Metabolic Adaptation and Protein Complexes in Prokaryotes




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
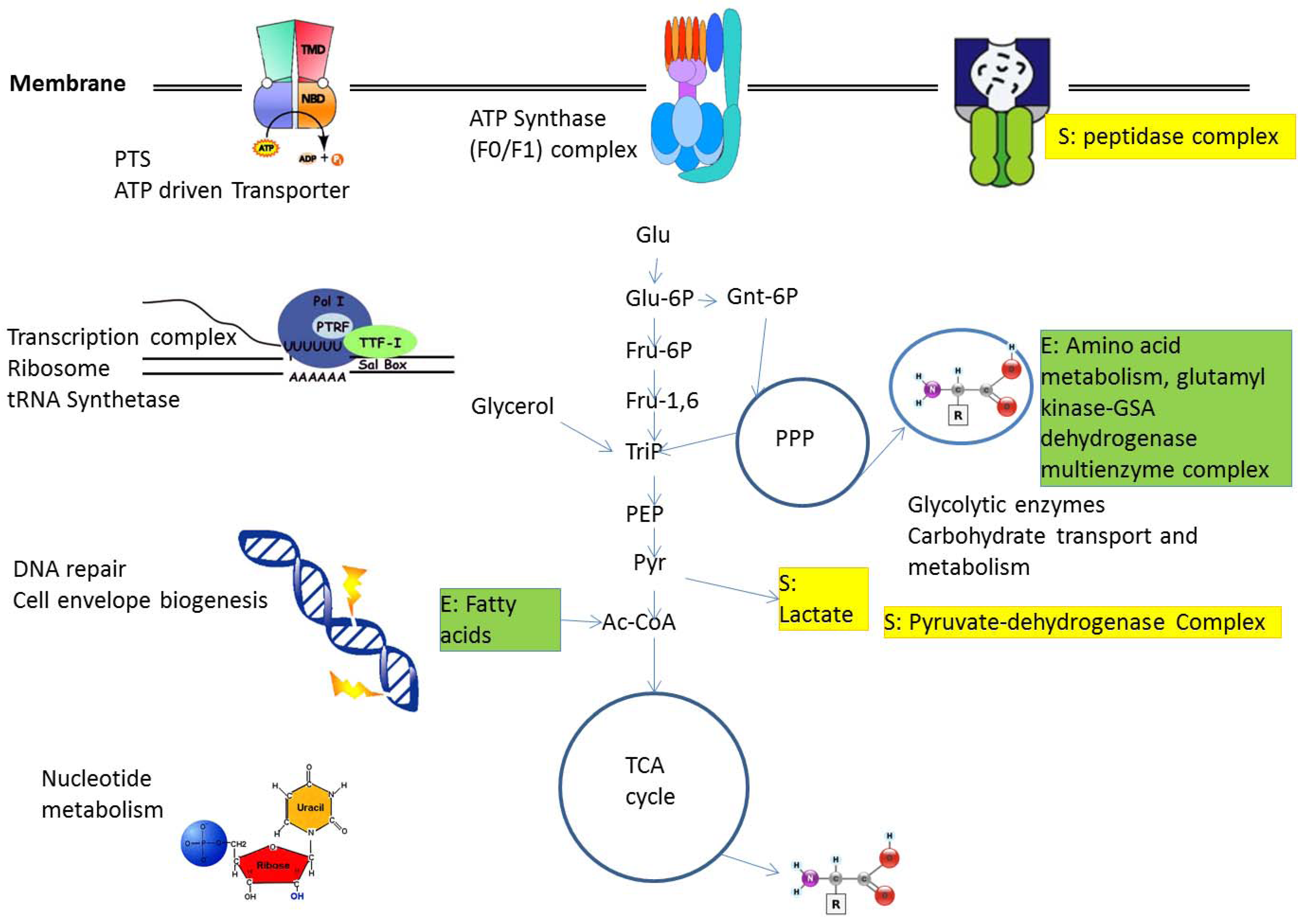
2.1. Protein Complexes in Central Metabolism
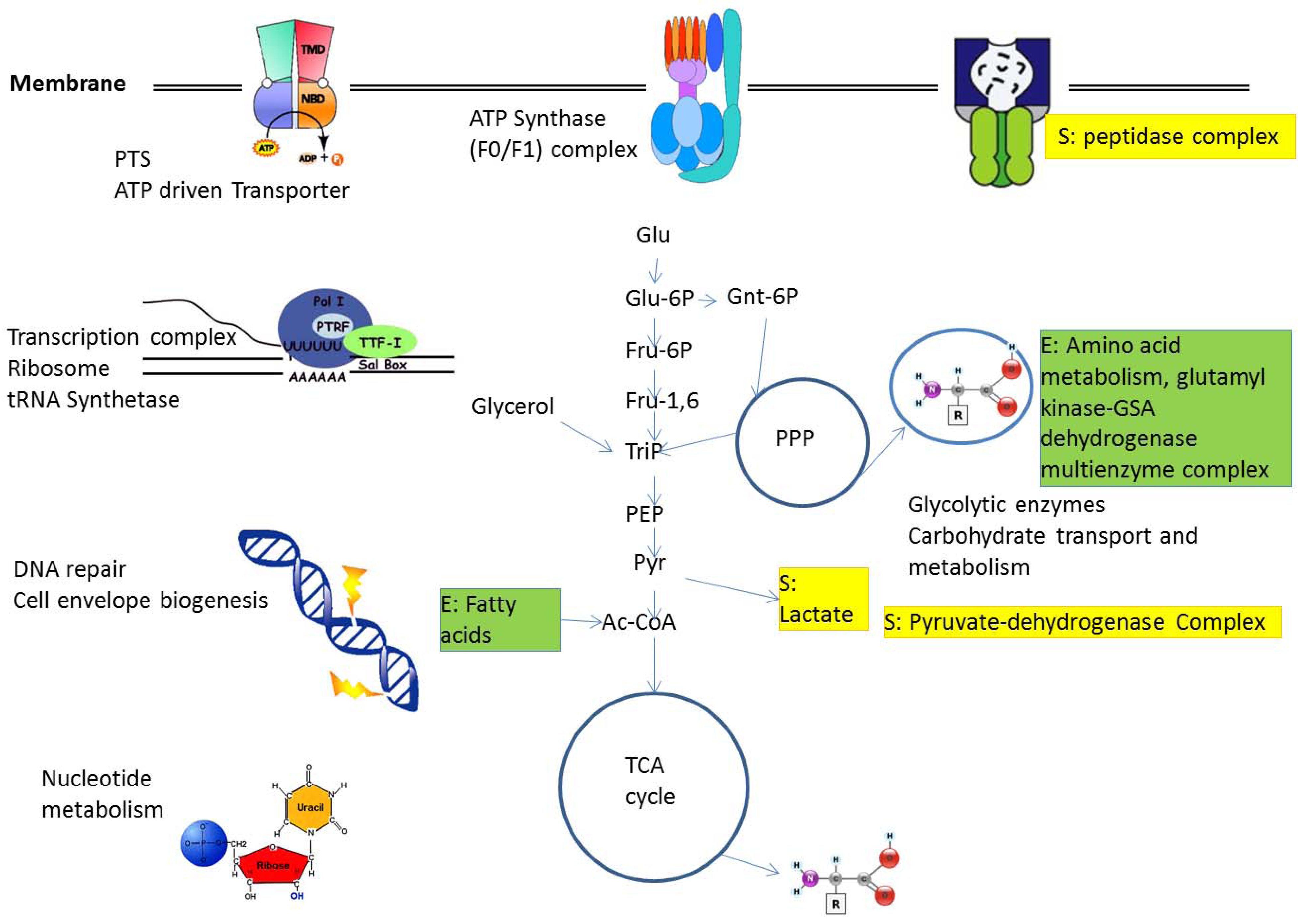
2.2. Metabolic Adaptation and Protein Complexes
2.2.1. Metabolic Adaptation in Intracellular Model Pathogens
2.2.2. Regulatory Strategies and Prokaryotic Protein Complexes
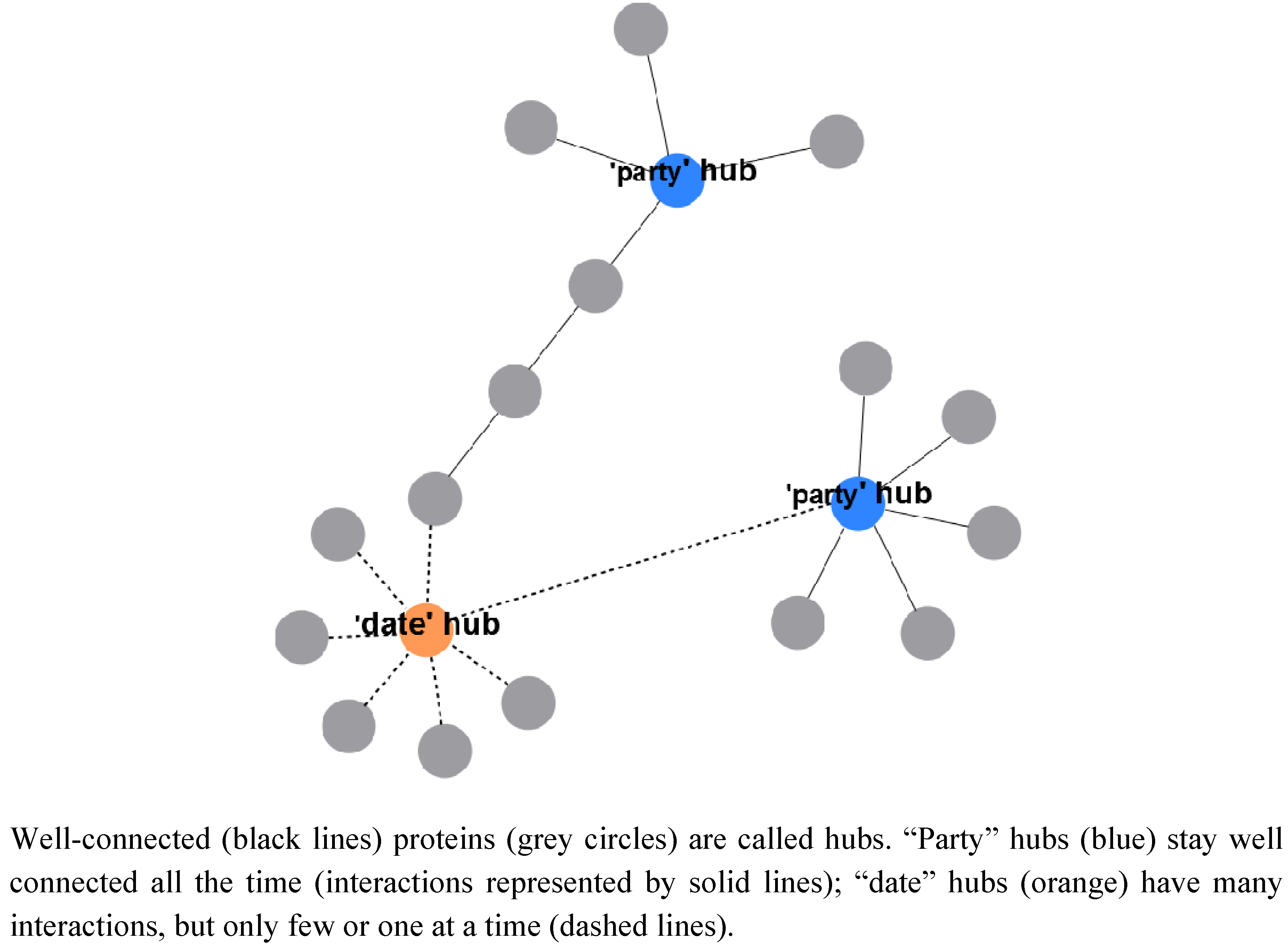
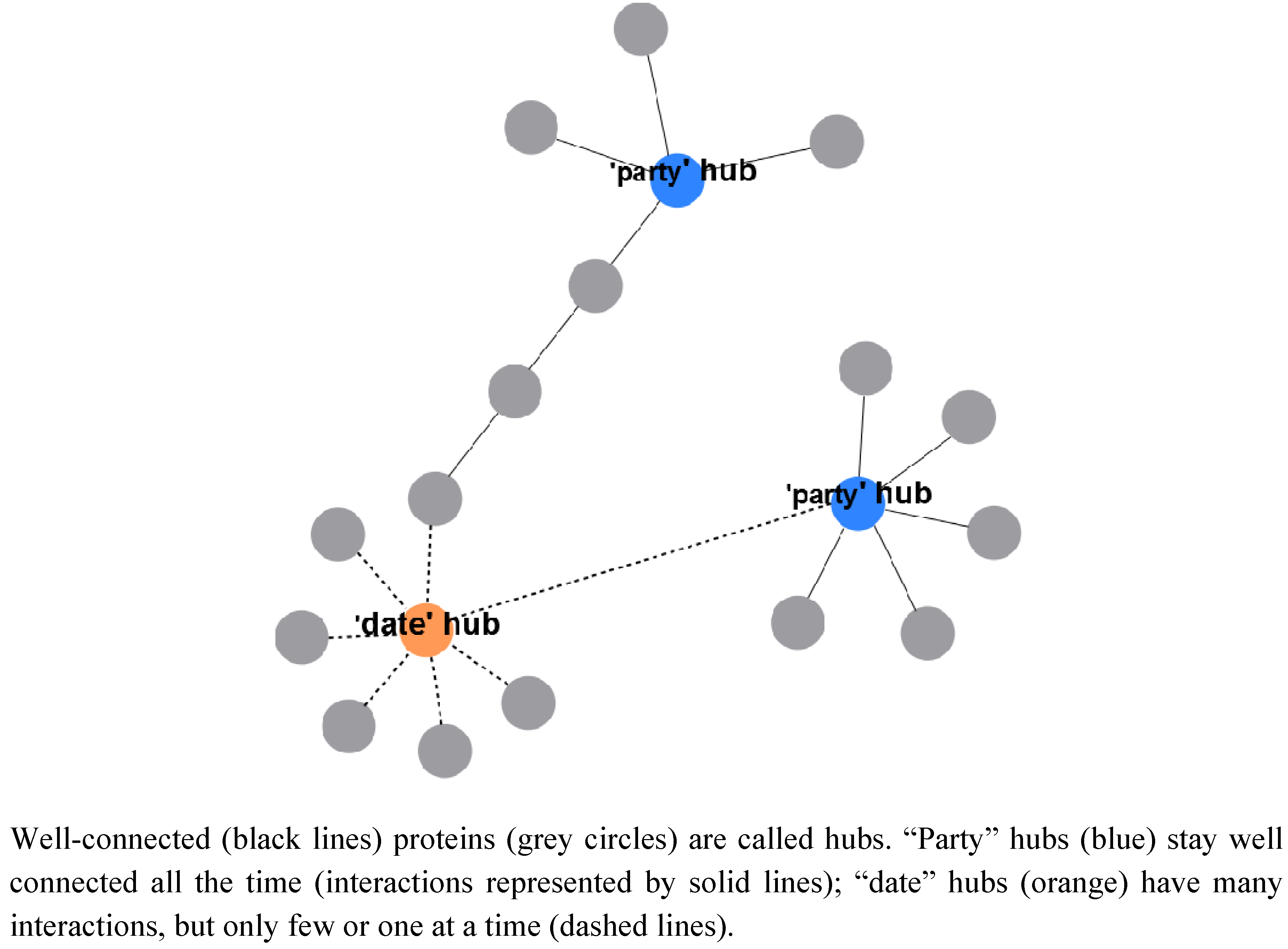
2.2.3. A System-Wide, Global View on Prokaryotic Protein Complexes
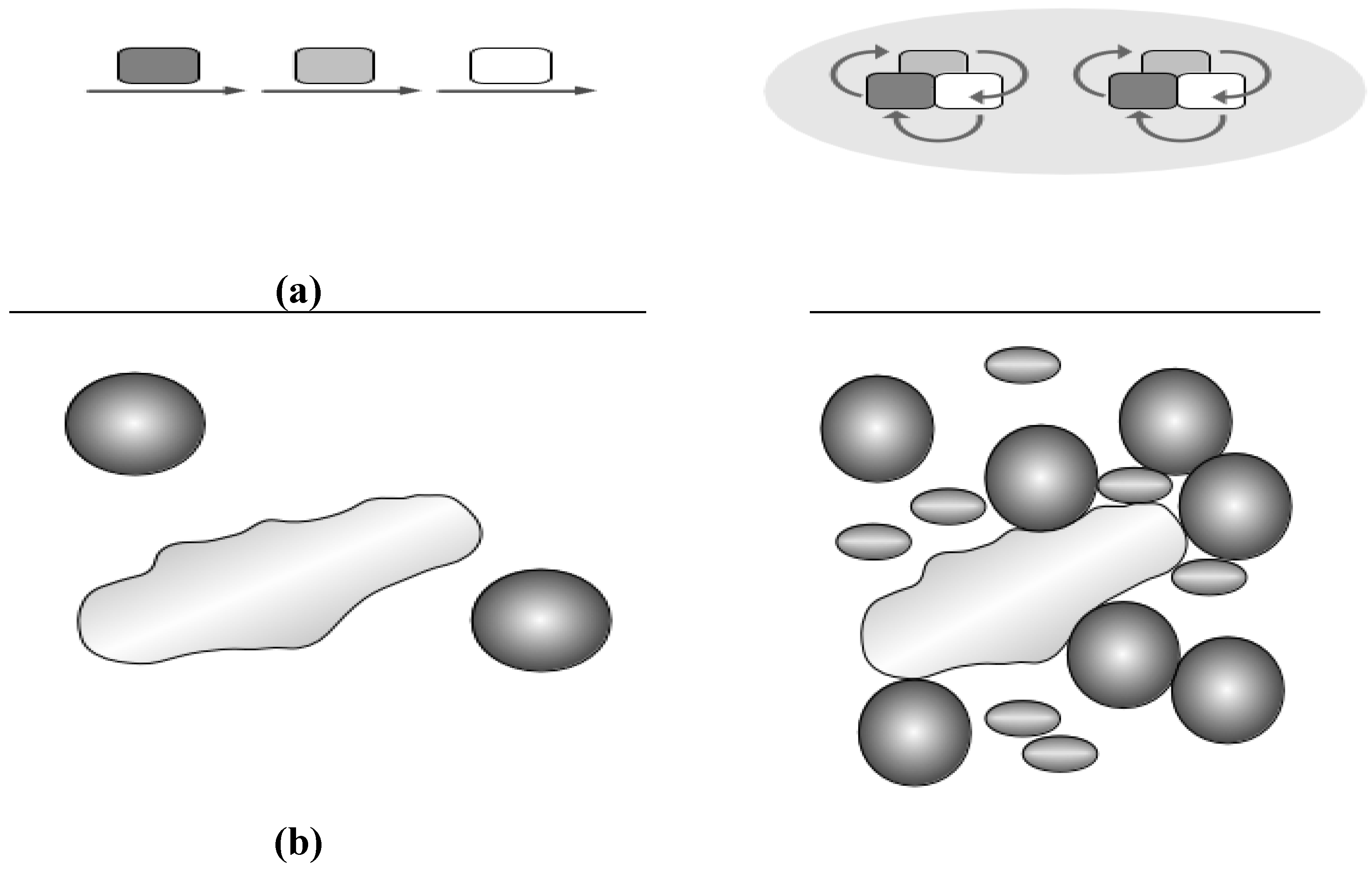
2.3. Crowding and Substrate Channeling for Metabolic Complexes
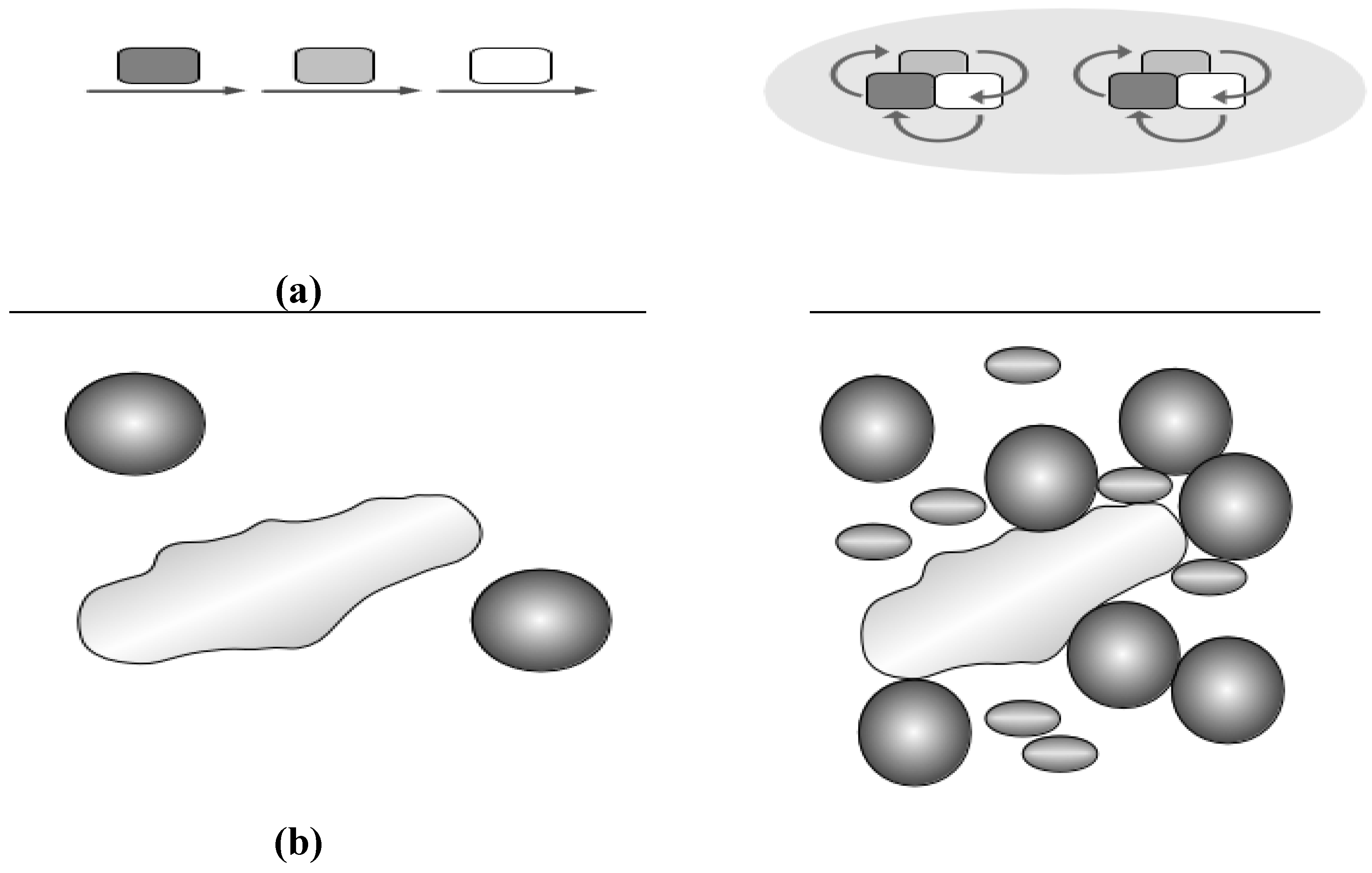
3. Conclusions
Acknowledgments
Conflict of Interest
References
- Eisenreich, W.; Dandekar, T.; Heesemann, J.; Goebel, W. Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat. Rev. Microbiol. 2010, 8, 401–412. [Google Scholar] [CrossRef]
- Klitgord, N.; Segrè, D. Environments that induce synthetic microbial ecosystems. PLoS Comput. Biol. 2010, 18, 6. [Google Scholar]
- Güell, M.; van Noort, V.; Yus, E.; Chen, W.H.; Leigh-Bell, J.; Michalodimitrakis, K.; Yamada, T.; Arumugam, M.; Doerks, T.; Kühner, S.; et al. Transcriptome complexity in a genome-reduced bacterium. Science 2009, 27, 1268–1271. [Google Scholar]
- Kühner, S.; van Noort, V.; Betts, M.J.; Leo-Macias, A.; Batisse, C.; Rode, M.; Yamada, T.; Maier, T.; Bader, S.; Beltran-Alvarez, P.; et al. Proteome organization in a genome-reduced bacterium. Science 2009, 27, 1235–1240. [Google Scholar]
- Yus, E.; Maier, T.; Michalodimitrakis, K.; van Noort, V.; Yamada, T.; Chen, W.H.; Wodke, J.A.; Güell, M.; Martínez, S.; Bourgeois, R.; et al. Impact of genome reduction on bacterial metabolism and its regulation. Science 2009, 326, 1263–1268. [Google Scholar] [CrossRef]
- Jozefczuk, S.; Klie, S.; Catchpole, G.; Szymanski, J.; Cuadros-Inostroza, A.; Steinhauser, D.; Selbig, J.; Willmitzer, L. Metabolomic and transcriptomic stress response of Escherichia coli. Mol. Syst. Biol. 2010, 6, 364. [Google Scholar]
- Kotte, O.; Zaugg, J.B.; Heinemann, M. Bacterial adaptation through distributed sensing of metabolic fluxes. Mol. Syst. Biol. 2010, 6, 355. [Google Scholar]
- Parrish, J.R.; Yu, J.; Liu, G.; Hines, J.A.; Chan, J.E.; Mangiola, B.A.; Zhang, H.; Pacifico, S.; Fotouhi, F.; DiRita, V.J.; Ideker, T.; Andrews, P.; Finley, R.L., Jr. A proteome-wide protein interaction map for Campylobacter jejuni. Genome Biol. 2007, 8, R130. [Google Scholar] [CrossRef]
- Commichau, F.M.; Stülke, J. Trigger enzymes: bifunctional proteins active in metabolism and in controlling gene expression. Mol. Microbiol. 2008, 67, 692–702. [Google Scholar] [CrossRef]
- Han, J.D.; Bertin, N.; Hao, T.; Goldberg, D.S.; Berriz, G.F.; Zhang, L.V.; Dupuy, D.; Walhout, A.J.; Cusick, M.E.; Roth, F.P.; et al. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature 2004, 430, 88–93. [Google Scholar] [CrossRef]
- Krause, R.; von Mering, C.; Bork, P.; Dandekar, T. Shared components of protein complexes—Versatile building blocks or biochemical artefacts? BioEssays 2004, 26, 1333–1343. [Google Scholar] [CrossRef]
- Jeong, H.; Tombor, B.; Albert, R.; Oltvai, Z.N.; Barabási, A.L. The large-scale organization of metabolic networks. Nature 2000, 407, 651–654. [Google Scholar] [CrossRef]
- Arita, M. The metabolic world of Escherichia coli is not small. Proc. Natl. Acad. Sci. USA 2004, 101, 1543–1547. [Google Scholar] [CrossRef]
- Schmidt, S.; Sunyaev, S.; Bork, P.; Dandekar, T. Metabolites: a helping hand for pathway evolution? Trends Biochem. Sci. 2003, 28, 336–341. [Google Scholar] [CrossRef]
- Ma, H.W.; Zeng, A.P. The connectivity structure; Giant strong component and centrality of metabolic networks. Bioinformatics 2003, 19, 1423–1430. [Google Scholar] [CrossRef]
- Lander, A.D. The edges of understanding. BMC Biol. 2010, 8, 40. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Slotine, J.J.; Barabási, A.L. Controllability of complex networks. Nature 2011, 473, 167–173. [Google Scholar]
- Butland, G.; Peregrín-Alvarez, J.M.; Li, J.; Yang, W.; Yang, X.; Canadien, V.; Starostine, A.; Richards, D.; Beattie, B.; Krogan, N.; et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 2005, 3, 531–537. [Google Scholar]
- Mäder, U.; Schmeisky, A.G.; Flórez, L.A.; Stülke, J. SubtiWiki—a comprehensive community resource for the model organism Bacillus subtilis. Nucleic Acids Res. 2012, 40, D1278–D1287. [Google Scholar] [CrossRef]
- Kelder, T.; van Iersel, M.P.; Hanspers, K.; Kutmon, M.; Conklin, B.R.; Evelo, C.; Pico, A.R. WikiPathways: Building research communities on biological pathways. Nucleic Acids Res. 2012, 40, D1301–D1307. [Google Scholar] [CrossRef]
- Lammers, C.R.; Flórez, L.A.; Schmeisky, A.G.; Roppel, S.F.; Mäder, U.; Hamoen, L.; Stülke, J. Connecting parts with processes: SubtiWiki and SubtiPathways integrate gene and pathway annotation for Bacillus subtilis. Microbiology 2010, 156, 849–859. [Google Scholar] [CrossRef] [Green Version]
- Van Noort, V.; Seebacher, J.; Bader, S.; Mohammed, S.; Vonkova, I.; Betts, M.J.; Kühner, S.; Kumar, R.; Maier, T.; O'Flaherty, M.; et al. Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol. Syst. Biol. 2012, 8, 571. [Google Scholar]
- Jers, C.; Pedersen, M.M.; Paspaliari, D.K.; Schütz, W.; Johnsson, C.; Soufi, B.; Macek, B.; Jensen, P.R.; Mijakovic, I. Bacillus subtilis BY-kinase PtkA controls enzyme activity and localization of its protein substrates. Mol. Microbiol. 2010, 77, 287–299. [Google Scholar] [CrossRef]
- Liebeke, M.; Meyer, H.; Donat, S.; Ohlsen, K.; Lalk, M. A metabolomic view of Staphylococcus aureus and its ser/thr kinase and phosphatase deletion mutants: Involvement in cell wall biosynthesis. Chem. Biol. 2010, 17, 820–830. [Google Scholar] [CrossRef]
- Friedman, D.B.; Stauff, D.L.; Pishchany, G.; Whitwell, C.W.; Torres, V.J.; Skaar, E.P. Staphylococcus aureus redirects central metabolism to increase iron availability. PLoS Pathogens 2006, 2, e87. [Google Scholar]
- Stülke, J.; Hillen, W. Regulation of carbon catabolism in Bacillus species. Annu. Rev. Microbiol. 2000, 54, 849–880. [Google Scholar] [CrossRef]
- Eisenreich, W.; Slaghuis, J.; Laupitz, R.; Bussemer, J.; Stritzker, J.; Schwarz, C.; Schwarz, R.; Dandekar, T.; Goebel, W.; Bacher, A. 13C isotopologue perturbation studies of Listeria monocytogenes carbon metabolism and its modulation by the virulence regulator PrfA. Proc. Natl. Acad. Sci. USA 2006, 103, 2040–2045. [Google Scholar]
- Eylert, E.; Schär, J.; Mertins, S.; Stoll, R.; Bacher, A.; Goebel, W.; Eisenreich, W. Carbon metabolism of Listeria monocytogenes growing inside macrophages. Mol. Microbiol. 2008, 69, 1008–1017. [Google Scholar] [CrossRef]
- Tchawa, Y.M.; Leatham, M.P.; Allen, J.H.; Laux, D.C.; Conway, T.; Cohen, P.S. Role of gluconeogenesis and the tricarboxylic acid cycle in the virulence of Salmonella enterica serovar Typhimurium in BALB/c mice. Infect. Immun. 2006, 74, 1130–1140. [Google Scholar] [CrossRef]
- Maier, T.; Schmidt, A.; Güell, M.; Kühner, S.; Gavin, A.C.; Aebersold, R.; Serrano, L. Quantification of mRNA and protein and integration with protein turnover in a bacterium. Mol. Syst. Biol. 2011, 7, 511. [Google Scholar]
- Stelling, J.; Klamt, S.; Bettenbrock, K.; Schuster, S.; Gilles, E.D. Metabolic network structure determines key aspects of functionality and regulation. Nature 2002, 420, 190–193. [Google Scholar] [CrossRef]
- Kaleta, C.; de Figueiredo, L.F.; Schuster, S. Can the whole be less than the sum of its parts? Pathway analysis in genome-scale metabolic networks using elementary flux patterns. Genome Res. 2009, 19, 1872–1883. [Google Scholar] [CrossRef]
- Wessely, F.; Bartl, M.; Guthke, R.; Li, P.; Schuster, S.; Kaleta, C. Optimal regulatory strategies for metabolic pathways in Escherichia coli depending on protein costs. Mol. Syst. Biol. 2011, 7, 515. [Google Scholar]
- Pagels, M.; Fuchs, S.; Pané-Farré, J.; Kohler, C.; Menschner, L.; Heckerm, M.; McNamarram, P.J.; Bauer, M.C.; von Wachenfeldt, C.; Liebeke, M.; et al. Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol. Microbiol. 2010, 76, 1142–1161. [Google Scholar] [CrossRef]
- Lee, T.I.; Rinaldi, N.J.; Robert, F.; Odom, D.T.; Bar-Joseph, Z.; Gerber, G.K.; Hannett, N.M.; Harbison, C.T.; Thompson, C.M.; Simon, I.; et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 2002, 298, 799–804. [Google Scholar]
- Zaman, S.; Lippman, S.I.; Zhao, X.; Broach, J.R. How Saccharomyces responds to nutrients. Annu. Rev. Genet. 2008, 42, 27. [Google Scholar] [CrossRef]
- Tuller, T.; Carmi, A.; Vestsigian, K.; Navon, S.; Dorfan, Y.; Zaborske, J.; Pan, T.; Dahan, O.; Furman, I.; Pilpel, Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 2010, 16, 344–354. [Google Scholar]
- Mitchell, A.; Romano, G.H.; Groisman, B.; Yona, A.; Dekel, E.; Kupiec, M.; Dahan, O.; Pilpel, Y. Adaptive prediction of environmental changes by microorganisms. Nature 2009, 460, 220. [Google Scholar]
- Ames, T.D.; Rodionov, D.A.; Weinberg, Z.; Breaker, R.R. A Eubacterial Riboswitch Class That Senses the Coenzyme Tetrahydrofolate. Chem. Biol. 2010, 17, 681–685. [Google Scholar] [CrossRef]
- Winkler, W.C.; Cohen-Chalamish, S.; Breaker, R.R. An mRNA structure that controls gene expression by binding FMN. Proc. Natl. Acad. Sci. USA 2002, 99, 15908–15913. [Google Scholar] [CrossRef]
- Liang, C.; Liebeke, M.; Schwarz, R.; Zühlke, D.; Fuchs, S.; Menschner, L.; Engelmann, S.; Wolz, C.; Jaglitz, S.; Bernhardt, J.; et al. Staphylococcus aureus physiological growth limitations: Insights from flux calculations built on proteomics and external metabolite data. Proteomics 2011, 11, 1915–1935. [Google Scholar] [CrossRef]
- Van der Pol, B. On “relaxation-oscillations”. The London, Edinburgh, and Dublin Philosophical Magazin and Journal of Science Series 7 1927, 2, 978–992. [Google Scholar]
- Zhang, Y.H. Substrate channeling and enzyme complexes for biotechnological applications. Biotechnol. Adv. 2011, 29, 715–725. [Google Scholar] [CrossRef]
- Cori, C.F.; Velick, S.F.; Cori, G.T. The combination of diphosphopyridine nucleotide with glyceraldehyde phosphate dehydrogenase. Biochim. Biophys. Acta 1950, 4, 160–169. [Google Scholar] [CrossRef]
- Srere, P.A.; Sumegi, B.; Sherry, A.D. Organizational aspects of the citric acid cycle. Biochem. Soc. Symp. 1987, 54, 173–178. [Google Scholar]
- Tombes, R.M.; Shapiro, B.M. Metabolite channeling: a phosphorylcreatine shuttle to mediate high energy phosphate transport between sperm mitochondrion and tail. Cell 1985, 41, 325–334. [Google Scholar] [CrossRef]
- Yang, S.Y.; Cuebas, D.; Schulz, H. Channeling of 3-hydroxy-4-trans-decenoyl coenzyme A on the bifunctional beta-oxidation enzyme from rat liver peroxisomes and on the large subunit of the fatty acid oxidation complex from Escherichia coli. J. Biol. Chem. 1986, 261, 15390–15395. [Google Scholar]
- Kholodenko, B.N.; Westerhoff, H.V.; Cascante, M. Effect of channelling on the concentration of bulk-phase intermediates as cytosolic proteins become more concentrated. Biochem. J. 1996, 313, 921–926. [Google Scholar]
- Miziorko, H.M.; Laib, F.E.; Behnke, C.E. Evidence for substrate channeling in the early steps of cholesterogenesis. J. Biol. Chem. 1990, 265, 9606–9609. [Google Scholar]
- Welch, G.R.; Easterby, J.S. Metabolic channeling versus free diffusion: Transition-time analysis. Trends Biochem. Sci. 1994, 19, 193–197. [Google Scholar] [CrossRef]
- Mendes, P.; Kell, D.B.; Westerhoff, H.V. Channelling can decrease pool size. Eur. J. Biochem. 1992, 204, 257–266. [Google Scholar] [CrossRef]
- Peuhkurinen, K.J.; Nuutinen, E.M.; Pietiläinen, E.P.; Hiltunen, J.K.; Hassinen, I.E. Role of pyruvate carboxylation in the energy-linked regulation of pool sizes of tricarboxylic acid-cycle intermediates in the myocardium. Biochem J. 1982, 208, 577–581. [Google Scholar]
- Bauler, P.; Huber, G.; Leyh, T.; McCammon, J.A. Channeling by Proximity: The Catalytic Advantages of Active Site Colocalization Using Brownian Dynamics. J. Phys. Chem. Lett. 2010, 1, 1332–1335. [Google Scholar] [CrossRef]
- McLysaght, A.; Conant, G.C. Patterns of indirect protein interactions suggest a spatial organization to metabolism. Mol. Biosyst. 2011, 7, 3056–3064. [Google Scholar] [CrossRef]
- Buch, A.; Archana, G.; Naresh, K.G. Metabolic channeling of glucose towards gluconate in phosphate-solubilizing Pseudomonas aeruginosa P4 under phosphorus deficiency. Res. Microbiol. 2008, 159, 635–642. [Google Scholar] [CrossRef]
- Huthmacher, C.; Gille, C.; Holzhütter, H.G. A computational analysis of protein interactions in metabolic networks reveals novel enzyme pairs potentially involved in metabolic channeling. J. Theor. Biol. 2008, 252, 456–464. [Google Scholar] [CrossRef]
- Miyoshi, D.; Sugimoto, N. Molecular crowding effects on structure and stability of DNA. Biochemie 2008, 90, 1040–1051. [Google Scholar] [CrossRef]
- Richter, K.; Nessling, M.; Lichter, P. Macromolecular crowding and its potential impact on nuclear function. Biochim. Biophys. Acta 2008, 1783, 2100–2107. [Google Scholar]
- Wang, S.; Tate, M.W.; Gruner, S.M. Protein crowding impedes pressure-induced unfolding of staphylococcal nuclease. Biochim. Biophys. Acta 2012, 1820, 957–961. [Google Scholar]
- Sanfelice, D.; Adrover, M.; Martorell, G.; Pastore, A.; Temussi, P.A. Crowding versus molecular seeding: NMR studies of protein aggregation in hen egg white. J. Phys. Condens. Matter 2012, 24, 244107. [Google Scholar] [CrossRef]
- McGuffee, S.R.; Elcock, A.H. Diffusion, crowding and protein stability in a dynamic molecular model of the bacterial cytoplasm. PLoS Comput. Biol. 2010, 6, e1000694. [Google Scholar] [CrossRef]
- Morelli, M.J.; Allen, R.J.; Wolde, P.R. Effects of macromolecular crowding on genetic networks. Biophys. J. 2011, 101, 2882–2891. [Google Scholar] [CrossRef]
- Norris, M.G.; Malys, N. What is the true enzyme kinetics in the biological system? An investigation of macromolecular crowding effect upon enzyme kinetics of glucose-6-phosphate dehydrogenase. Biochem. Biophys. Res. Commun. 2011, 405, 388–392. [Google Scholar] [CrossRef]
- Echeverria, C.; Kapral, R. Molecular crowding and protein enzymatic dynamics. Phys. Chem. Chem. Phys. 2012, 4, 6755–6763. [Google Scholar] [CrossRef]
- Dong, H.; Qin, S.; Zhou, H.X. Effects of macromolecular crowding on protein conformational changes. PLoS Comput. Biol. 2010, 6, e1000833. [Google Scholar] [CrossRef]
- Ando, T.; Skolnick, J. Crowding and hydrodynamic interactions likely dominate in vivo macromolecular motion. Proc. Natl. Acad. Sci. USA 2010, 107, 18457–18462. [Google Scholar] [CrossRef]
- Kim, Y.C.; Best, R.B.; Mittal, J. Macromolecular crowding effects on protein-protein binding affinity and specificity. J. Chem. Phys. 2010, 133, 205101. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krüger, B.; Liang, C.; Prell, F.; Fieselmann, A.; Moya, A.; Schuster, S.; Völker, U.; Dandekar, T. Metabolic Adaptation and Protein Complexes in Prokaryotes. Metabolites 2012, 2, 940-958. https://doi.org/10.3390/metabo2040940
Krüger B, Liang C, Prell F, Fieselmann A, Moya A, Schuster S, Völker U, Dandekar T. Metabolic Adaptation and Protein Complexes in Prokaryotes. Metabolites. 2012; 2(4):940-958. https://doi.org/10.3390/metabo2040940
Chicago/Turabian StyleKrüger, Beate, Chunguang Liang, Florian Prell, Astrid Fieselmann, Andres Moya, Stefan Schuster, Uwe Völker, and Thomas Dandekar. 2012. "Metabolic Adaptation and Protein Complexes in Prokaryotes" Metabolites 2, no. 4: 940-958. https://doi.org/10.3390/metabo2040940
APA StyleKrüger, B., Liang, C., Prell, F., Fieselmann, A., Moya, A., Schuster, S., Völker, U., & Dandekar, T. (2012). Metabolic Adaptation and Protein Complexes in Prokaryotes. Metabolites, 2(4), 940-958. https://doi.org/10.3390/metabo2040940