

Multiple Reaction Monitoring (MRM)-Based Targeted Kidney Metabolite Profiling of a Mouse Model of Hyperuricemia

 ,
,
Abstract
1. Introduction
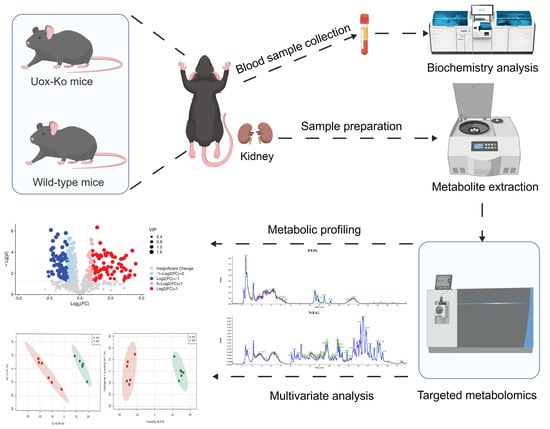
2. Materials and Methods
2.1. Bioethics, Experimental Design, and Sample Collection
2.2. Biochemistry Analysis of the Serum
2.3. Histopathological Analysis of the Kidneys
2.4. MRM Targeted Metabolomic Analysis
2.4.1. Chemicals and Reagents
2.4.2. Sample Preparation for Metabolite Profiling
2.4.3. LC-MS/MS Analysis
2.4.4. Data Processing for Metabolomic Analysis
2.5. Statistical Analysis
3. Results
3.1. Biochemistry and Histopathological Analysis
3.2. Evaluation of the Reproducibility of the Metabolomic Analysis Method
3.3. Multivariate Analysis
3.4. Metabolomic Profiling from Kidney Tissues
3.5. Pathway Enrichment Analysis of the Differential Metabolites
3.6. Kidney Potential Metabolic Biomarkers for CUN
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| UA | uric acid |
| HUA | hyperuricemia |
| CKD | chronic kidney disease |
| DKD | diabetes kidney disease |
| AKI | acute kidney injury |
| MRM | multiple reaction monitoring |
| IgAN | immunoglobulin A nephropathy |
| UN | urate nephropathy |
| CUN | chronic urate nephropathy |
| KO | knockout |
| Uox | urate oxidase |
| WT | wild type |
| PCR | polymerase chain reaction |
| CRE | creatinine |
| BUN | urea nitrogen |
| GLU | glucose |
| TG | triglycerides |
| TC | total cholesterol |
| ALT | alanine aminotransferase |
| AST | aspertate aminotransferase |
| HDL | high-density lipoprotein |
| LDL | low-density lipoprotein |
| ALB | albumin |
| H & E | Hematoxylin-eosin |
| MT | Masson’s trichrome |
| TIS | tubular injury score |
| LC-MS | liquid chromatography-mass spectrometry |
| QC | quality control |
| UPLC | ultraperformance liquid chromatography |
| ESI | electrospray ionization |
| RT | retention time |
| PCA | principle component analysis |
| PLS-DA | partial least squares discriminant analysis |
| VIP | variable important for the projection |
| cAMP | 3′, 5′-cyclic AMP |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| ROC | receiver operating characteristic |
| AUC | area under the curve |
| CV | coefficient of variation |
| FA | fatty acids |
| LPE | lysophosphatidylethanolamines |
| TRP | Tryptophan |
| 5-HIAA | 5-OH-indoleacetate |
| EET | epoxyeicosatrienoic acids |
| DHET | dihydroxyeicosatrienoic acids |
| TCA | tricarboxylic acid |
| IL | interleukin |
| TNF | tumor necrosis factor |
| 5-HT | 5-hydroxytryptamine |
| AA | arachidonic acid |
| AAM | arachidonic acid metabolism |
| EMT | epithelial–mesenchymal transition |
| sEH | soluble epoxide hydrolase |
References
- Ramazzina, I.; Folli, C.; Secchi, A.; Berni, R.; Percudani, R. Completing the uric acid degradation pathway through phylogenetic comparison of whole genomes. Nat. Chem. Biol. 2006, 2, 144–148. [Google Scholar] [CrossRef]
- Keebaugh, A.C.; Thomas, J.W. The Evolutionary Fate of the Genes Encoding the Purine Catabolic Enzymes in Hominoids, Birds, and Reptiles. Mol. Biol. Evol. 2010, 27, 1359–1369. [Google Scholar] [CrossRef]
- Liu, R.; Han, C.; Wu, D.; Xia, X.H.; Gu, J.Q.; Guan, H.X.; Shan, Z.Y.; Teng, W.P. Prevalence of Hyperuricemia and Gout in Mainland China from 2000 to 2014: A Systematic Review and Meta-Analysis. BioMed Res. Int. 2015, 2015, 762820. [Google Scholar] [CrossRef]
- Chen-Xu, M.; Yokose, C.; Rai, S.K.; Pillinger, M.H.; Choi, H.K. Contemporary Prevalence of Gout and Hyperuricemia in the United States and Decadal Trends: The National Health and Nutrition Examination Survey, 2007–2016. Arthritis Rheumatol. 2019, 71, 991–999. [Google Scholar] [CrossRef]
- Kuo, C.F.; Grainge, M.J.; Zhang, W.; Doherty, M. Global epidemiology of gout: Prevalence, incidence and risk factors. Nat. Rev. Rheumatol. 2015, 11, 649–662. [Google Scholar] [CrossRef]
- Dalbeth, N.; Merriman, T.R.; Stamp, L.K. Gout. Lancet 2016, 388, 2039–2052. [Google Scholar] [CrossRef]
- Richette, P.; Perez-Ruiz, F.; Doherty, M.; Jansen, T.L.; Nuki, G.; Pascual, E.; Punzi, L.; So, A.K.; Bardin, T. Improving cardiovascular and renal outcomes in gout: What should we target? Nat. Rev. Rheumatol. 2014, 10, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.; Garcia-Arroyo, F.; Sasai, F.; Rodriguez-Iturbe, B.; Sanchez-Lozada, L.G.; Lanaspa, M.A.; Johnson, R.J. Mini Review: Reappraisal of Uric Acid in Chronic Kidney Disease. Am. J. Nephrol. 2021, 52, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Goicoechea, M.; de Vinuesa, S.G.; Verdalles, U.; Ruiz-Caro, C.; Ampuero, J.; Rincon, A.; Arroyo, D.; Luno, J. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin. J. Am. Soc. Nephrol. 2010, 5, 1388–1393. [Google Scholar] [CrossRef]
- Goicoechea, M.; Garcia de Vinuesa, S.; Verdalles, U.; Verde, E.; Macias, N.; Santos, A.; Perez de Jose, A.; Cedeno, S.; Linares, T.; Luno, J. Allopurinol and progression of CKD and cardiovascular events: Long-term follow-up of a randomized clinical trial. Am. J. Kidney Dis. 2015, 65, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Bartakova, V.; Kuricova, K.; Pacal, L.; Nova, Z.; Dvorakova, V.; Svrckova, M.; Maluskova, D.; Svobodova, I.; Rehorova, J.; Svojanovsky, J.; et al. Hyperuricemia contributes to the faster progression of diabetic kidney disease in type 2 diabetes mellitus. J. Diabetes. Complicat. 2016, 30, 1300–1307. [Google Scholar] [CrossRef]
- Xu, X.; Hu, J.; Song, N.; Chen, R.; Zhang, T.; Ding, X. Hyperuricemia increases the risk of acute kidney injury: A systematic review and meta-analysis. BMC Nephrol. 2017, 18, 27. [Google Scholar] [CrossRef]
- Patel, C.; Wilson, C.P.; Ahmed, N.; Hattab, Y. Acute Uric Acid Nephropathy following Epileptic Seizures: Case Report and Review. Case Rep. Nephrol. 2019, 2019, 4890287. [Google Scholar] [CrossRef]
- Hsu, Y.-H. Chronic urate nephropathy. Incont. Pelvic. Floor. Dysfunct. 2012, 6, 89. [Google Scholar]
- Wiederkehr, M.R.; Moe, O.W. Uric Acid Nephrolithiasis: A Systemic Metabolic Disorder. Clin. Rev. Bone Miner. Metab. 2011, 9, 207–217. [Google Scholar] [CrossRef]
- Fathallah-Shaykh, S.A.; Cramer, M.T. Uric acid and the kidney. Pediatr. Nephrol. 2014, 29, 999–1008. [Google Scholar] [CrossRef]
- Shekarriz, B.; Stoller, M.L. Uric acid nephrolithiasis: Current concepts and controversies. J. Urol. 2002, 168, 1307–1314. [Google Scholar] [CrossRef]
- Christiansen, C.; Maus, D.; Hoppenz, E.; Murillo-Leon, M.; Hoffmann, T.; Scholz, J.; Melerowicz, F.; Steinfeldt, T.; Seeber, F.; Blume, M. In vitro maturation of Toxoplasma gondii bradyzoites in human myotubes and their metabolomic characterization. Nat. Commun. 2022, 13, 1168. [Google Scholar] [CrossRef]
- Holbrook-Smith, D.; Durot, S.; Sauer, U. High-throughput metabolomics predicts drug-target relationships for eukaryotic proteins. Mol. Syst. Biol. 2022, 18, e10767. [Google Scholar] [CrossRef]
- Li, H.; Zhang, H.; Yan, F.; He, Y.; Ji, A.; Liu, Z.; Li, M.; Ji, X.; Li, C. Kidney and plasma metabolomics provide insights into the molecular mechanisms of urate nephropathy in a mouse model of hyperuricemia. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166374. [Google Scholar] [CrossRef]
- Missawi, O.; Venditti, M.; Cappello, T.; Zitouni, N.; Marco, G.; Boughattas, I.; Bousserrhine, N.; Belbekhouche, S.; Minucci, S.; Maisano, M.; et al. Autophagic event and metabolomic disorders unveil cellular toxicity of environmental microplastics on marine polychaete Hediste diversicolor. Environ. Pollut. 2022, 302, 119106. [Google Scholar] [CrossRef] [PubMed]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted Metabolomics Strategies-Challenges and Emerging Directions. J. Am. Soc. Mass Spectr. 2016, 27, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Gertsman, I.; Barshop, B.A. Promises and pitfalls of untargeted metabolomics. J. Inherit. Metab. Dis. 2018, 41, 355–366. [Google Scholar] [CrossRef]
- Cai, Y.P.; Weng, K.; Guo, Y.; Peng, J.; Zhu, Z.J. An integrated targeted metabolomic platform for high-throughput metabolite profiling and automated data processing. Metabolomics 2015, 11, 1575–1586. [Google Scholar] [CrossRef]
- Xie, Z.E.; Ferreira, C.R.; Virequ, A.A.; Cooks, R.G. Multiple reaction monitoring profiling (MRM profiling): Small molecule exploratory analysis guided by chemical functionality. Chem. Phys. Lipids 2021, 235, 105048. [Google Scholar] [CrossRef]
- Xia, J.F.; Liang, Q.L.; Liang, X.P.; Wang, Y.M.; Hu, P.; Li, P.; Luo, G.A. Ultraviolet and tandem mass spectrometry for simultaneous quantification of 21 pivotal metabolites in plasma from patients with diabetic nephropathy. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 1930–1936. [Google Scholar] [CrossRef]
- Sas, K.M.; Nair, V.; Byun, J.; Kayampilly, P.; Zhang, H.; Saha, J.; Brosius, F.C.; Kretzler, M.; Pennathur, S. Targeted lipidomic and transcriptomic analysis identifies dysregulated renal ceramide metabolism in a mouse model of diabetic kidney disease. J. Proteom. Bioinform. 2015, s 14, 002. [Google Scholar]
- Ademowo, O.S.; Sharma, P.; Cockwell, P.; Reis, A.; Chapple, I.L.; Griffiths, H.R.; Dias, I.H.K. Distribution of plasma oxidised phosphatidylcholines in chronic kidney disease and periodontitis as a co-morbidity. Free Radic. Biol. Med. 2020, 146, 130–138. [Google Scholar] [CrossRef]
- Andre, C.; Bennis, Y.; Titeca-Beauport, D.; Caillard, P.; Cluet, Y.; Kamel, S.; Choukroun, G.; Maizel, J.; Liabeuf, S.; Bodeau, S. Two rapid, accurate liquid chromatography tandem mass spectrometry methods for the quantification of seven uremic toxins: An application for describing their accumulation kinetic profile in a context of acute kidney injury. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1152, 122234. [Google Scholar] [CrossRef]
- Deng, Y.; Wu, Q.; Chen, W.; Zhu, L.; Liu, W.; Xia, F.; Sun, L.; Lin, X.; Zeng, R. Lipidomics reveals association of circulating lipids with body mass index and outcomes in IgA nephropathy patients. J. Mol. Cell Biol. 2021, 13, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Gao, Z.; Chen, G.; Peng, C.; Sun, Y.; Jiang, B.; Zhou, H.; Cheng, Y.; Hu, F.; Zhang, Q. An integrative proteomics metabolomics based strategy reveals the mechanisms involved in wasp sting induced acute kidney injury. Toxicon 2022, 205, 1–10. [Google Scholar] [CrossRef]
- Lu, J.; Hou, X.; Yuan, X.; Cui, L.L.; Liu, Z.; Li, X.D.; Ma, L.D.; Cheng, X.Y.; Xin, Y.; Wang, C.; et al. Knockout of the urate oxidase gene provides a stable mouse model of hyperuricemia associated with metabolic disorders. Kidney Int. 2018, 93, 69–80. [Google Scholar] [CrossRef]
- Wang, R.C.; Wu, G.L.; Dai, T.T.; Lang, Y.T.; Chi, Z.C.; Yang, S.L.; Dong, D.S. Naringin attenuates renal interstitial fibrosis by regulating the TGF-beta/Smad signaling pathway and inflammation. Exp. Ther. Med. 2021, 21, 66. [Google Scholar] [CrossRef]
- Wang, B.; Liu, D.; Zhu, Q.H.; Li, M.; Chen, H.; Guo, Y.; Fan, L.P.; Yue, L.S.; Li, L.Y.; Zhao, M. Rutin ameliorates kidney interstitial fibrosis in rats with obstructive nephropathy. Int. Immunopharmacol. 2016, 35, 77–84. [Google Scholar] [CrossRef]
- Liu, C.; Cai, T.; Cheng, Y.; Bai, J.; Li, M.; Gu, B.; Huang, M.; Fu, W. Postbiotics Prepared Using Lactobacillus reuteri Ameliorates Ethanol-Induced Liver Injury by Regulating the FXR/SHP/SREBP-1c Axis. Mol. Nutr. Food Res. 2024, 68, e2300927. [Google Scholar] [CrossRef]
- Zhou, X.; Ji, S.; Chen, L.; Liu, X.; Deng, Y.; You, Y.; Wang, M.; He, Q.; Peng, B.; Yang, Y.; et al. Gut microbiota dysbiosis in hyperuricaemia promotes renal injury through the activation of NLRP3 inflammasome. Microbiome 2024, 12, 109. [Google Scholar] [CrossRef]
- Page, T.; Coleman, M. Purine metabolism abnormalities in a hyperuricosuric subclass of autism. Biochim. Biophys. Acta 2000, 1500, 291–296. [Google Scholar] [CrossRef]
- Ansoleaga, B.; Jove, M.; Schluter, A.; Garcia-Esparcia, P.; Moreno, J.; Pujol, A.; Pamplona, R.; Portero-Otin, M.; Ferrer, I. Deregulation of purine metabolism in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 68–80. [Google Scholar] [CrossRef]
- Garcia-Esparcia, P.; Hernandez-Ortega, K.; Ansoleaga, B.; Carmona, M.; Ferrer, I. Purine metabolism gene deregulation in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2015, 41, 926–940. [Google Scholar] [CrossRef]
- Yu, M.; Cui, F.X.; Jia, H.M.; Zhou, C.; Yang, Y.; Zhang, H.W.; Ding, G.; Zou, Z.M. Aberrant purine metabolism in allergic asthma revealed by plasma metabolomics. J. Pharm. Biomed. 2016, 120, 181–189. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Duley, J.A.; Christodoulou, J. Inborn errors of purine metabolism: Clinical update and therapies. J. Inherit. Metab. Dis. 2014, 37, 669–686. [Google Scholar] [CrossRef] [PubMed]
- Levillain, O.; Hus-Citharel, A.; Garvi, S.; Peyrol, S.; Reymond, I.; Mutin, M.; Morel, F. Ornithine metabolism in male and female rat kidney: Mitochondrial expression of ornithine aminotransferase and arginase II. Am. J. Physiol. Renal Physiol. 2004, 286, F727–F738. [Google Scholar] [CrossRef]
- Shin, S.; Gombedza, F.C.; Bandyopadhyay, B.C. l-ornithine activates Ca(2+) signaling to exert its protective function on human proximal tubular cells. Cell Signal. 2020, 67, 109484. [Google Scholar] [CrossRef]
- Popolo, A.; Adesso, S.; Pinto, A.; Autore, G.; Marzocco, S. L-Arginine and its metabolites in kidney and cardiovascular disease. Amino Acids 2014, 46, 2271–2286. [Google Scholar] [CrossRef]
- Cherla, G.; Jaimes, E.A. Role of L-arginine in the pathogenesis and treatment of renal disease. J. Nutr. 2004, 134, 2801–2806. [Google Scholar] [CrossRef]
- Schramm, L.; La, M.; Heidbreder, E.; Hecker, M.; Beckman, J.S.; Lopau, K.; Zimmermann, J.; Rendl, J.; Reiners, C.; Winderl, S.; et al. L-arginine deficiency and supplementation in experimental acute renal failure and in human kidney transplantation. Kidney Int. 2002, 61, 1423–1432. [Google Scholar] [CrossRef]
- Schneider, R.; Raff, U.; Vornberger, N.; Schmidt, M.; Freund, R.; Reber, M.; Schramm, L.; Gambaryan, S.; Wanner, C.; Schmidt, H.H.; et al. L-Arginine counteracts nitric oxide deficiency and improves the recovery phase of ischemic acute renal failure in rats. Kidney Int. 2003, 64, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Han, P.; Ma, S.; Peng, R.; Wang, C.; Kong, W.; Cong, L.; Fu, J.; Zhang, Z.; Yu, H.; et al. Abnormal metabolism of gut microbiota reveals the possible molecular mechanism of nephropathy induced by hyperuricemia. Acta Pharm. Sin. B 2020, 10, 249–261. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Zhang, S.W.; Wang, G.X. Metabolomic biomarkers in diabetic kidney diseases—A systematic review. J. Diabetes Complicat. 2015, 29, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ouyang, S.; Xie, Y.; Gong, Z.; Du, J. Characterizing the gut microbiota in patients with chronic kidney disease. Postgrad. Med. 2020, 132, 495–505. [Google Scholar] [CrossRef]
- Long, Y.; Nie, J. Homocysteine in Renal Injury. Kidney Dis. 2016, 2, 80–87. [Google Scholar] [CrossRef]
- Cohen, E.; Margalit, I.; Shochat, T.; Goldberg, E.; Krause, I. The relationship between the concentration of plasma homocysteine and chronic kidney disease: A cross sectional study of a large cohort. J. Nephrol. 2019, 32, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, Q.; Jiang, Y.; Zhao, H.; Zhao, T.; Cao, Y.; Li, P.; Niu, W. Genetically elevated circulating homocysteine concentrations increase the risk of diabetic kidney disease in Chinese diabetic patients. J. Cell. Mol. Med. 2019, 23, 2794–2800. [Google Scholar] [CrossRef]
- Jardine, M.J.; Kang, A.; Zoungas, S.; Navaneethan, S.D.; Ninomiya, T.; Nigwekar, S.U.; Gallagher, M.P.; Cass, A.; Strippoli, G.; Perkovic, V. The effect of folic acid based homocysteine lowering on cardiovascular events in people with kidney disease: Systematic review and meta-analysis. BMJ 2012, 344, e3533. [Google Scholar] [CrossRef]
- Cheung, G.T.; Siow, Y.L.; O, K. Homocysteine stimulates monocyte chemoattractant protein-1 expression in mesangial cells via NF-kappaB activation. Can. J. Physiol. Pharmacol. 2008, 86, 88–96. [Google Scholar] [CrossRef]
- Holecek, M. Histidine in Health and Disease. Metabolism, Physiological Importance, and Use as a Supplement. Nutrients 2020, 12, 848. [Google Scholar] [CrossRef]
- Watanabe, M.; Suliman, M.E.; Qureshi, A.R.; Garcia-Lopez, E.; Barany, P.; Heimburger, O.; Stenvinkel, P.; Lindholm, B. Consequences of low plasma histidine in chronic kidney disease patients: Associations with inflammation, oxidative stress, and mortality. Am. J. Clin. Nutr. 2008, 87, 1860–1866. [Google Scholar] [CrossRef]
- Vera-Aviles, M.; Vantana, E.; Kardinasari, E.; Koh, N.L.; Latunde-Dada, G.O. Protective role of histidine supplementation against oxidative stress damage in the management of anemia of chronic kidney disease. Pharmaceuticals 2018, 11, 111. [Google Scholar] [CrossRef]
- Li, Z.; Li, A.; Gao, J.; Li, H.; Qin, X. Kidney Tissue Targeted Metabolic Profiling of Unilateral Ureteral Obstruction Rats by NMR. Front. Pharmacol. 2016, 7, 307. [Google Scholar] [CrossRef]
- Mohammad-Zadeh, L.F.; Moses, L.; Gwaltney-Brant, S.M. Serotonin. A review. J. Vet. Pharmacol. Ther. 2008, 31, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Hirowatari, Y.; Shimura, Y.; Takahashi, H. Serotonin levels in platelet-poor plasma and whole blood in people with type 2 diabetes with chronic kidney disease. Diabetes Res. Clin. Pract. 2011, 94, 167–171. [Google Scholar] [CrossRef]
- Kobayashi, S.; Satoh, M.; Namikoshi, T.; Haruna, Y.; Fujimoto, S.; Arakawa, S.; Komai, N.; Tomita, N.; Sasaki, T.; Kashihara, N. Blockade of serotonin 2A receptor improves glomerular endothelial function in rats with streptozotocin-induced diabetic nephropathy. Clin. Exp. Nephrol. 2008, 12, 119–125. [Google Scholar] [CrossRef]
- Barzegar-Fallah, A.; Alimoradi, H.; Asadi, F.; Dehpour, A.R.; Asgari, M.; Shafiei, M. Tropisetron ameliorates early diabetic nephropathy in streptozotocin-induced diabetic rats. Clin. Exp. Pharmacol. Physiol. 2015, 42, 361–368. [Google Scholar] [CrossRef]
- Lameire, N.H. Serotonin and the regulation of renal blood flow in acute renal failure. Am. J. Kidney Dis. 1999, 33, LII–LIV. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, Q.; Xia, T.; Fu, S.; Tao, X.; Wen, Y.; Chan, S.; Gao, S.; Xiong, X.; Chen, W. Diagnostic value of plasma tryptophan and symmetric dimethylarginine levels for acute kidney injury among tacrolimus-treated kidney transplant patients by targeted metabolomics analysis. Sci. Rep. 2018, 8, 14688. [Google Scholar] [CrossRef]
- Konje, V.C.; Rajendiran, T.M.; Bellovich, K.; Gadegbeku, C.A.; Gipson, D.S.; Afshinnia, F.; Mathew, A.V.; Michigan Kidney Translational Core CIG. Tryptophan levels associate with incident cardiovascular disease in chronic kidney disease. Clin. Kidney J. 2021, 14, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Dankers, A.C.; Mutsaers, H.A.; Dijkman, H.B.; van den Heuvel, L.P.; Hoenderop, J.G.; Sweep, F.C.; Russel, F.G.; Masereeuw, R. Hyperuricemia influences tryptophan metabolism via inhibition of multidrug resistance protein 4 (MRP4) and breast cancer resistance protein (BCRP). Biochim. Biophys. Acta 2013, 1832, 1715–1722. [Google Scholar] [CrossRef]
- Chen, H.; Chen, L.; Liu, D.; Chen, D.Q.; Vaziri, N.D.; Yu, X.Y.; Zhang, L.; Su, W.; Bai, X.; Zhao, Y.Y. Combined Clinical Phenotype and Lipidomic Analysis Reveals the Impact of Chronic Kidney Disease on Lipid Metabolism. J. Proteome Res. 2017, 16, 1566–1578. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A.; Farooqui, T. Glycerophospholipids in brain: Their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem. Phys. Lipids 2000, 106, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, Z.; Qin, M.; Zhang, B.; Lin, L.; Ma, Q.; Liu, C.; Chen, X.; Li, H.; Lai, W.; et al. Comprehensive Metabolomics Identified the Prominent Role of Glycerophospholipid Metabolism in Coronary Artery Disease Progression. Front. Mol. Biosci. 2021, 8, 632950. [Google Scholar] [CrossRef]
- Wang, S.; Tang, K.; Lu, Y.; Tian, Z.; Huang, Z.; Wang, M.; Zhao, J.; Xie, J. Revealing the role of glycerophospholipid metabolism in asthma through plasma lipidomics. Clin. Chim. Acta 2021, 513, 34–42. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, B.; Huang, S.; Wang, F.; Zheng, L.; Lu, J.; Zeng, Y.; Chen, J.; Li, S. Metabolomics Analysis Reveals the Protection Mechanism of Huangqi-Danshen Decoction on Adenine-Induced Chronic Kidney Disease in Rats. Front. Pharmacol. 2019, 10, 992. [Google Scholar] [CrossRef]
- Zhang, Z.H.; He, J.Q.; Qin, W.W.; Zhao, Y.Y.; Tan, N.H. Biomarkers of obstructive nephropathy using a metabolomics approach in rat. Chem.-Biol. Interact. 2018, 296, 229–239. [Google Scholar] [CrossRef]
- Liu, N.; Sun, Q.; Xu, H.; Yu, X.; Chen, W.; Wei, H.; Jiang, J.; Xu, Y.; Lu, W. Hyperuricemia induces lipid disturbances mediated by LPCAT3 upregulation in the liver. FASEB J. 2020, 34, 13474–13493. [Google Scholar] [CrossRef]
- Cao, H.; Zhang, A.; Sun, H.; Zhou, X.; Guan, Y.; Liu, Q.; Kong, L.; Wang, X. Metabolomics-proteomics profiles delineate metabolic changes in kidney fibrosis disease. Proteomics 2015, 15, 3699–3710. [Google Scholar] [CrossRef]
- Xiang, Z.; Sun, H.; Cai, X.; Chen, D. The study on serum and urine of renal interstitial fibrosis rats induced by unilateral ureteral obstruction based on metabonomics and network analysis methods. Anal. Bioanal. Chem. 2016, 408, 2607–2619. [Google Scholar] [CrossRef]
- Ren, L.; Guo, X.Y.; Gao, F.; Jin, M.L.; Song, X.N. Identification of the Perturbed Metabolic Pathways Associating with Renal Fibrosis and Evaluating Metabolome Changes of Pretreatment with Astragalus polysaccharide Through Liquid Chromatography Quadrupole Time-Of-Flight Mass Spectrometry. Front. Pharmacol. 2020, 10, 1623. [Google Scholar] [CrossRef]
- Wang, T.; Fu, X.; Chen, Q.; Patra, J.K.; Wang, D.; Wang, Z.; Gai, Z. Arachidonic Acid Metabolism and Kidney Inflammation. Int. J. Mol. Sci. 2019, 20, 3683. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef]
- Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 3285. [Google Scholar] [CrossRef]
- Imig, J.D. Epoxyeicosatrienoic acids, 20-hydroxyeicosatetraenoic acid, and renal microvascular function. Prostaglandins Other Lipid Mediat. 2013, 104, 2–7. [Google Scholar] [CrossRef]
- Sharma, M.; McCarthy, E.T.; Reddy, D.S.; Patel, P.K.; Savin, V.J.; Medhora, M.; Falck, J.R. 8,9-Epoxyeicosatrienoic acid protects the glomerular filtration barrier. Prostaglandins Other Lipid Mediat. 2009, 89, 43–51. [Google Scholar] [CrossRef]
- Imig, J.D. Epoxyeicosatrienoic acids, hypertension, and kidney injury. Hypertension 2015, 65, 476–482. [Google Scholar] [CrossRef]
- Hoff, U.; Bubalo, G.; Fechner, M.; Blum, M.; Zhu, Y.; Pohlmann, A.; Hentschel, J.; Arakelyan, K.; Seeliger, E.; Flemming, B.; et al. A synthetic epoxyeicosatrienoic acid analogue prevents the initiation of ischemic acute kidney injury. Acta Physiol. 2019, 227, e13297. [Google Scholar] [CrossRef]
- Wang, W.H.; Zhang, C.; Lin, D.H.; Wang, L.; Graves, J.P.; Zeldin, D.C.; Capdevila, J.H. Cyp2c44 epoxygenase in the collecting duct is essential for the high K+ intake-induced antihypertensive effect. Am. J. Physiol. Renal Physiol. 2014, 307, F453–F460. [Google Scholar] [CrossRef]
- Yeboah, M.M.; Hye Khan, M.A.; Chesnik, M.A.; Sharma, A.; Paudyal, M.P.; Falck, J.R.; Imig, J.D. The epoxyeicosatrienoic acid analog PVPA ameliorates cyclosporine-induced hypertension and renal injury in rats. Am. J. Physiol. Renal Physiol. 2016, 311, F576–F585. [Google Scholar] [CrossRef]
- Hercule, H.C.; Schunck, W.H.; Gross, V.; Seringer, J.; Leung, F.P.; Weldon, S.M.; da Costa Goncalves, A.; Huang, Y.; Luft, F.C.; Gollasch, M. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arter. Thromb. Vasc. Biol. 2009, 29, 54–60. [Google Scholar] [CrossRef]
- Wang, D.; Borrego-Conde, L.J.; Falck, J.R.; Sharma, K.K.; Wilcox, C.S.; Umans, J.G. Contributions of nitric oxide, EDHF, and EETs to endothelium-dependent relaxation in renal aerent arterioles. Kidney Int. 2003, 63, 2187–2193. [Google Scholar] [CrossRef]
- Skibba, M.; Hye Khan, M.A.; Kolb, L.L.; Yeboah, M.M.; Falck, J.R.; Amaradhi, R.; Imig, J.D. Epoxyeicosatrienoic Acid Analog Decreases Renal Fibrosis by Reducing Epithelial-to-Mesenchymal Transition. Front. Pharmacol. 2017, 8, 406. [Google Scholar] [CrossRef]
- Mota-Zamorano, S.; Robles, N.R.; Lopez-Gomez, J.; Cancho, B.; Gonzalez, L.M.; Garcia-Pino, G.; Navarro-Perez, M.L.; Gervasini, G. Plasma and urinary concentrations of arachidonic acid-derived eicosanoids are associated with diabetic kidney disease. EXCLI J. 2021, 20, 698–708. [Google Scholar]
- Peng, L.; Sun, B.; Liu, Y.; Huang, J.; Chen, G.; Zhang, X.; Chen, C.; Wang, D.; Wang, G. Increased lipoxygenase and decreased cytochrome P450s metabolites correlated with the incidence of diabetic nephropathy: Potential role of eicosanoids from metabolomics in type 2 diabetic patients. Clin. Exp. Pharmacol. Physiol. 2021, 48, 679–685. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite Name | VIP | Fold-Change | p-Value | Compound Class | Label | Confidence Level |
|---|---|---|---|---|---|---|
| Pyroglutamic acid | 1.6018337 | 0.28835 | 2.11 × 10−6 | Amino acids, peptides, and analogs | down | Level 1 |
| Fructose | 1.5954118 | 2.6474 | 4.68 × 10−7 | Sugar and derivatives | up | Level 1 |
| Riboflavin | 1.5712089 | 0.43611 | 1.05 × 10−5 | Indoles and heterocyclic compounds | down | Level 1 |
| Dimethyl-L-arginine | 1.5542553 | 0.37179 | 3.44 × 10−5 | Amino acids, peptides, and analogs | down | Level 2 |
| Glucaric acid | 1.4796976 | 5.8869 | 0.0001739 | Organic acids and derivatives | up | Level 2 |
| Indoxyl sulfate | 1.4441036 | 5.7386 | 0.0001343 | Indoles and heterocyclic compounds | up | Level 1 |
| Palmitoylethanolamide | 1.3687033 | 0.40007 | 0.0004409 | Amines, choline, and organonitrogen compounds | down | Level 2 |
| Trimethylamine N-oxide | 1.3166437 | 9.0069 | 0.0012998 | Amines, choline, and organonitrogen compounds | up | Level 2 |
| 3-Hydroxyanthranilic acid | 1.2637099 | 4.8648 | 0.0024075 | Organic acids and derivatives | up | Level 1 |
| Spermidine | 1.2554446 | 3.1751 | 0.0050288 | Amines, choline, and organonitrogen compounds | up | Level 2 |
| Hippuric acid | 1.1131107 | 15.761 | 0.022992 | Organic acids and derivatives | up | Level 1 |
| Metabolites | UA | CRE | BUN | TIS |
|---|---|---|---|---|
| pyroglutamic acid | −0.713 * | −0.796 ** | −0.741 ** | −0.916 *** |
| fructose | 0.797 ** | 0.663 * | 0.797 ** | 0.756 ** |
| riboflavin | −0.832 ** | −0.818 ** | −0.839 ** | −0.869 ** |
| dimethyl-L-arginine | −0.797 ** | −0.782 ** | −0.769 ** | −0.869 ** |
| glucaric acid | 0.769 ** | 0.811 ** | 0.762 ** | 0.869 ** |
| indoxyl sulfate | 0.797 ** | 0.828 ** | 0.797 ** | 0.869 ** |
| palmitoylethanolamide | −0.706 * | −0.737 ** | −0.699 * | −0.869 ** |
| trimethylamine N-oxide | 0.734 ** | 0.726 * | 0.748 ** | 0.869 ** |
| 3-hydroxyanthranilic acid | 0.79 ** | 0.779 ** | 0.832 ** | 0.869 ** |
| spermidine | 0.692 * | 0.74 ** | 0.699 * | 0.869 ** |
| hippuric acid | 0.755 ** | 0.789 ** | 0.734 ** | 0.869 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Li, H.; Tang, T.; Zhang, Q.; Song, T.; Zhao, Z.; Zhu, L.; Chen, Q.; Zhang, H.; Zhang, Y.; Kong, J. Multiple Reaction Monitoring (MRM)-Based Targeted Kidney Metabolite Profiling of a Mouse Model of Hyperuricemia. Metabolites 2026, 16, 362. https://doi.org/10.3390/metabo16060362
Li H, Tang T, Zhang Q, Song T, Zhao Z, Zhu L, Chen Q, Zhang H, Zhang Y, Kong J. Multiple Reaction Monitoring (MRM)-Based Targeted Kidney Metabolite Profiling of a Mouse Model of Hyperuricemia. Metabolites. 2026; 16(6):362. https://doi.org/10.3390/metabo16060362
Chicago/Turabian StyleLi, Hailong, Tingting Tang, Qingli Zhang, Tingting Song, Zichu Zhao, Lei Zhu, Qu Chen, Haili Zhang, Yan Zhang, and Jingjing Kong. 2026. "Multiple Reaction Monitoring (MRM)-Based Targeted Kidney Metabolite Profiling of a Mouse Model of Hyperuricemia" Metabolites 16, no. 6: 362. https://doi.org/10.3390/metabo16060362
APA StyleLi, H., Tang, T., Zhang, Q., Song, T., Zhao, Z., Zhu, L., Chen, Q., Zhang, H., Zhang, Y., & Kong, J. (2026). Multiple Reaction Monitoring (MRM)-Based Targeted Kidney Metabolite Profiling of a Mouse Model of Hyperuricemia. Metabolites, 16(6), 362. https://doi.org/10.3390/metabo16060362