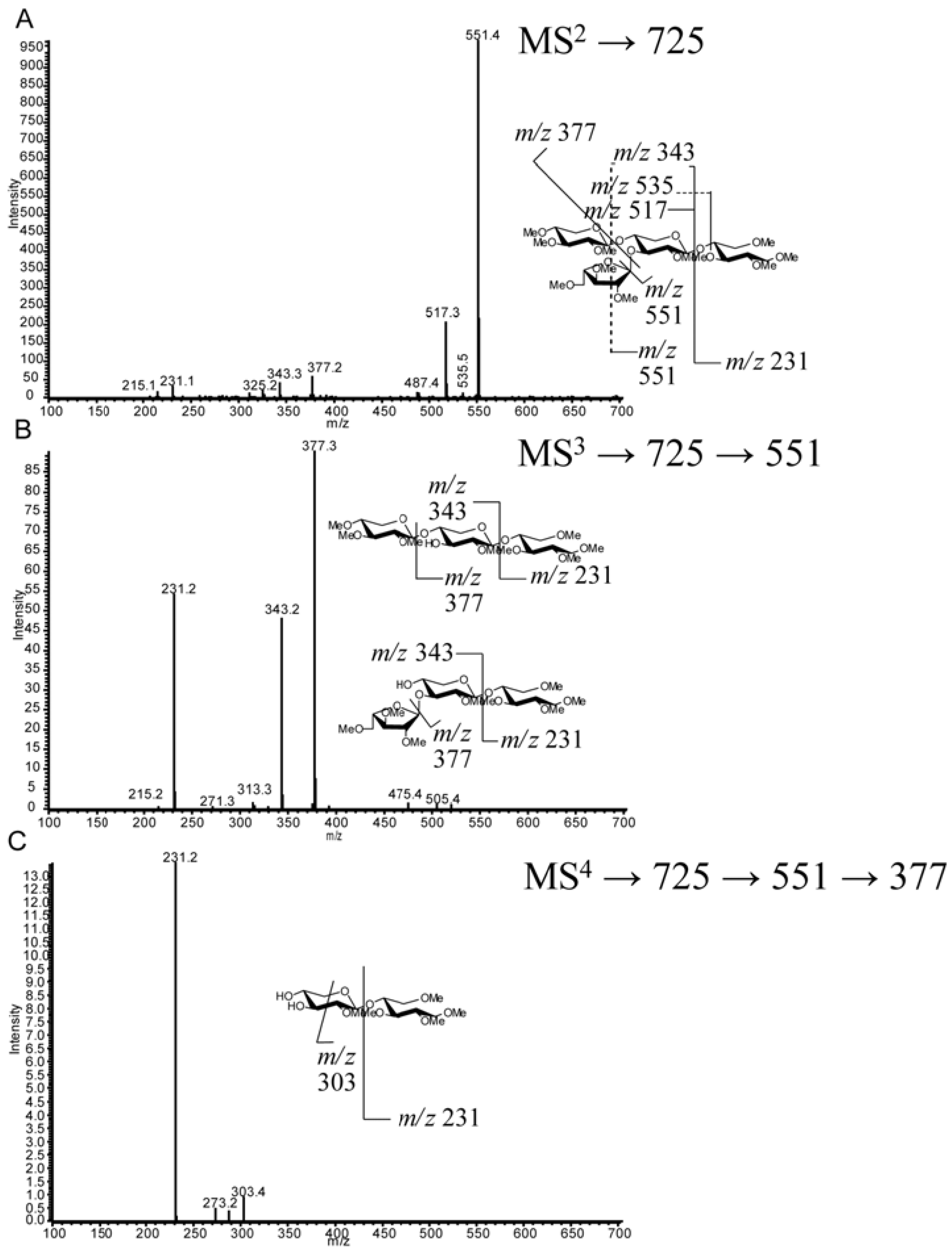
| VI | 4 (23.5) | C20H36O17 | 725.4 | MS n Fragmentation | T. viride and T. reesei digestion | [12,13,18,45] |
| VII | 4 (25.1) | C20H36O17 | 725.4 | MS n Fragmentation | T. viride digestion | Linear sequence that does not correlate with Xyl4 standard |
| VIII | 5 (28.5) | C25H44O21 | 885.4 | MS n Fragmentation | T. viride digestion | [12,13,18,20,45] |
| IX | 6 (32.4) | C30H52O25 | 1045.5 | MS n Fragmentation | T. viride digestion | Tentative assignment may be X |
| X | 6 (33.6) | C30H52O25 | 1045.5 | MS n Fragmentation | T. viride digestion | [20] Peak may be IX |
| XI | 6 (35.5) | C30H52O25 | 1045.5 | MS n Fragmentation | T. viride and T. reesei digestion | [12,18] |
| XII | 7 (37.5) | C35H60O29 | 1205.6 | MS n Fragmentation | T. viride digestion | |
| XIII | 7 (39.9) | C35H60O29 | 1205.6 | MS n Fragmentation | T. viride digestion | |
| XIV | 7 (40.7) | C35H60O29 | 1205.6 | MS n Fragmentation | T. viride digestion | |
| XV | 8 (44.9) | C40H68O33 | 1365.7 | MS n Fragmentation | T. viride digestion | [18] |
| XVI | 9 (48.6) | C45H76O37 | 1525.7 | Retention Time | T. viride digestion | |
| XVII | 5 (27.5) | C25H44O21 | 885.4 | MS n Fragmentation | T. reesei digestion | [27] |
| XVIII | 6 (32.8) | C30H52O25 | 1045.5 | MS n Fragmentation | T. reesei digestion | |
| XIX | 7 (38.9) | C35H60O29 | 1205.6 | MS n Fragmentation | T. reesei digestion | |
| XX | 7 (40.9) | C35H60O29 | 1205.6 | MS n Fragmentation | T. reesei digestion | |
| XXI | 7 (42.9) | C35H60O29 | 1205.6 | MS n Fragmentation | T. reesei digestion | |
| XXII | 8 (43.8) | C40H68O33 | 1365.7 | Retention Time | T. reesei digestion | |
| XXIII | 8 (44.8) | C40H68O33 | 1365.7 | Retention Time | T. reesei digestion | |
| XXIV | 9 (47.5) | C45H76O37 | 1525.7 | Retention Time | T. reesei digestion | |
| (Glc)4-Internal Standard | C24H44O21 | 901.5 |

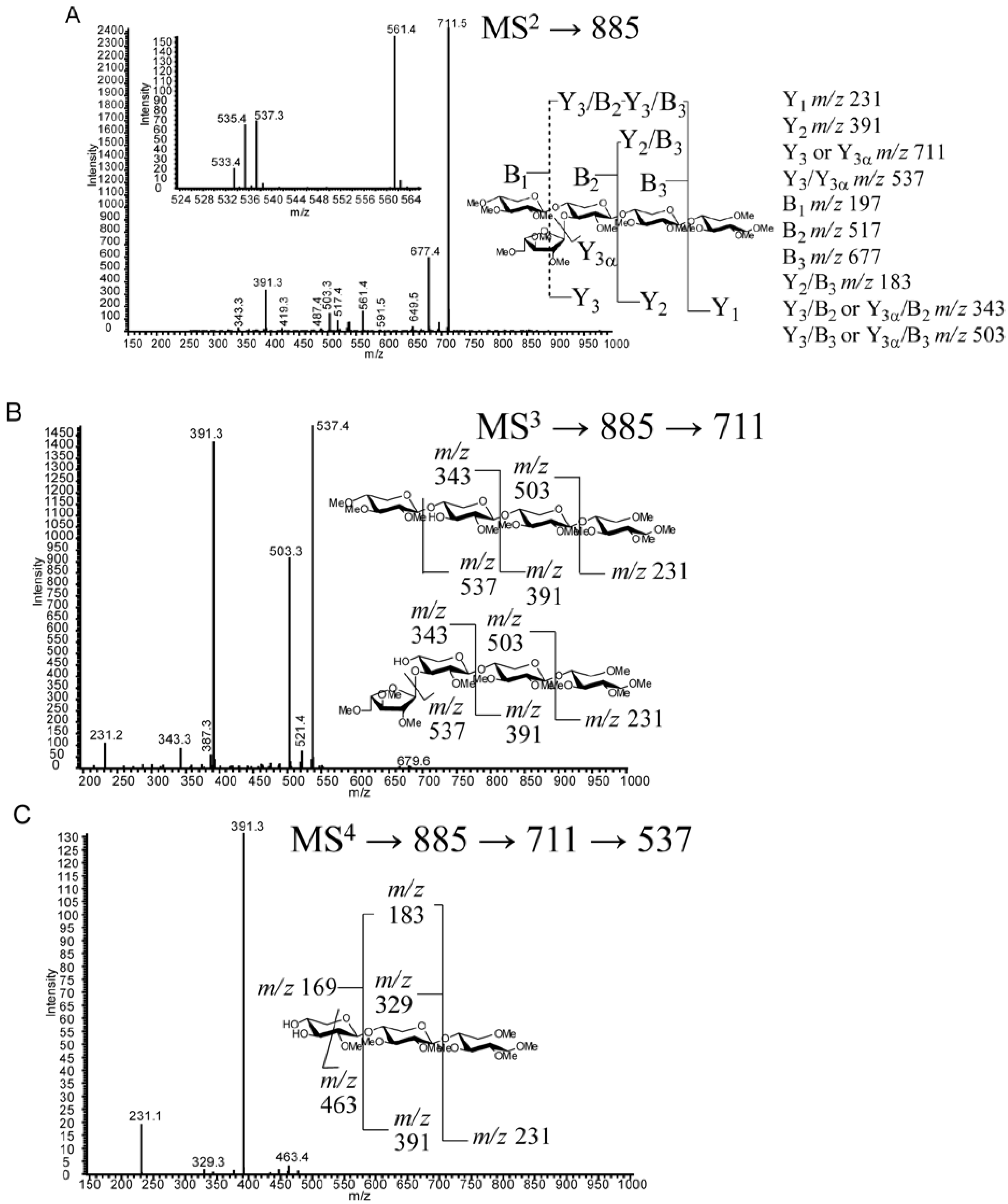
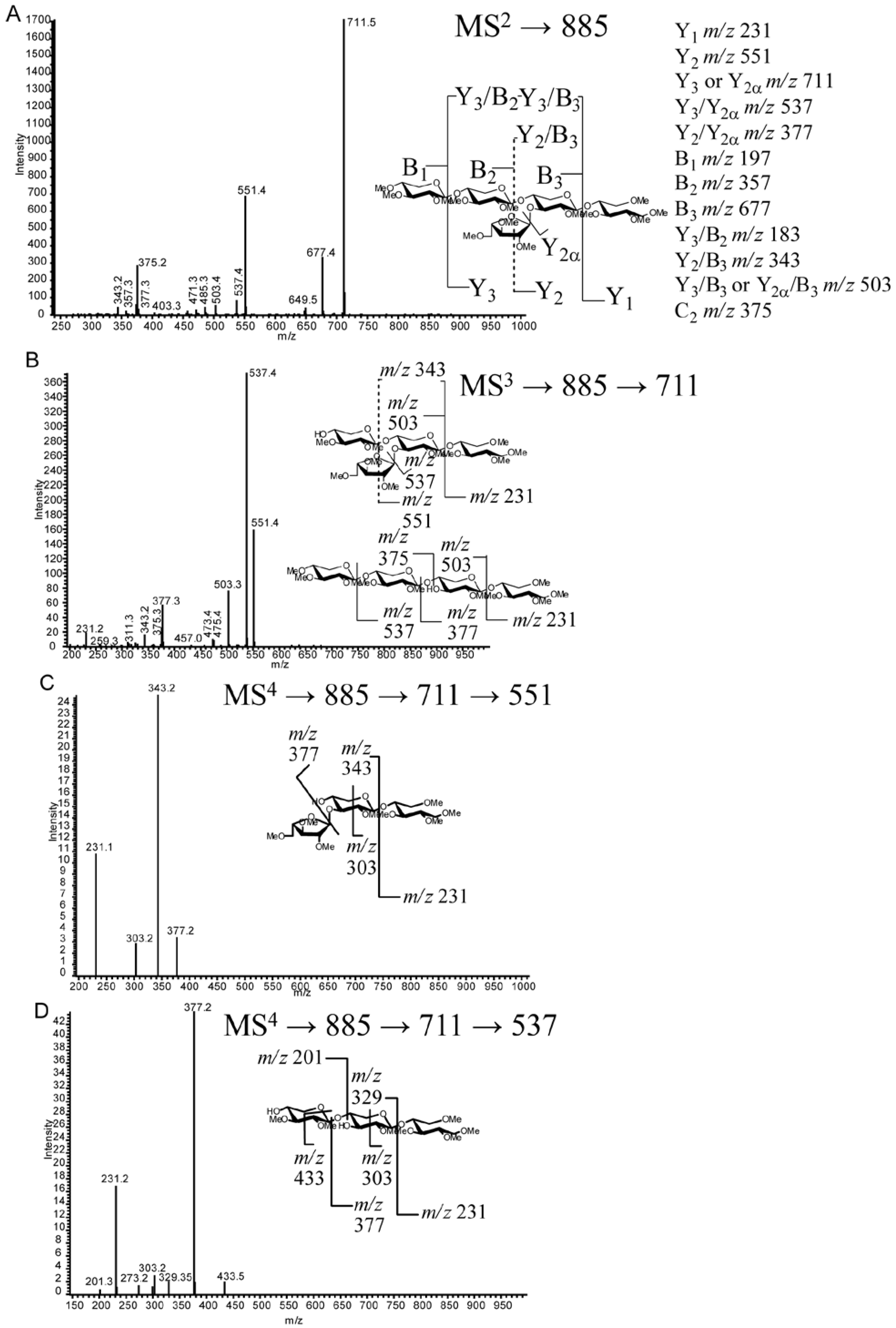
2.4. LC-MS2

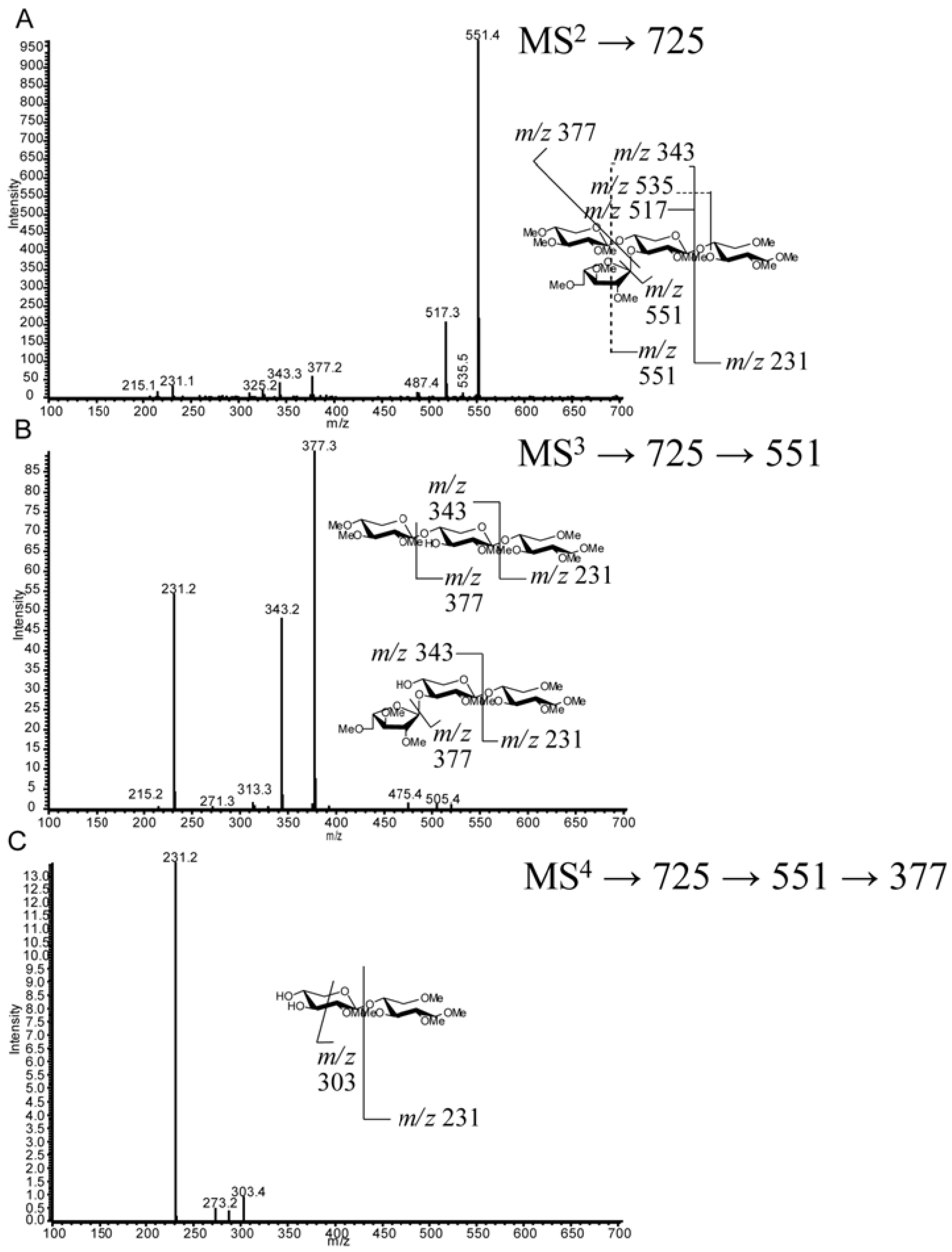
2.5. LC-MSn
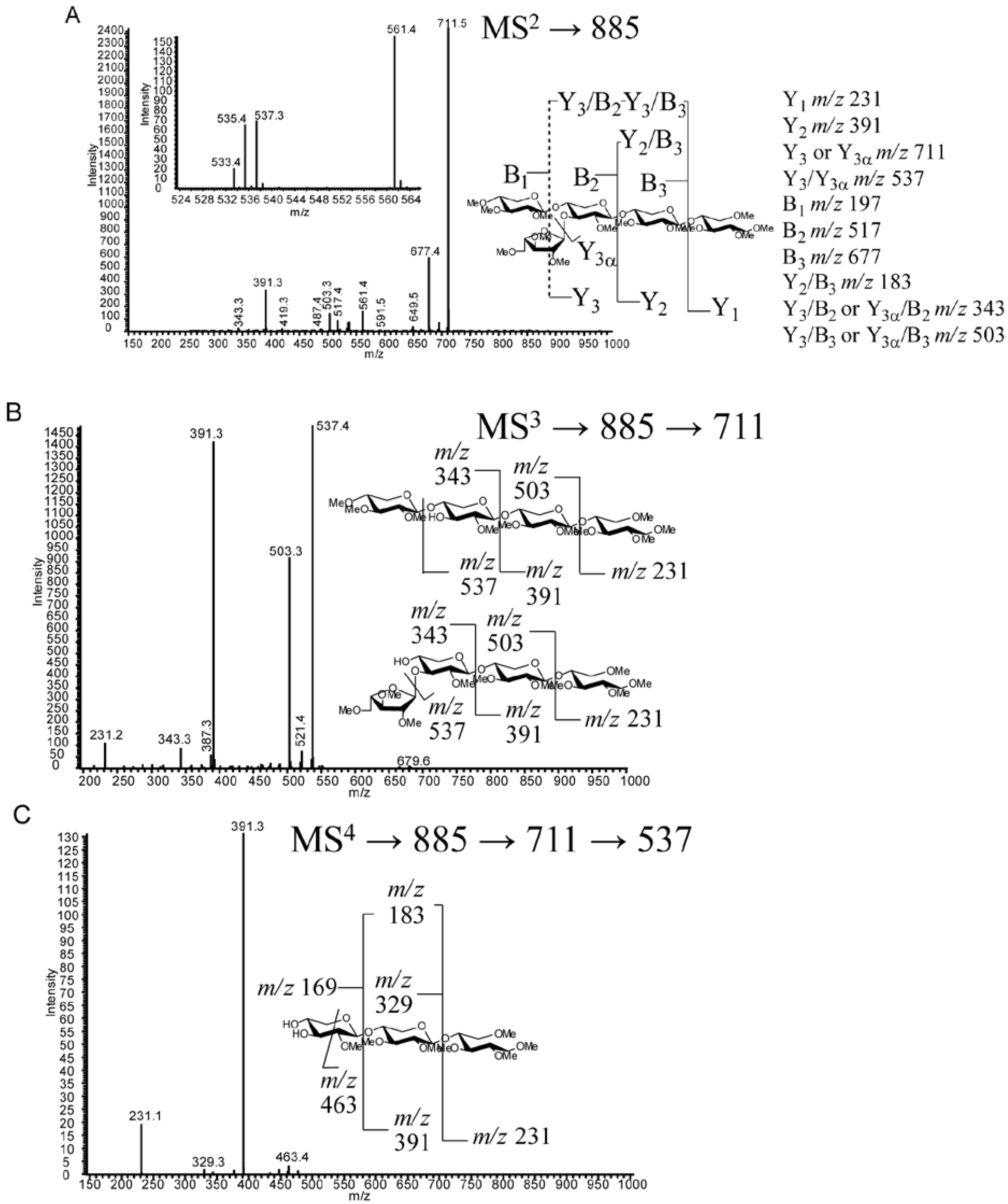

2.6. Discussion
3. Experimental Section
3.1. Materials
3.2. Xylan Sample Preparation
3.3. Permethylation Reaction of Enzymatic Products
3.4. C18-LC-MSn.
MSn Method
4. Conclusions
Supplementary Materials
Acknowledgments
Conflict of Interest
References
- Walsh, M.E.; Ugarte, D.G.D.; Shapouri, H.; Slinksky, S.P. Bioenergy crop production in the United States: Potential quantities, land use changes, and economic impacts on the agricultural sector. Environ. Resource Econ. 2003, 24, 313–333. [Google Scholar] [CrossRef]
- Vogel, J. Unique aspects of the grass cell wall. Curr. Opin. Plant. Biol. 2008, 11, 301–307. [Google Scholar] [CrossRef]
- Qing, Q.; Yang, B.; Wyman, C.E. Xylooligomers are strong inhibitors of cellulose hydrolysis by enzymes. Bioresour. Technol. 2010, 101, 9624–9630. [Google Scholar] [CrossRef]
- Kabel, M.A.; van den Borne, H.; Vincken, J.P.; Voragen, A.G.J.; Schols, H.A. Structural differences of xylans affect their interaction with cellulose. Carbohydr. Polym. 2007, 69, 94–105. [Google Scholar] [CrossRef]
- Gírio, F.M.; Fonseca, C.; Carvalheiro, F.; Duarte, L.C.; Marques, S.; Bogel-Łukasik, R. Hemicelluloses for fuel ethanol: A review. Bioresour. Technol. 2010, 101, 4775–4800. [Google Scholar] [CrossRef]
- Selinger, L.B.; Forsberg, C.W.; Cheng, K.J. The rumen: A unique source of enzymes for enhancing livestock production. Anaerobe 1996, 2, 263–284. [Google Scholar] [CrossRef]
- Faik, A. Xylan biosynthesis: news from the grass. Plant. Physiol. 2010, 153, 396–402. [Google Scholar] [CrossRef]
- Mosier, N.; Wyman, C.; Dale, B.; Elander, R.; Lee, Y.Y.; Holtzapple, M.; Ladisch, M. Features of promising technologies for pretreatment of lignocellulosic biomass. Bioresour. Technol. 2005, 96, 673–686. [Google Scholar] [CrossRef]
- Esteghlalian, A.; Hashimoto, A.G.; Fenske, J.J.; Penner, M.H. Modeling and optimization of the dilute-sulfuric-acid pretreatment of corn stover, poplar and switchgrass. Bioresour. Technol. 1997, 59, 129–136. [Google Scholar] [CrossRef]
- Dervilly-Pinel, G.; Tran, V.; Saulnier, L. Investigation of the distribution of arabinose residues on the xylan backbone of water-soluble arabinoxylans from wheat flour. Carbohydr. Polym. 2004, 55, 171–177. [Google Scholar] [CrossRef]
- Gruppen, H.; Kormelink, F.J.M.; Voragen, A.G.J. Water-Unextractable Cell-Wall Material from Wheat-Flour . 3. A Structural Model for Arabinoxylans. J. Cereal Sci. 1993, 18, 111–128. [Google Scholar] [CrossRef]
- Ordaz-Ortiz, J.J.; Devaux, M.F.; Saulnier, L. Classification of wheat varieties based on structural features of arabinoxylans as revealed by endoxylanase treatment of flour and grain. J. Agric. Food Chem. 2005, 53, 8349–8356. [Google Scholar] [CrossRef]
- Ordaz-Ortiz, J.J.; Saulnier, L. Structural variability of arabinoxylans from wheat flour. Comparison of water-extractable and xylanase-extractable arabinoxylans. J. Cereal Sci. 2005, 42, 119–125. [Google Scholar] [CrossRef]
- Vietor, R.J.; Kormelink, F.J.M.; Angelino, S.A.G.F.; Voragen, A.G.J. Substitution Patterns of Water-Unextractable Arabinoxylans from Barley and Malt. Carbohydr. Polym. 1994, 24, 113–118. [Google Scholar] [CrossRef]
- Rantanen, H.; Virkki, L.; Tuomainen, P.; Kabel, M.; Schols, H.; Tenkanen, M. Preparation of arabinoxylobiose from rye xylan using family 10 Aspergillus aculeatus endo-1,4-β-D-xylanase. Carbohydr. Polym. 2007, 68, 350–359. [Google Scholar] [CrossRef]
- Westphal, Y.; Kuhnel, S.; de Waard, P.; Hinz, S.W.; Schols, H.A.; Voragen, A.G.; Gruppen, H. Branched arabino-oligosaccharides isolated from sugar beet arabinan. Carbohydr. Res. 2010, 345, 1180–1189. [Google Scholar] [CrossRef]
- Fernández, M.L.E.; Obel, N.; Scheller, H.V.; Roepstorff, P. Differentiation of isomeric oligosaccharide structures by ESI tandem MS and GC-MS. Carbohydr. Res. 2004, 339, 655–664. [Google Scholar] [CrossRef]
- Quéméner, B.; Ordaz-Ortiz, J.J.; Saulnier, L. Structural characterization of underivatized arabino-xylo-oligosaccharides by negative-ion electrospray mass spectrometry. Carbohydr. Res. 2006, 341, 1834–1847. [Google Scholar] [CrossRef]
- Correia, M.A.; Mazumder, K.; Bras, J.L.; Firbank, S.J.; Zhu, Y.; Lewis, R.J.; York, W.S.; Fontes, C.M.; Gilbert, H.J. Structure and function of an arabinoxylan-specific xylanase. J. Biol. Chem. 2011, 286, 22510–22520. [Google Scholar]
- Mazumder, K.; York, W.S. Structural analysis of arabinoxylans isolated from ball-milled switchgrass biomass. Carbohydr. Res. 2010, 345, 2183–2193. [Google Scholar] [CrossRef]
- Brown, D.M.; Goubet, F.; Wong, V.W.; Goodacre, R.; Stephens, E.; Dupree, P.; Turner, S.R. Comparison of five xylan synthesis mutants reveals new insight into the mechanisms of xylan synthesis. Plant. J. 2007, 52, 1154–1168. [Google Scholar]
- Maslen, S.L.; Goubet, F.; Adam, A.; Dupree, P.; Stephens, E. Structure elucidation of arabinoxylan isomers by normal phase HPLC-MALDI-TOF/TOF-MS/MS. Carbohydr. Res. 2007, 342, 724–735. [Google Scholar] [CrossRef]
- Ridlova, G.; Mortimer, J.C.; Maslen, S.L.; Dupree, P.; Stephens, E. Oligosaccharide relative quantitation using isotope tagging and normal-phase liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 2723–2730. [Google Scholar]
- Zaia, J. Mass spectrometry of oligosaccharides. Mass Spectrom. Rev. 2004, 23, 161–227. [Google Scholar] [CrossRef]
- Ruhaak, L.R.; Zauner, G.; Huhn, C.; Bruggink, C.; Deelder, A.M.; Wuhrer, M. Glycan labeling strategies and their use in identification and quantification. Anal. Bioanal. Chem. 2010, 397, 3457–3481. [Google Scholar] [CrossRef]
- Bauer, S. Mass spectrometry for characterizing plant cell wall polysaccharides. Front. Plant. Sci. 2012, 3, 45. [Google Scholar]
- Bowman, M.J.; Dien, B.S.; Hector, R.E.; Sarath, G.; Cotta, M.A. Liquid chromatography-mass spectrometry investigation of enzyme-resistant xylooligosaccharide structures of switchgrass associated with ammonia pretreatment, enzymatic saccharification, and fermentation. Bioresour. Technol. 2012, 110, 437–447. [Google Scholar] [CrossRef]
- Bowman, M.J.; Dien, B.S.; O'Bryan, P.J.; Sarath, G.; Cotta, M.A. Selective chemical oxidation and depolymerization of switchgrass (Panicum. virgatum L.) xylan with oligosaccharide product analysis by mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 941–950. [Google Scholar]
- Dien, B.; O'Bryan, P.J.; Hector, R.; Iten, L.; Cotta, M.A. Conversion of switchgrass to sugars and ethanol using dilute ammonium hydroxide pretreatment. In Proceedings of the 31st Symposium on Biotechnology for Fuels and Chemicals, San Francisco, CA, USA, 3 May 2009; p. 93.
- Dien, B.S.; Jung, H.J.G.; Vogel, K.P.; Casler, M.D.; Lamb, J.F.S.; Iten, L.; Mitchell, R.B.; Sarath, G. Chemical composition and response to dilute-acid pretreatment and enzymatic saccharification of alfalfa, reed canarygrass, and switchgrass. Biomass Bioenergy 2006, 30, 880–891. [Google Scholar] [CrossRef]
- Jung, H.J.G.; Vogel, K.P. Lignification of Switchgrass (Panicum Virgatum) and Big Bluestem (AndropogonGerardii) Plant-Parts during Maturation and Its Effect on Fiber Degradability. J. Sci. Food Agric. 1992, 59, 169–176. [Google Scholar]
- Whistler, R.L.; Bachrach, J.; Bowman, D.R. Preparation and properties of corn cob holocellulose. Arch. Biochem. 1948, 19, 25–33. [Google Scholar]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Crocker, D. Determination of structural carbohydrates and lignin in biomass; Technical Report NREL/TP-510-42618. 2005. Available online: http://www.nrel.gov/biomass/pdfs/42618.pdf accessed on 14 November 2012.
- Packett, L.V.; Plumlee, M.L.; Barnes, R.; Mott, G.O. Influence of Hemicellulose A and B on Cellulose Digestion Volatile Fatty Acid Production and Forage Nutritive Evaluation. J. Nutr. 1965, 85, 89–101. [Google Scholar]
- Paës, G.; Berrin, J.G.; Beaugrand, J. GH11 xylanases: Structure/function/properties relationships and applications. Biotechnol. Adv. 2012, 30, 564–592. [Google Scholar] [CrossRef]
- Pollet, A.; Delcour, J.A.; Courtin, C.M. Structural determinants of the substrate specificities of xylanases from different glycoside hydrolase families. Crit. Rev. Biotechnol. 2010, 30, 176–191. [Google Scholar] [CrossRef]
- Collins, T.; Gerday, C.; Feller, G. Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol. Rev. 2005, 29, 3–23. [Google Scholar] [CrossRef]
- Parkkinen, T.; Hakulinen, N.; Tenkanen, M.; Siika-aho, M.; Rouvinen, J. Crystallization and preliminary X-ray analysis of a novel Trichoderma reesei xylanase IV belonging to glycoside hydrolase family 5. Acta Crystallogr., Sect. D 2004, 60, 542–544. [Google Scholar]
- Tenkanen, M.; Puls, J.; Poutanen, K. Two Major Xylanases of Trichoderma Reesei. Enzyme Microb. Technol. 1992, 14, 566–574. [Google Scholar] [CrossRef]
- Berlin, A.; Maximenko, V.; Gilkes, N.; Saddler, J. Optimization of enzyme complexes for lignocellulose hydrolysis. Biotechnol. Bioeng. 2007, 97, 287–296. [Google Scholar] [CrossRef]
- Saha, B.C.; Iten, L.B.; Cotta, M.A.; Wu, Y.V. Dilute acid pretreatment, enzymatic saccharification and fermentation of wheat straw to ethanol. Process. Biochemistry 2005, 40, 3693–3700. [Google Scholar] [CrossRef]
- Costello, C.E.; Contado-Miller, J.M.; Cipollo, J.F. A glycomics platform for the analysis of permethylated oligosaccharide alditols. J. Am. Soc. Mass Spectrom. 2007, 18, 1799–1812. [Google Scholar] [CrossRef]
- Hu, Y.; Mechref, Y. Comparing MALDI-MS, RP-LC-MALDI-MS and RP-LC-ESI-MS glycomic profiles of permethylated N-glycans derived from model glycoproteins and human blood serum. Electrophoresis 2012, 33, 1768–1777. [Google Scholar] [CrossRef]
- Huang, R.; Pomin, V.H.; Sharp, J.S. LC-MSn analysis of isomeric chondroitin sulfate oligosaccharides using a chemical derivatization strategy. J. Am. Soc. Mass Spectrom. 2011, 22, 1577–1587. [Google Scholar] [CrossRef]
- Izydorczyk, M.S.; Biliaderis, C.G. Studies on the Structure of Wheat-Endosperm Arabinoxylans. Carbohydr. Polym. 1994, 24, 61–71. [Google Scholar] [CrossRef]
- Domon, B.; Costello, C.E. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconjugate J. 1988, 5, 397–409. [Google Scholar] [CrossRef]
- Jordan, D.B.; Bowman, M.J.; Braker, J.D.; Dien, B.S.; Hector, R.E.; Lee, C.C.; Mertens, J.A.; Wagschal, K. Plant cell walls to ethanol. Biochem. J. 2012, 442, 241–252. [Google Scholar] [CrossRef]
- Chundawat, S.P.; Vismeh, R.; Sharma, L.N.; Humpula, J.F.; da Costa Sousa, L.; Chambliss, C.K.; Jones, A.D.; Balan, V.; Dale, B.E. Multifaceted characterization of cell wall decomposition products formed during ammonia fiber expansion (AFEX) and dilute acid based pretreatments. Bioresour. Technol. 2010, 101, 8429–8438. [Google Scholar]
- Ciucanu, I.; Kerek, F. A simple and rapid method for the permethylation of carbohydrates. Carbohydr. Res. 1984, 131, 209–217. [Google Scholar] [CrossRef]
- Ciucanu, I.; Costello, C.E. Elimination of oxidative degradation during the per-O-methylation of carbohydrates. J. Am. Chem. Soc. 2003, 125, 16213–16219. [Google Scholar] [CrossRef]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bowman, M.J.; Dien, B.S.; O'Bryan, P.J.; Sarath, G.; Cotta, M.A. Comparative Analysis of End Point Enzymatic Digests of Arabino-Xylan Isolated from Switchgrass (Panicum virgatum L) of Varying Maturities using LC-MSn. Metabolites 2012, 2, 959-982. https://doi.org/10.3390/metabo2040959
Bowman MJ, Dien BS, O'Bryan PJ, Sarath G, Cotta MA. Comparative Analysis of End Point Enzymatic Digests of Arabino-Xylan Isolated from Switchgrass (Panicum virgatum L) of Varying Maturities using LC-MSn. Metabolites. 2012; 2(4):959-982. https://doi.org/10.3390/metabo2040959
Chicago/Turabian StyleBowman, Michael J., Bruce S. Dien, Patricia J. O'Bryan, Gautam Sarath, and Michael A. Cotta. 2012. "Comparative Analysis of End Point Enzymatic Digests of Arabino-Xylan Isolated from Switchgrass (Panicum virgatum L) of Varying Maturities using LC-MSn" Metabolites 2, no. 4: 959-982. https://doi.org/10.3390/metabo2040959
APA StyleBowman, M. J., Dien, B. S., O'Bryan, P. J., Sarath, G., & Cotta, M. A. (2012). Comparative Analysis of End Point Enzymatic Digests of Arabino-Xylan Isolated from Switchgrass (Panicum virgatum L) of Varying Maturities using LC-MSn. Metabolites, 2(4), 959-982. https://doi.org/10.3390/metabo2040959