
Investigating Multi-Omic Signatures of Ethnicity and Dysglycaemia in Asian Chinese and European Caucasian Adults: Cross-Sectional Analysis of the TOFI_Asia Study at 4-Year Follow-Up

 , , , , , , , , and
, , , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Background
Ethics Approval and Trial Registration
2.2. Sample Collection
2.2.1. Faecal Sample Collection
2.2.2. Anthropometric, Clinical, and Biochemical Assessments
2.3. Sample Analysis
2.3.1. Shotgun Metagenomics
2.3.2. Untargeted Metabolomics
2.4. Data Preprocessing
2.4.1. Clinical and Biochemical Characteristic Preprocessing
2.4.2. Shotgun Metagenomics Sequence Processing
2.4.3. Metabolomics Data Processing
2.5. Bioinformatics and Statistical Analysis
2.5.1. Univariate Analysis of the Multi-Omic Dataset
2.5.2. Integrated Analysis of Clinical, Metagenomic, and Metabolomic Data Using Mixomics
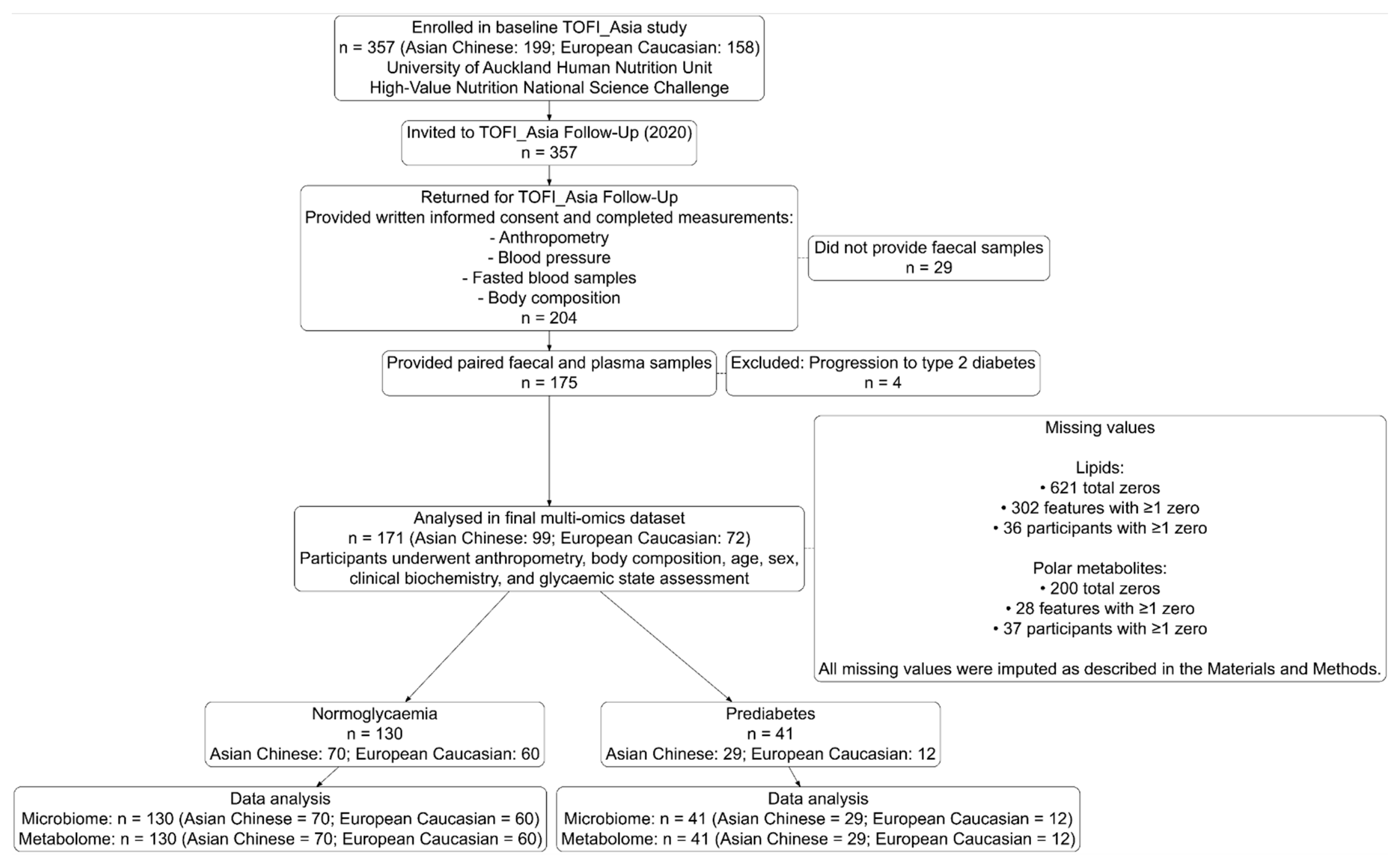
3. Results
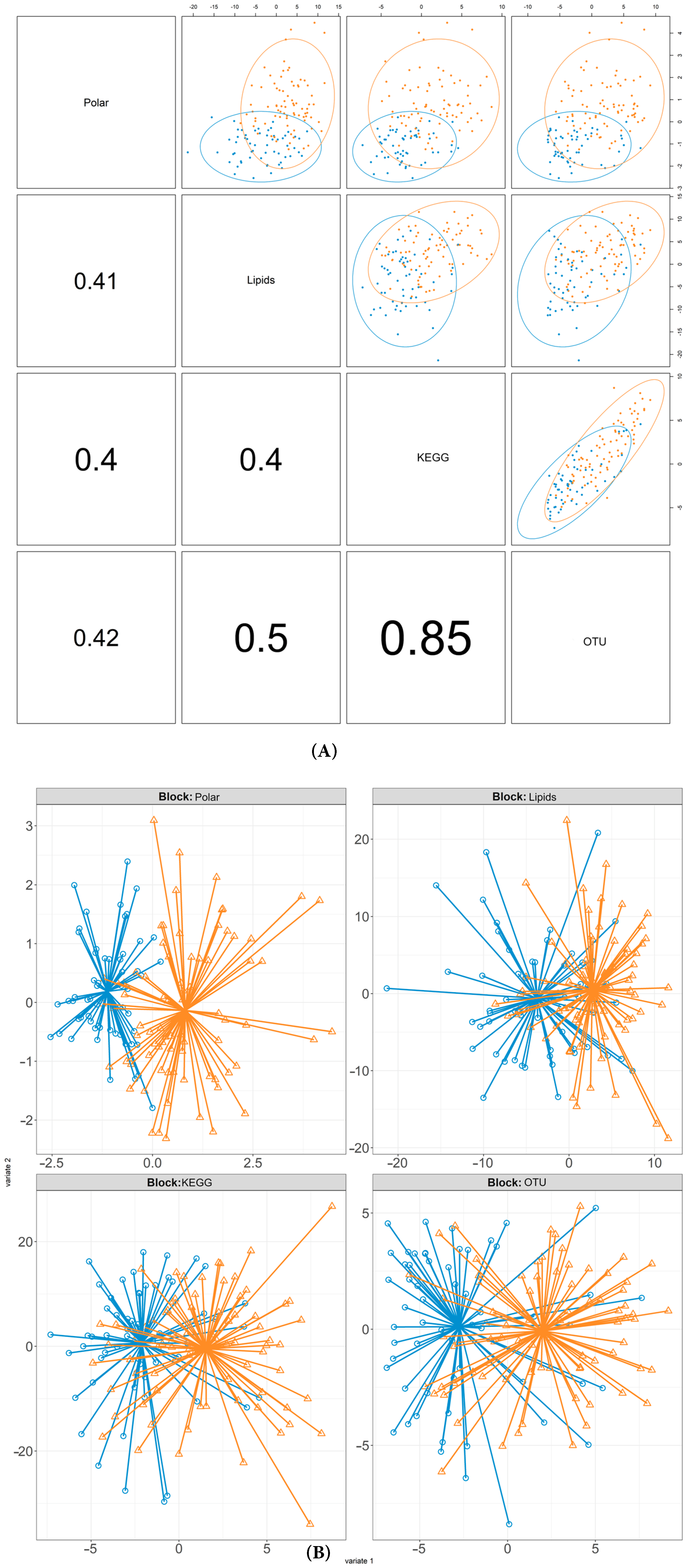
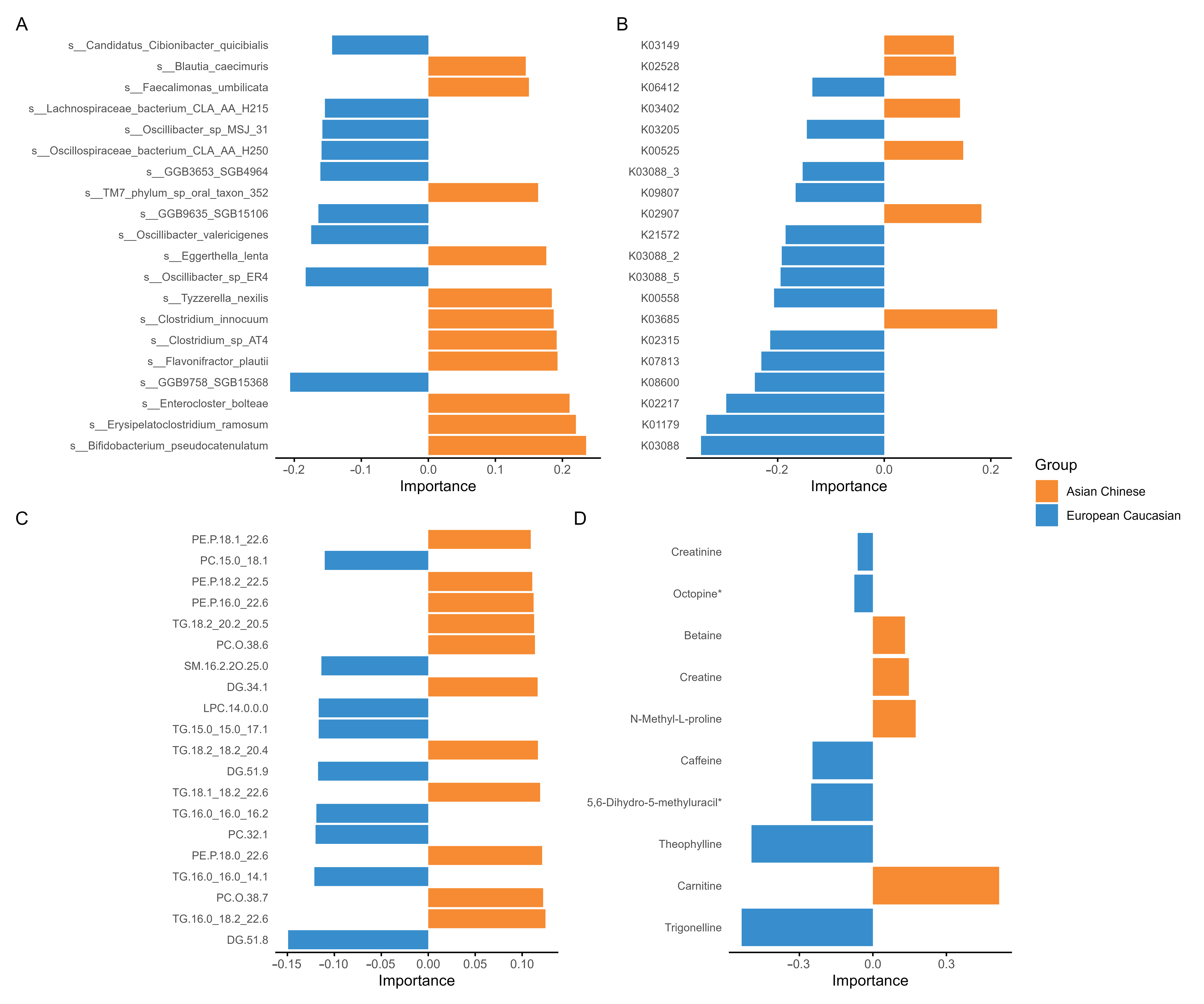
3.1. Multi-Omic Signatures of Ethnicity
3.2. Cross-Omic Microbial–Metabolite Hubs in Ethnicity-Stratified Analysis
3.3. Influence of Glycaemic Status on Multi-Omics Datasets
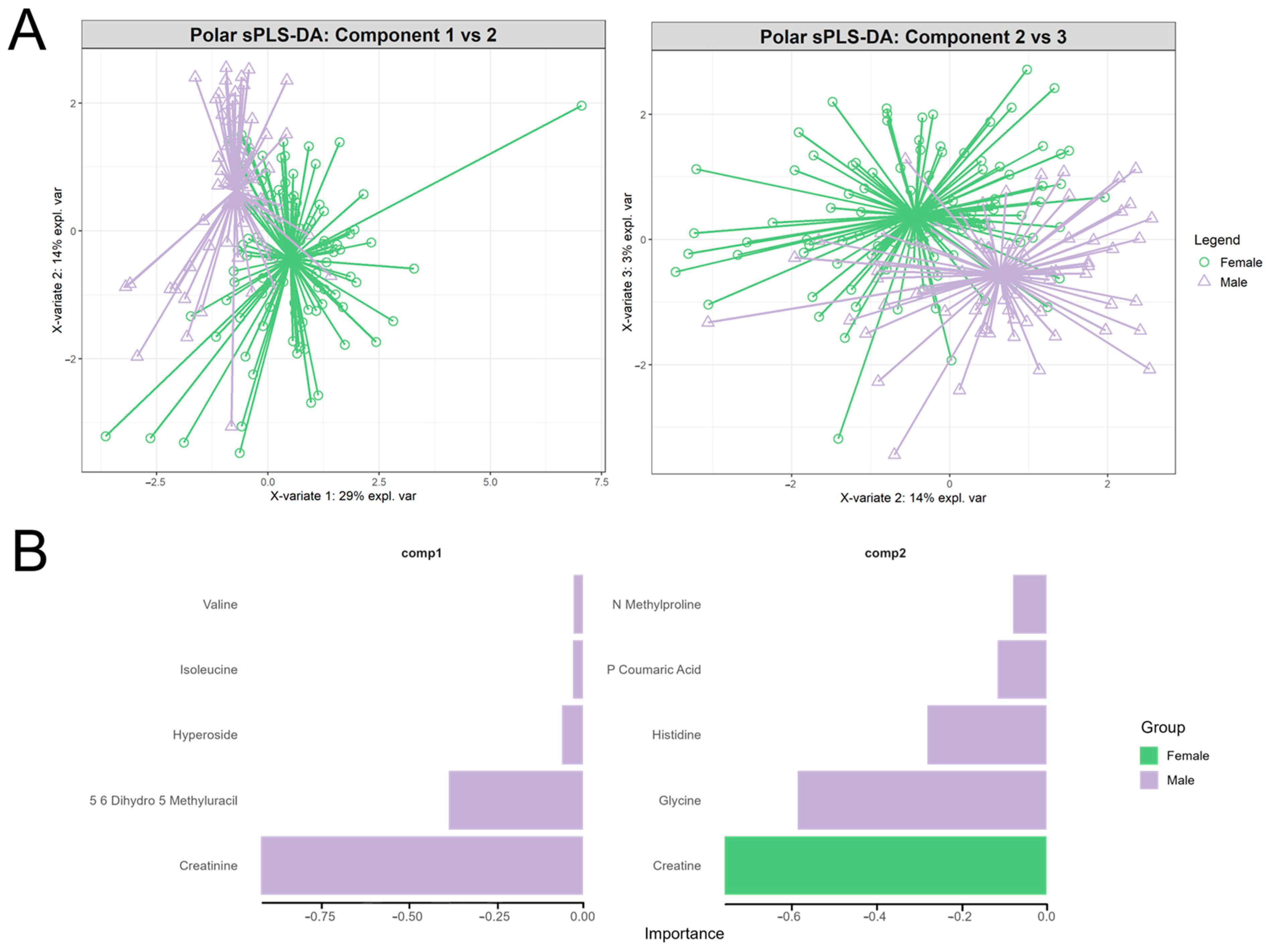
3.4. Assessing Block-Specific Discrimination: Polar Metabolite-Driven Sex Separation
4. Discussion
4.1. Metagenomic Distinctions Are Primarily Driven by Ethnicity and Not Glycaemic Status
4.2. Cross-Omic Correlation Structure Reveals Metabolic and Microbial Hubs
4.3. Metabolomic Datasets Reinforce Ethnicity over Metabolic Status Separation
4.4. Sex-Associated Variation in Polar Metabolite Signatures
4.5. Methodological Considerations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IDF. Diabetes Atlas 2021, 10th ed.; IDF: Brussels, Belgium, 2021. [Google Scholar]
- Bergman, M. Prediabetes remission in people with obesity. Lancet Diabetes Endocrinol. 2024, 12, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.Z.; Magliano, D.J.; Herman, W.H.; Shaw, J.E. Diabetes: A 21st century challenge. Lancet Diabetes Endocrinol. 2014, 2, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.H.; Lee, J.H.; Kim, J.W.; Cho, J.H.; Choi, Y.H.; Ko, S.H.; Zimmet, P.; Son, H.Y. Epidemic obesity and type 2 diabetes in Asia. Lancet 2006, 368, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- WOF. Obesity Atlas 2024; World Obesity Federation: London, UK, 2024. [Google Scholar]
- Pan, X.-F.; Wang, L.; Pan, A. Epidemiology and determinants of obesity in China. Lancet Diabetes Endocrinol. 2021, 9, 373–392. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Chamukuttan, S.; Shetty, S.A.; Arun, N.; Susairaj, P. Obesity in Asia—Is it different from rest of the world. Diabetes/Metab. Res. Rev. 2012, 28 (Suppl. S2), 47–51. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.L.; Frost, G.; Taylor-Robinson, S.D.; Bell, J.D. Excess body fat in obese and normal-weight subjects. Nutr. Res. Rev. 2012, 25, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.L.; Hollingsworth, K.; Aribisala, B.; Chen, M.J.; Mathers, J.; Taylor, R. Reversal of type 2 diabetes: Normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011, 54, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Lingvay, I.; Esser, V.; Legendre, J.L.; Price, A.L.; Wertz, K.M.; Adams-Huet, B.; Zhang, S.; Unger, R.H.; Szczepaniak, L.S. Noninvasive quantification of pancreatic fat in humans. J. Clin. Endocrinol. Metab. 2009, 94, 4070–4076. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, I.R.; Yip, W.C.; Lu, L.W.; Jiang, Y.; Murphy, R.; Plank, L.D.; Cooper, G.J.; Peters, C.N.; Lu, J.; Hollingsworth, K.G.; et al. Pancreas Fat, an Early Marker of Metabolic Risk? A Magnetic Resonance Study of Chinese and Caucasian Women: TOFI_Asia Study. Front. Physiol. 2022, 13, 819606. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, L.E.; Bäckhed, F. The Metabolic Role and Therapeutic Potential of the Microbiome. Endocr. Rev. 2022, 43, 907–926. [Google Scholar] [CrossRef] [PubMed]
- Haak, B.W.; Prescott, H.C.; Wiersinga, W.J. Therapeutic Potential of the Gut Microbiota in the Prevention and Treatment of Sepsis. Front. Immunol. 2018, 9, 2042. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Pamp Sünje, J.; Hill Jonathan, A.; Surana Neeraj, K.; Edelman Sanna, M.; Troy Erin, B.; Reading, N.C.; Wang, S.; Mora, J.R.; Umesaki, Y. Gut Immune Maturation Depends on Colonization with a Host-Specific Microbiota. Cell 2012, 149, 1578–1593. [Google Scholar] [CrossRef] [PubMed]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef] [PubMed]
- Rios-Morales, M.; Vieira-Lara, M.A.; Homan, E.; Langelaar-Makkinje, M.; Gerding, A.; Li, Z.; Huijkman, N.; Rensen, P.C.N.; Wolters, J.C.; Reijngoud, D.-J.; et al. Butyrate oxidation attenuates the butyrate-induced improvement of insulin sensitivity in myotubes. Biochim. Et. Biophys. Acta (BBA)—Mol. Basis Dis. 2022, 1868, 166476. [Google Scholar] [CrossRef] [PubMed]
- Hernández, M.A.G.; Canfora, E.E.; Jocken, J.W.E.; Blaak, E.E. The Short-Chain Fatty Acid Acetate in Body Weight Control and Insulin Sensitivity. Nutrients 2019, 11, 1943. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Mathrani, A.; Lu, L.W.; Sequeira-Bisson, I.R.; Silvestre, M.P.; Hoggard, M.; Barnett, D.; Fogelholm, M.; Raben, A.; Poppitt, S.D.; Taylor, M.W. Gut microbiota profiles in two New Zealand cohorts with overweight and prediabetes: A Tū Ora/PREVIEW comparative study. Front. Microbiol. 2023, 14, 1244179. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gou, W.; Su, C.; Du, W.; Zhang, J.; Miao, Z.; Xiao, C.; Jiang, Z.; Wang, Z.; Fu, Y.; et al. Association of gut microbiota with glycaemic traits and incident type 2 diabetes, and modulation by habitual diet: A population-based longitudinal cohort study in Chinese adults. Diabetologia 2022, 65, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.E.; Fraser, K.; Kruger, M.C.; Sequeira, I.R.; Yip, W.; Lu, L.W.; Plank, L.D.; Murphy, R.; Cooper, G.J.S.; Martin, J.-C.; et al. Metabolomic signatures for visceral adiposity and dysglycaemia in Asian Chinese and Caucasian European adults: The cross-sectional TOFI_Asia study. Nutr. Metab. 2020, 17, 95. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, I.R.; Yip, W.; Lu, L.; Jiang, Y.; Murphy, R.; Plank, L.; Zhang, S.; Liu, H.; Chuang, C.L.; Vazhoor-Amarsingh, G.; et al. Visceral Adiposity and Glucoregulatory Peptides are Associated with Susceptibility to Type 2 Diabetes: The TOFI_Asia Study. Obesity 2020, 28, 2368–2378. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Lê Cao, K.-A. DIABLO: An integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [CrossRef] [PubMed]
- Rohart, F.G.B.; Singh, A.; Le Cao, K.-A. mixOmics: An R package for ’omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S. Beyond the X Factor: Relevance of Sex Hormones in NAFLD Pathophysiology. Cells 2021, 10, 2502. [Google Scholar] [CrossRef] [PubMed]
- Decsi, T.; Kennedy, K. Sex-specific differences in essential fatty acid metabolism12345. Am. J. Clin. Nutr. 2011, 94, S1914–S1919. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, F.B.; Shen, W.-J. Hormone-sensitive lipase. J. Lipid Res. 2002, 43, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Esbjörnsson, M.; Norman, B.; Dahlström, M.; Gierup, J.; Jansson, E. Metabolic and morphological profile in skeletal muscle of healthy boys and girls. Physiol. Rep. 2022, 10, e15414. [Google Scholar] [CrossRef] [PubMed]
- Pisoni, S.; Marrachelli, V.G.; Morales, J.M.; Maestrini, S.; Di Blasio, A.M.; Monleón, D. Sex Dimorphism in the Metabolome of Metabolic Syndrome in Morbidly Obese Individuals. Metabolites 2022, 12, 419. [Google Scholar] [CrossRef] [PubMed]
- Escarcega, R.D.; MJ, V.K.; Kyriakopoulos, V.E.; Ortiz, G.J.; Gusdon, A.M.; Fan, H.; Peesh, P.; Blasco Conesa, M.P.; Colpo, G.D.; Ahnstedt, H.W.; et al. Serum metabolome profiling in patients with mild cognitive impairment reveals sex differences in lipid metabolism. Neurobiol. Dis. 2025, 204, 106747. [Google Scholar] [CrossRef] [PubMed]
- Association, A.D. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2012, 36, S67–S74. [Google Scholar] [CrossRef] [PubMed]
- Joblin-Mills, A.; Wu, Z.E.; Sequeira-Bisson, I.R.; Miles-Chan, J.L.; Poppitt, S.D.; Fraser, K. Utilising a Clinical Metabolomics LC-MS Study to Determine the Integrity of Biological Samples for Statistical Modelling after Long Term −80 °C Storage: A TOFI_Asia Sub-Study. Metabolites 2024, 14, 313. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef] [PubMed]
- Handley, S.A. Virus+ Sequence Masked Human Reference Genome (hg19); Zenodo: Geneva, Switzerland, 2021. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2024. [Google Scholar]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Peddada, S.D. Multigroup analysis of compositions of microbiomes with covariate adjustments and repeated measures. Nat. Methods 2024, 21, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Eggesbo, M.; Peddada, S.D. Linear and nonlinear correlation estimators unveil undescribed taxa interactions in microbiome data. Nat. Commun. 2022, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hastie, T.; Robert, T.; Balasubramanian, N.; Gilbert, C. Impute: Imputation for Microarray Data; R: Vienna, Austria, 2024. [Google Scholar]
- van den Berg, R.A.; Hoefsloot, H.C.J.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Max, K.a. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Lahti, L.; Shetty, S. Microbiome R Package; R: Vienna, Austria, 2012. [Google Scholar]
- Lê Cao, K.-A.; Costello, M.-E.; Lakis, V.A.; Bartolo, F.; Chua, X.-Y.; Brazeilles, R.; Rondeau, P. MixMC: A Multivariate Statistical Framework to Gain Insight into Microbial Communities. PLoS ONE 2016, 11, e0160169. [Google Scholar] [CrossRef] [PubMed]
- Mandić, A.D.; Woting, A.; Jaenicke, T.; Sander, A.; Sabrowski, W.; Rolle-Kampcyk, U.; von Bergen, M.; Blaut, M. Clostridium ramosum regulates enterochromaffin cell development and serotonin release. Sci. Rep. 2019, 9, 1177. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Sun, Y.; Li, Y.; Li, M.; Cao, Z.; Huang, Z.; Zhang, F.; Yan, P.; Wang, L.; Luo, J.; et al. Landscape of the gut archaeome in association with geography, ethnicity, urbanization, and diet in the Chinese population. Microbiome 2022, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Camara, A.; Konate, S.; Tidjani Alou, M.; Kodio, A.; Togo, A.H.; Cortaredona, S.; Henrissat, B.; Thera, M.A.; Doumbo, O.K.; Raoult, D.; et al. Clinical evidence of the role of Methanobrevibacter smithii in severe acute malnutrition. Sci. Rep. 2021, 11, 5426. [Google Scholar] [CrossRef] [PubMed]
- Tamanai-Shacoori, Z.; Smida, I.; Bousarghin, L.; Loreal, O.; Meuric, V.; Fong, S.B.; Bonnaure-Mallet, M.; Jolivet-Gougeon, A. Roseburia spp.: A marker of health? Future Microbiol. 2017, 12, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mao, B.; Gu, J.; Wu, J.; Cui, S.; Wang, G.; Zhao, J.; Zhang, H.; Chen, W. Blautia-a new functional genus with potential probiotic properties? Gut Microbes 2021, 13, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, M.; Ikeyama, N.; Yuki, M.; Ohkuma, M. Draft Genome Sequence of Faecalimonas umbilicata JCM 30896, an Acetate-Producing Bacterium Isolated from Human Feces. Microbiol. Resour. Announc. 2018, 7, 1110–1128. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.W.; Priya, S.; Blekhman, R.; Bordenstein, S.R. Gut microbiota diversity across ethnicities in the United States. PLoS Biol. 2018, 16, e2006842. [Google Scholar] [CrossRef] [PubMed]
- Dwiyanto, J.; Hussain, M.H.; Reidpath, D.; Ong, K.S.; Qasim, A.; Lee, S.W.H.; Lee, S.M.; Foo, S.C.; Chong, C.W.; Rahman, S. Ethnicity influences the gut microbiota of individuals sharing a geographical location: A cross-sectional study from a middle-income country. Sci. Rep. 2021, 11, 2618. [Google Scholar] [CrossRef] [PubMed]
- Mallott, E.K.; Sitarik, A.R.; Leve, L.D.; Cioffi, C.; Camargo, C.A., Jr.; Hasegawa, K.; Bordenstein, S.R. Human microbiome variation associated with race and ethnicity emerges as early as 3 months of age. PLoS Biol. 2023, 21, e3002230. [Google Scholar] [CrossRef] [PubMed]
- Umirah, F.; Neoh, C.F.; Ramasamy, K.; Lim, S.M. Differential gut microbiota composition between type 2 diabetes mellitus patients and healthy controls: A systematic review. Diabetes Res. Clin. Pract. 2021, 173, 108689. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, S.M.; Carson, T.; Lowe, J.; Ramaraj, T.; Leff, J.W.; Luo, L.; Bell, C.J.; Shah, V.O. Composition, Diversity and Abundance of Gut Microbiome in Prediabetes and Type 2 Diabetes. Diabetes Obes. Metab. 2015, 2, 1–7. [Google Scholar] [CrossRef]
- Hu, J.; Ding, J.; Li, X.; Li, J.; Zheng, T.; Xie, L.; Li, C.; Tang, Y.; Guo, K.; Huang, J.; et al. Distinct signatures of gut microbiota and metabolites in different types of diabetes: A population-based cross-sectional study. eClinicalMedicine 2023, 62, 102132. [Google Scholar] [CrossRef] [PubMed]
- Chung The, H.; Nguyen Ngoc Minh, C.; Tran Thi Hong, C.; Nguyen Thi Nguyen, T.; Pike, L.J.; Zellmer, C.; Pham Duc, T.; Tran, T.A.; Ha Thanh, T.; Van, M.P.; et al. Exploring the Genomic Diversity and Antimicrobial Susceptibility of Bifidobacterium pseudocatenulatum in a Vietnamese Population. Microbiol. Spectr. 2021, 9, e0052621. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Peng, Y.; Huang, Y.; Xie, M.; Dai, Z.; Cai, H.; Dong, W.; Xu, W.; Xie, Z.; Chen, D.; et al. Fluoride induced leaky gut and bloom of Erysipelatoclostridium ramosum mediate the exacerbation of obesity in high-fat-diet fed mice. J. Adv. Res. 2023, 50, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Magdy Wasfy, R.; Mbaye, B.; Borentain, P.; Tidjani Alou, M.; Murillo Ruiz, M.L.; Caputo, A.; Andrieu, C.; Armstrong, N.; Million, M.; Gerolami, R. Ethanol-Producing Enterocloster bolteae Is Enriched in Chronic Hepatitis B-Associated Gut Dysbiosis: A Case–Control Culturomics Study. Microorganisms 2023, 11, 2437. [Google Scholar] [CrossRef] [PubMed]
- Frame, N.W.; Allas, M.J.; Pequegnat, B.; Vinogradov, E.; Liao, V.C.H.; Al-Abdul-Wahid, S.; Arroyo, L.; Allen-Vercoe, E.; Lowary, T.L.; Monteiro, M.A. Structure and synthesis of a vaccine and diagnostic target for Enterocloster bolteae, an autism-associated gut pathogen-Part II. Carbohydr. Res. 2023, 526, 108805. [Google Scholar] [CrossRef] [PubMed]
- Gophna, U.; Konikoff, T.; Nielsen, H.B. Oscillospira and related bacteria—From metagenomic species to metabolic features. Environ. Microbiol. 2017, 19, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Støving, R.K.; Berreira Ibraim, S.; Hyötyläinen, T.; Thirion, F.; Arora, T.; Lyu, L.; Stankevic, E.; Hansen, T.H.; Déchelotte, P.; et al. The gut microbiota contributes to the pathogenesis of anorexia nervosa in humans and mice. Nat. Microbiol. 2023, 8, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Browning, K.R.; Merrikh, H. Pathogenic bacteria experience pervasive RNA polymerase backtracking during infection. mBio 2024, 15, e02737-23. [Google Scholar] [CrossRef] [PubMed]
- Moraïs, S.; Winkler, S.; Zorea, A.; Levin, L.; Nagies, F.S.P.; Kapust, N.; Lamed, E.; Artan-Furman, A.; Bolam, D.N.; Yadav, M.P.; et al. Cryptic diversity of cellulose-degrading gut bacteria in industrialized humans. Science 2024, 383, eadj9223. [Google Scholar] [CrossRef] [PubMed]
- Rosell-Díaz, M.; Elena, S.-G.; Anna, M.-A.; Lluís, R.-T.; Josep, G.-O.; Vicente, P.-B.; Andrés, M.; Mariona, J.; Reinald, P.; Josep, P.; et al. Gut microbiota links to serum ferritin and cognition. Gut Microbes 2023, 15, 2290318. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yin, J.; Wan, D.; Yin, Y. The Role of Iron in Intestinal Mucus: Perspectives from Both the Host and Gut Microbiota. Adv. Nutr. 2024, 15, 100307. [Google Scholar] [CrossRef] [PubMed]
- Dostal, A.; Lacroix, C.; Bircher, L.; Pham, V.T.; Follador, R.; Zimmermann, M.B.; Chassard, C. Iron Modulates Butyrate Production by a Child Gut Microbiota In Vitro. mBio 2015, 6, e01453-15. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yang, Y.; Chen, X.; Bei, C.; Gao, Q.; Chao, Y.; Wang, C. An RNase III-processed sRNA coordinates sialic acid metabolism of Salmonella enterica during gut colonization. Proc. Natl. Acad. Sci. USA 2025, 122, e2414563122. [Google Scholar] [CrossRef] [PubMed]
- Lejars, M.; Hajnsdorf, E. Bacterial RNase III: Targets and physiology. Biochimie 2024, 217, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Weidner, U.; Geier, S.; Ptock, A.; Friedrich, T.; Leif, H.; Weiss, H. The Gene Locus of the Proton-translocating NADH: Ubiquinone Oxidoreductase in Escherichia coli: Organization of the 14 Genes and Relationship Between the Derived Proteins and Subunits of Mitochondrial Complex, I. J. Mol. Biol. 1993, 233, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Ohnishi, S.T.; Salerno, J.C. Five decades of research on mitochondrial NADH-quinone oxidoreductase (complex I). Biol. Chem. 2018, 399, 1249–1264. [Google Scholar] [CrossRef] [PubMed]
- Blachier, F.; Andriamihaja, M.; Blais, A. Sulfur-Containing Amino Acids and Lipid Metabolism. J. Nutr. 2020, 150, 2524S–2531S. [Google Scholar] [CrossRef] [PubMed]
- Woting, A.; Pfeiffer, N.; Loh, G.; Klaus, S.; Blaut, M. Clostridium ramosum promotes high-fat diet-induced obesity in gnotobiotic mouse models. mBio 2014, 5, e01530-14. [Google Scholar] [CrossRef] [PubMed]
- Lotankar, M.; Houttu, N.; Benchraka, C.; Lahti, L.; Laitinen, K. Links between gut microbiota with specific serum metabolite groups in pregnant women with overweight or obesity. Nutr. Metab. Cardiovasc. Dis. 2025, 104095. [Google Scholar] [CrossRef] [PubMed]
- Manghi, P.; Bhosle, A.; Wang, K.; Marconi, R.; Selma-Royo, M.; Ricci, L.; Asnicar, F.; Golzato, D.; Ma, W.; Hang, D.; et al. Coffee consumption is associated with intestinal Lawsonibacter asaccharolyticus abundance and prevalence across multiple cohorts. Nat. Microbiol. 2024, 9, 3120–3134. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Gao, J.; Chen, S.; Wang, G.; Liu, H.; Wang, X.; Sun, P.; Yu, X.; Yang, Q.; Wu, S.; et al. Triglyceride levels and risk of cardiovascular disease and all-cause mortality in Chinese adults younger than 40 years old: A prospective cohort study. Cardiovasc. Diagn. Ther. 2024, 14, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Aberra, T.; Peterson, E.D.; Pagidipati, N.J.; Mulder, H.; Wojdyla, D.M.; Philip, S.; Granowitz, C.; Navar, A.M. The association between triglycerides and incident cardiovascular disease: What is optimal? J. Clin. Lipidol. 2020, 14, 438–447.e433. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, A.; Shai, I.; Bitzur, R.; Kochba, I.; Tekes-Manova, D.; Israeli, E.; Shochat, T.; Rudich, A. Changes in triglyceride levels over time and risk of type 2 diabetes in young men. Diabetes Care 2008, 31, 2032–2037. [Google Scholar] [CrossRef] [PubMed]
- Borgo, F.; Garbossa, S.; Riva, A.; Severgnini, M.; Luigiano, C.; Benetti, A.; Pontiroli, A.E.; Morace, G.; Borghi, E. Body Mass Index and Sex Affect Diverse Microbial Niches within the Gut. Front. Microbiol. 2018, 9, 213. [Google Scholar] [CrossRef] [PubMed]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shiraki, K.; Ito, M.; Takei, Y.; et al. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 2015, 15, 100. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cheng, Y.-W.; Shao, L.; Sun, S.-H.; Wu, J.; Song, Q.-H.; Zou, H.-S.; Ling, Z.-X. Gut microbiota dysbiosis in Chinese children with type 1 diabetes mellitus: An observational study. World J. Gastroenterol. 2021, 27, 2394. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, T.; Liang, X.; Zhu, L.; Fang, Y.; Dong, L.; Zheng, Y.; Xu, X.; Li, M.; Cai, T.; et al. A decrease in Flavonifractor plautii and its product, phytosphingosine, predisposes individuals with phlegm-dampness constitution to metabolic disorders. Cell Discov. 2025, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Yamashita, T.; Osone, T.; Hosooka, T.; Shinohara, M.; Kitahama, S.; Sasaki, K.; Sasaki, D.; Yoneshiro, T.; Suzuki, T.; et al. Bacteroides spp. promotes branched-chain amino acid catabolism in brown fat and inhibits obesity. iScience 2021, 24, 103342. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cantos, M.V.; Babu, A.F.; Hanhineva, K.; Kuipers, O.P. Identification of metabolites produced by six gut commensal Bacteroidales strains using non-targeted LC-MS/MS metabolite profiling. Microbiol. Res. 2024, 283, 127700. [Google Scholar] [CrossRef] [PubMed]
- Shalon, D.; Culver, R.N.; Grembi, J.A.; Folz, J.; Treit, P.V.; Shi, H.; Rosenberger, F.A.; Dethlefsen, L.; Meng, X.; Yaffe, E.; et al. Profiling the human intestinal environment under physiological conditions. Nature 2023, 617, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Carmody, R.N.; Bisanz, J.E. Roles of the gut microbiome in weight management. Nat. Rev. Microbiol. 2023, 21, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lan, R.; Qiao, L.; Lin, X.; Hu, D.; Zhang, S.; Yang, J.; Zhou, J.; Ren, Z.; Li, X.; et al. Bacteroides vulgatus Ameliorates Lipid Metabolic Disorders and Modulates Gut Microbial Composition in Hyperlipidemic Rats. Microbiol. Spectr. 2023, 11, e02517-22. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yin, J.; Liu, X.; Liu, Y.; Jin, X. Gut commensal bacterium Bacteroides vulgatus exacerbates helminth-induced cardiac fibrosis through succinate accumulation. PLoS Pathog. 2025, 21, e1013069. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Xiao, H.-M.; Liu, H.-M.; Lv, W.-Q.; Greenbaum, J.; Gong, R.; Zhang, Q.; Chen, Y.-C.; Peng, C.; Xu, X.-J.; et al. Gut microbiota impacts bone via Bacteroides vulgatus-valeric acid-related pathways. Nat. Commun. 2023, 14, 6853. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Feng, X.; Xin, F.; An, R.; Huang, H.; Mao, L.; Liu, P.; Zhang, J.; Huang, H.; Liu, X.; et al. B. vulgatus ameliorates high-fat diet-induced obesity through modulating intestinal serotonin synthesis and lipid absorption in mice. Gut Microbes 2024, 16, 2423040. [Google Scholar] [CrossRef] [PubMed]
- Jiarpinijnun, A.; Benjakul, S.; Pornphatdetaudom, A.; Shibata, J.; Okazaki, E.; Osako, K. High Arachidonic Acid Levels in the Tissues of Herbivorous Fish Species (Siganus fuscescens, Calotomus japonicus and Kyphosus bigibbus). Lipids 2017, 52, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, H. Intake of arachidonic acid-containing lipids in adult humans: Dietary surveys and clinical trials. Lipids Health Dis. 2019, 18, 101. [Google Scholar] [CrossRef] [PubMed]
- Guesnet, P.; Alessandri, J.-M. Docosahexaenoic acid (DHA) and the developing central nervous system (CNS)—Implications for dietary recommendations. Biochimie 2011, 93, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, W.B. Fatty aldehyde and fatty alcohol metabolism: Review and importance for epidermal structure and function. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2014, 1841, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Honsho, M.; Fujiki, Y. Plasmalogen homeostasis—regulation of plasmalogen biosynthesis and its physiological consequence in mammals. FEBS Lett. 2017, 591, 2720–2729. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-S.; Tian, F.-W.; Zhao, J.-X.; Zhang, H.; Zhai, Q.-X.; Chen, W. The influence of dietary patterns on gut microbiome and its consequences for nonalcoholic fatty liver disease. Trends Food Sci. Technol. 2020, 96, 135–144. [Google Scholar] [CrossRef]
- Uranga, J.A.; López-Miranda, V.; Lombó, F.; Abalo, R. Food, nutrients and nutraceuticals affecting the course of inflammatory bowel disease. Pharmacol. Rep. 2016, 68, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhu, W.; Chen, C.; Yan, B.; Zhu, L.; Chen, X.; Peng, C. The mechanisms of lysophosphatidylcholine in the development of diseases. Life Sci. 2020, 247, 117443. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Lauterbach, M.; Latz, E. Western Diet and the Immune System: An Inflammatory Connection. Immunity 2019, 51, 794–811. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Baden, M.Y.; Guasch-Ferré, M.; Wittenbecher, C.; Li, J.; Li, Y.; Wan, Y.; Bhupathiraju, S.N.; Tobias, D.K.; Clish, C.B.; et al. Plasma metabolite profiles related to plant-based diets and the risk of type 2 diabetes. Diabetologia 2022, 65, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Bhandari, U.; Panda, B.P.; Dubey, K.; Khan, W.; Ahmad, S. Trigonelline inhibits intestinal microbial metabolism of choline and its associated cardiovascular risk. J. Pharm. Biomed. Anal. 2018, 159, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhang, N.; Song, Y.; Liu, L.; Li, J.; Zhang, Y.; Huo, Y.; Liu, X.; Duan, Y.; Wang, B.; et al. Serum L-Carnitine Levels Are Associated With First Stroke in Chinese Adults With Hypertension. Stroke 2022, 53, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhou, Q.; Wu, Y.; Chen, X.; Zhong, F. Properties and Mechanisms of Flavin-Dependent Monooxygenases and Their Applications in Natural Product Synthesis. Int. J. Mol. Sci. 2022, 23, 2622. [Google Scholar] [CrossRef] [PubMed]
- Kalagi, N.A.; Thota, R.N.; Stojanovski, E.; Alburikan, K.A.; Garg, M.L. Association between Plasma Trimethylamine N-Oxide Levels and Type 2 Diabetes: A Case Control Study. Nutrients 2022, 14, 2093. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, W.H.W.; Buffa, J.A.; Fu, X.; Britt, E.B.; Koeth, R.A.; Levison, B.S.; Fan, Y.; Wu, Y.; Hazen, S.L. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur. Heart J. 2014, 35, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Zheng, S.; Zhou, L.; Liu, X.; Chen, T.; Wang, X.; Li, Q.; Zhao, Y.; Wang, D.; Xu, G. High-Coverage Metabolomics Reveals Gut Microbiota-Related Metabolic Traits of Type-2 Diabetes in Serum. J. Proteome Res. 2025, 24, 1649–1661. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, J.G.; Wong, F.; Reddy, K.R.; Garcia-Tsao, G.; Kamath, P.S.; Biggins, S.W.; Fallon, M.B.; Subramanian, R.M.; Maliakkal, B.; Thacker, L.; et al. Gender-Specific Differences in Baseline, Peak, and Delta Serum Creatinine: The NACSELD Experience. Dig. Dis. Sci. 2017, 62, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.-M.; Wang, J.; Li, Z.-Y.; Tuo, J.-Y.; Tan, Y.-T.; Li, H.-L.; Xiang, Y.-B. Sex-Specific Correlation Analysis of Branched-Chain Amino Acids in Dietary Intakes and Plasma among Chinese Adults. J. Nutr. 2023, 153, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Smith-Ryan, A.E.; Cabre, H.E.; Eckerson, J.M.; Candow, D.G. Creatine Supplementation in Women’s Health: A Lifespan Perspective. Nutrients 2021, 13, 877. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Asian Chinese (n = 99) | European Caucasian (n = 72) | p-Value | Normo-Glycaemia (n = 130) | Prediabetes (n = 41) | p-Value |
|---|---|---|---|---|---|---|
| M:F ratio | 40:59 | 30:42 | 0.99 | 43:87 | 27:14 | <0.01 |
| Age (y) | 46.6 ± 12.8 | 51.2 ± 15.6 | 0.05 | 46.9 ± 14.2 | 53.8 ± 13.1 | <0.01 |
| Body weight (kg) | 73.2 ± 13 | 81.3 ± 16.7 | <0.01 | 75.2 ± 15.6 | 81.1 ± 12.8 | <0.01 |
| Height (m) | 1.7 ± 0.1 | 1.7 ± 0.1 | <0.01 | 1.7 ± 0.1 | 1.7 ± 0.1 | 0.27 |
| BMI (kg m−2) | 26.7 ± 3.7 | 27.1 ± 4.7 | 0.77 | 26.5 ± 4.4 | 28.1 ± 3.3 | 0.01 |
| Waist circumf. (cm) | 89.3 ± 10.4 | 93.2 ± 13.7 | 0.09 | 89.1 ± 11.8 | 96.8 ± 11.1 | <0.01 |
| Hip circumf. (cm) | 102.1 ± 6.6 | 107.6 ± 10 | <0.01 | 104.1 ± 8.9 | 105.3 ± 7.6 | 0.42 |
| SBP (mm Hg) | 123 ± 17.3 | 117.5 ± 16.7 | 0.02 | 119.1 ± 17.3 | 125.7 ± 16.1 | <0.01 |
| DBP (mm Hg) | 70 ± 11.1 | 65.4 ± 8.5 | <0.01 | 67.5 ± 10.2 | 70 ± 10.5 | 0.18 |
| TBF (kg) | 24.5 ± 7.2 | 28.1 ± 11.1 | 0.05 | 25.8 ± 9.5 | 26.8 ± 7.8 | 0.39 |
| TBF (%) | 34.7 ± 7 | 35.2 ± 9.2 | 0.86 | 35.1 ± 8.2 | 34.3 ± 7.4 | 0.29 |
| AAT (kg) | 2.3 ± 0.9 | 2.6 ± 1.4 | 0.15 | 2.3 ± 1.2 | 2.8 ± 1 | <0.01 |
| AAT (%) | 40.1 ± 9.2 | 39.8 ± 12.3 | 0.89 | 39.2 ± 11 | 42.4 ± 8.6 | 0.14 |
| VAT (kg) | 1.1 ± 0.7 | 1.1 ± 0.9 | 0.87 | 0.9 ± 0.7 | 1.5 ± 0.7 | <0.01 |
| VAT (%) | 43.4 ± 17.3 | 38.8 ± 20.4 | 0.08 | 37.2 ± 17.8 | 54.9 ± 14.9 | <0.01 |
| SAT (kg) | 1.2 ± 0.5 | 1.5 ± 0.8 | 0.03 | 1.4 ± 0.7 | 1.2 ± 0.6 | 0.11 |
| SAT (%) | 56.6 ± 17.3 | 61.2 ± 20.4 | 0.08 | 62.8 ± 17.8 | 45.1 ± 14.9 | <0.01 |
| VAT:SAT ratio | 1.0 ± 1.0 | 0.9 ± 1.0 | 0.08 | 0.8 ± 0.8 | 1.6 ± 1.3 | <0.01 |
| HbA1c (mmol mol−1) | 36.7 ± 4 | 34.5 ± 3.3 | <0.01 | 35.0 ± 3.6 | 38.3 ± 3.7 | <0.01 |
| FPG (mmol L−1) | 5.4 ± 0.5 | 5.2 ± 0.5 | 0.04 | 5.2 ± 0.5 | 5.8 ± 0.4 | <0.01 |
| ALT (U L−1) | 23.8 ± 19.9 | 16.5 ± 10.5 | <0.01 | 19.9 ± 16.7 | 23.4 ± 17.7 | 0.03 |
| AST (U L−1) | 24.7 ± 12.9 | 21.8 ± 5.1 | 0.58 | 23.4 ± 11.3 | 23.9 ± 7.4 | 0.2 |
| ALP (U L−1) | 60.9 ± 14.4 | 59.4 ± 14.6 | 0.56 | 60.1 ± 14.6 | 60.8 ± 14.1 | 0.74 |
| GGT (U L−1) | 30.6 ± 24.8 | 27.7 ± 27.1 | 0.26 | 26.5 ± 21.6 | 38.5 ± 34.8 | <0.01 |
| Total cholesterol (mmol L−1) | 5.2 ± 1 | 5.3 ± 1.1 | 0.44 | 5.2 ± 1.1 | 5.1 ± 1 | 0.4 |
| HDL-C (mmol L−1) | 1.5 ± 0.4 | 1.7 ± 0.5 | 0.05 | 1.6 ± 0.4 | 1.4 ± 0.3 | <0.01 |
| TG (mmol L−1) | 1.4 ± 0.9 | 1.1 ± 0.6 | 0.02 | 1.2 ± 0.7 | 1.6 ± 1 | <0.01 |
| LDL-C (mmol L−1) | 3 ± 0.8 | 3.1 ± 0.9 | 0.53 | 3.1 ± 0.9 | 3 ± 0.9 | 0.59 |
| Amylin (pg mL−1) | 31.2 ± 28.9 | 28.7 ± 17.4 | 0.76 | 28.9 ± 25.6 | 34.3 ± 21.3 | 0.07 |
| C-peptide (pg mL−1) | 1206.4 ± 633.7 | 1257.9 ± 555.2 | 0.49 | 1186.5 ± 596.2 | 1359.9 ± 603.3 | 0.02 |
| GIP (pg mL−1) | 63.2 ± 34.5 | 59.9 ± 27.7 | 0.84 | 60.4 ± 32.3 | 66.3 ± 29.9 | 0.19 |
| GLP-1 (pg mL−1) | 201.6 ± 98.3 | 193.5 ± 78.5 | 0.62 | 191.1 ± 92.4 | 220.7 ± 80.3 | <0.01 |
| Glucagon (pg mL−1) | 64.2 ± 31.3 | 57.7 ± 32 | 0.05 | 58.4 ± 31.3 | 71.1 ± 31.2 | <0.01 |
| Fasting insulin (pg mL−1) | 758.9 ± 695.3 | 766.4 ± 657.5 | 0.88 | 726 ± 640.4 | 876.3 ± 782.1 | 0.2 |
| HOMA2-IR | 2.3 ± 1.6 | 2.3 ± 1.8 | 0.85 | 2.2 ± 1.7 | 2.6 ± 1.8 | 0.13 |
| HOMA2-B | 140.9 ± 77.1 | 150.7 ± 83.1 | 0.6 | 148.2 ± 81.9 | 135.1 ± 72 | 0.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faraj, S.; Joblin-Mills, A.; Sequeira-Bisson, I.R.; Leiu, K.H.; Tung, T.; Wallbank, J.A.; Fraser, K.; Miles-Chan, J.L.; Poppitt, S.D.; Taylor, M.W. Investigating Multi-Omic Signatures of Ethnicity and Dysglycaemia in Asian Chinese and European Caucasian Adults: Cross-Sectional Analysis of the TOFI_Asia Study at 4-Year Follow-Up. Metabolites 2025, 15, 522. https://doi.org/10.3390/metabo15080522
Faraj S, Joblin-Mills A, Sequeira-Bisson IR, Leiu KH, Tung T, Wallbank JA, Fraser K, Miles-Chan JL, Poppitt SD, Taylor MW. Investigating Multi-Omic Signatures of Ethnicity and Dysglycaemia in Asian Chinese and European Caucasian Adults: Cross-Sectional Analysis of the TOFI_Asia Study at 4-Year Follow-Up. Metabolites. 2025; 15(8):522. https://doi.org/10.3390/metabo15080522
Chicago/Turabian StyleFaraj, Saif, Aidan Joblin-Mills, Ivana R. Sequeira-Bisson, Kok Hong Leiu, Tommy Tung, Jessica A. Wallbank, Karl Fraser, Jennifer L. Miles-Chan, Sally D. Poppitt, and Michael W. Taylor. 2025. "Investigating Multi-Omic Signatures of Ethnicity and Dysglycaemia in Asian Chinese and European Caucasian Adults: Cross-Sectional Analysis of the TOFI_Asia Study at 4-Year Follow-Up" Metabolites 15, no. 8: 522. https://doi.org/10.3390/metabo15080522
APA StyleFaraj, S., Joblin-Mills, A., Sequeira-Bisson, I. R., Leiu, K. H., Tung, T., Wallbank, J. A., Fraser, K., Miles-Chan, J. L., Poppitt, S. D., & Taylor, M. W. (2025). Investigating Multi-Omic Signatures of Ethnicity and Dysglycaemia in Asian Chinese and European Caucasian Adults: Cross-Sectional Analysis of the TOFI_Asia Study at 4-Year Follow-Up. Metabolites, 15(8), 522. https://doi.org/10.3390/metabo15080522