



The Complexity of Oxidative Stress in Human Age-Related Diseases—A Review

Abstract
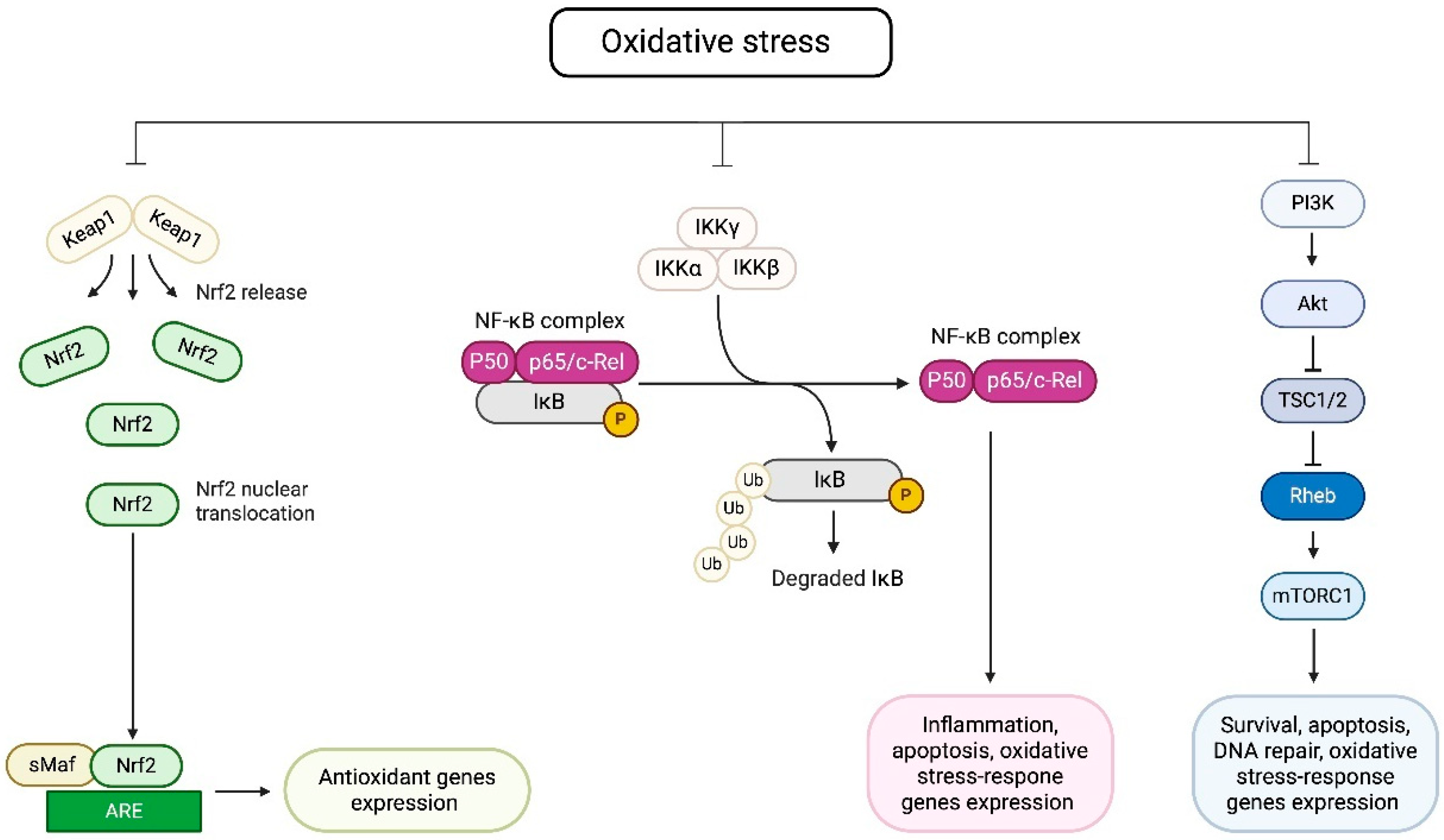
1. Introduction
2. Oxidative Stress
- −
- −
- −
- Impaired insulin signaling and increased oxidative burden in adipose tissue [40];
- −
- And DNA damage, contributing to telomere attrition and apoptosis.
3. Antioxidant Mechanisms
4. Age-Related Metabolic Challenges for the Antioxidant System
5. Antioxidant Enzyme Activities
5.1. SOD
- −
- Nuclear Factor-KappaB (NF-κB), sensitive to changes in the redox state in the cell [65];
- −
- Specificity Protein 1 (SP1 protein) [66];
- −
- Activator Protein-1 (AP-1), sensitive to, among others, cytokines and oxidative stress, described in the processes of cell proliferation and neoplastic transformation; in relation to SOD1, it is supposed to reduce gene transcription [67];
- −
- Activator Protein-2 (AP-2)—family of proteins through which ginsenoside Rb2 (active substance from the root of Panax ginseng) can increase sod1 transcription [68];
- −
- Proteins binding to the regulatory and enhancing sequence CCAAT, the so-called C/EBP (CCAAT-Enhancer-Binding Proteins), necessary for the basic transcription of the SOD1 [69].
5.2. CAT and GPX
5.3. PON1
6. Genetic Predisposition to Oxidative Stress Disturbances
6.1. Cardiovascular Disorders
6.2. Type 2 Diabetes Mellitus
6.3. Cancer
6.4. Accelerated Aging Diseases
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- United Nations. World Population Prospects—2017 Revision: Global Population | Nations Unies. Available online: https://www.un.org/fr/desa/world-population-prospects-2017-revision-global-population (accessed on 19 February 2025).
- Ageing. Available online: https://www.who.int/health-topics/ageing (accessed on 19 February 2025).
- GUS. Zdrowie i Ochrona Zdrowia w 2022 Roku. stat.gov.pl. Available online: https://stat.gov.pl/obszary-tematyczne/zdrowie/zdrowie/zdrowie-i-ochrona-zdrowia-w-2022-roku,1,13.html (accessed on 19 February 2025).
- Saeed, A.; Lopez, O.; Cohen, A.; Reis, S.E. Cardiovascular Disease and Alzheimer’s Disease: The Heart–Brain Axis. J. Am. Heart Assoc. 2023, 12, e030780. [Google Scholar] [CrossRef]
- Cleeland, C.; Pipingas, A.; Scholey, A.; White, D. Neurochemical changes in the aging brain: A systematic review. Neurosci. Biobehav. Rev. 2019, 98, 306–319. [Google Scholar] [CrossRef]
- Hong, S.; Choi, K.M. Sarcopenic Obesity, Insulin Resistance, Their Implications in Cardiovascular and Metabolic Consequences. Int. J. Mol. Sci. 2020, 21, 494. [Google Scholar] [CrossRef]
- Bao, Q.; Pan, J.; Qi, H.; Wang, L.; Qian, H.; Jiang, F.; Shao, Z.; Xu, F.; Tao, Z.; Ma, Q.; et al. Aging and age-related diseases—From endocrine therapy to target therapy. Mol. Cell. Endocrinol. 2014, 394, 115–118. [Google Scholar] [CrossRef]
- Koutsaliaris, I.K.; Moschonas, I.C.; Pechlivani, L.M.; Tsouka, A.N.; Tselepis, A.D. Inflammation, Oxidative Stress, Vascular Aging and Atherosclerotic Ischemic Stroke. Curr. Med. Chem. 2022, 29, 5496–5509. [Google Scholar] [CrossRef] [PubMed]
- Glassock, R.J.; Winearls, C. Ageing and the glomerular filtration rate: Truths and consequences. Trans. Am. Clin. Climatol. Assoc. 2009, 120, 419–428. [Google Scholar] [PubMed]
- Gallagher, J.C. Vitamin D and aging. Endocrinol. Metab. Clin. N. Am. 2013, 42, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Hage, C.; Salvatori, R. Growth Hormone and Aging. Endocrinol. Metab. Clin. N. Am. 2023, 52, 245–257. [Google Scholar] [CrossRef]
- Fraze, E.; Chiou, Y.A.; Chen, Y.D.; Reaven, G.M. Age-related changes in postprandial plasma glucose, insulin, and free fatty acid concentrations in nondiabetic individuals. J. Am. Geriatr. Soc. 1987, 35, 224–228. [Google Scholar] [CrossRef]
- Fleming, K.M.; West, J.; Aithal, G.P.; Fletcher, A.E. Abnormal liver tests in people aged 75 and above: Prevalence and association with mortality. Aliment. Pharmacol. Ther. 2011, 34, 324–334. [Google Scholar] [CrossRef]
- van Heemst, D. The ageing thyroid: Implications for longevity and patient care. Nat. Rev. Endocrinol. 2024, 20, 5–15. [Google Scholar] [CrossRef]
- Deyrup, A.T.; D’Ambrosio, D.; Muir, J.; Deyrup, A.; Knollmann-Ritschel, B.; Scordino, T.; Kraswoski, M.; Cao, L.; Shah, K.; Zepf, J.; et al. Essential laboratory tests for medical education. Acad. Pathol. 2022, 9, 100046. [Google Scholar] [CrossRef]
- Kruger, A. 1 The limits of normality in elderly patients. Baillières Clin. Haematol. 1987, 1, 271–289. [Google Scholar] [CrossRef]
- Nakamura, R.M.; Tan, E.M. Recent advances in laboratory tests and the significance of autoantibodies to nuclear antigens in systemic rheumatic diseases. Clin. Lab. Med. 1986, 6, 41–53. [Google Scholar] [CrossRef]
- Holt, P.R. Intestinal malabsorption in the elderly. Dig. Dis. 2007, 25, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Chew, J.; Tay, L.; Lim, J.P.; Leung, B.P.; Yeo, A.; Yew, S.; Ding, Y.Y.; Lim, W.S. Serum Myostatin and IGF-1 as Gender-Specific Biomarkers of Frailty and Low Muscle Mass in Community-Dwelling Older Adults. J. Nutr. Health Aging 2019, 23, 979–986. [Google Scholar] [CrossRef]
- Carcaillon, L.; García-García, F.J.; Tresguerres, J.A.F.; Avila, G.G.; Kireev, R.; Rodríguez-Mañas, L. Higher levels of endogenous estradiol are associated with frailty in postmenopausal women from the toledo study for healthy aging. J. Clin. Endocrinol. Metab. 2012, 97, 2898–2906. [Google Scholar] [CrossRef] [PubMed]
- Petermann-Rocha, F.; Chen, M.; Gray, S.R.; Ho, F.K.; Pell, J.P.; Celis-Morales, C. Factors associated with sarcopenia: A cross-sectional analysis using UK Biobank. Maturitas 2020, 133, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Arosio, B.; Calvani, R.; Ferri, E.; Coelho-Junior, H.J.; Carandina, A.; Campanelli, F.; Ghiglieri, V.; Marzetti, E.; Picca, A. Sarcopenia and Cognitive Decline in Older Adults: Targeting the Muscle–Brain Axis. Nutrients 2023, 15, 1853. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, B.; Yu, Y.; Gao, W.; Liu, W.; Chen, L.; Xia, Z.; Cao, Q. Vascular Aging in Ischemic Stroke. J. Am. Heart Assoc. 2024, 13, e033341. [Google Scholar] [CrossRef]
- Harman, D. Free radical theory of aging: An update: Increasing the functional life span. Ann. N. Y. Acad. Sci. 2006, 1067, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Dzięgielewska-Gęsiak, S.; Wyszomirska, K.; Fatyga, E.; Wysocka, E.; Muc-Wierzgoń, M. The role of oxidant-antioxidant markers and resistin in metabolic syndrome elderly individuals. Sci. Prog. 2021, 104, 00368504211006510. [Google Scholar] [CrossRef]
- Brivio, P.; Paladini, M.S.; Racagni, G.; Riva, M.A.; Calabrese, F.; Molteni, R. From Healthy Aging to Frailty: In Search of the Underlying Mechanisms. Curr. Med. Chem. 2019, 26, 3685–3701. [Google Scholar] [CrossRef]
- Dzięgielewska-Gęsiak, S.; Muc-Wierzgoń, M. Inflammation and Oxidative Stress in Frailty and Metabolic Syndromes—Two Sides of the Same Coin. Metabolites 2023, 13, 475. [Google Scholar] [CrossRef]
- The Relation of Inflammaging with Skeletal Muscle Properties in Elderly Men—PMC. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC6448117/ (accessed on 21 February 2025).
- Goldszmid, R.S.; Trinchieri, G. The price of immunity. Nat. Immunol. 2012, 13, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, A.; Monti, D.; Lanzarini, C.; Conte, M.; Pirazzini, C.; Giulia Bacalini, M.; Garagnani, P.; Giuliani, C.; Fontanesi, E.; Ostan, R.; et al. Immune System, Cell Senescence, Aging and Longevity—Inflamm-Aging Reappraised. Curr. Pharm. Des. 2013, 19, 1675–1679. [Google Scholar] [CrossRef]
- Raucci, A.; Di Maggio, S.; Scavello, F.; D’Ambrosio, A.; Bianchi, M.E.; Capogrossi, M.C. The Janus face of HMGB1 in heart disease: A necessary update. Cell. Mol. Life Sci. CMLS 2018, 76, 211–229. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.S.; Burt, K.G.; Jacobsen, T.; Fernandes, T.D.; Alipui, D.O.; Weber, K.T.; Levine, M.; Chavan, S.S.; Yang, H.; Tracey, K.J.; et al. High mobility group box-1 induces pro-inflammatory signaling in human nucleus pulposus cells via toll-like receptor 4-dependent pathway. J. Orthop. Res. 2019, 37, 220–231. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Bai, G.; Wang, Y.; Mak, J.K.L.; Ericsson, M.; Hägg, S.; Jylhävä, J. Is Frailty Different in Younger Adults Compared to Old? Prevalence, Characteristics, and Risk Factors of Early-Life and Late-Life Frailty in Samples from Sweden and UK. Gerontology 2023, 69, 1385–1393. [Google Scholar] [CrossRef]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, B.; Rahman, A.A.; Lee, S.; Malhotra, R. The Implications of Aging on Vascular Health. Int. J. Mol. Sci. 2024, 25, 11188. [Google Scholar] [CrossRef]
- Zhang, J.; Li, C.; Zhang, Y.; Wu, J.; Huang, Z. Therapeutic potential of nitric oxide in vascular aging due to the promotion of angiogenesis. Chem. Biol. Drug Des. 2023, 102, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in insulin resistance: Insights into mechanisms and therapeutic strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu-Wittel, M.; Gómez-Díaz, R.; González-Molina, Á.; Vidal-Serrano, S.; Díez-Manglano, J.; Salgado, F.; Soto-Martín, M.; Ollero-Baturone, M.; Proteo Researchers. Oxidative Stress, Telomere Shortening, and Apoptosis Associated to Sarcopenia and Frailty in Patients with Multimorbidity. J. Clin. Med. 2020, 9, 2669. [Google Scholar] [CrossRef]
- Jamal, A.; Brettle, H.; Jamil, D.A.; Tran, V.; Diep, H.; Bobik, A.; van der Poel, C.; Vinh, A.; Drummond, G.R.; Thomas, C.J.; et al. Reduced Insulin Resistance and Oxidative Stress in a Mouse Model of Metabolic Syndrome following Twelve Weeks of Citrus Bioflavonoid Hesperidin Supplementation: A Dose–Response Study. Biomolecules 2024, 14, 637. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- BTrist, G.; Hilton, J.B.; Hare, D.J.; Crouch, P.J.; Double, K.L. Superoxide Dismutase 1 in Health and Disease: How a Frontline Antioxidant Becomes Neurotoxic. Angew. Chem. Int. Ed. 2021, 60, 9215–9246. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide Dismutase. J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef] [PubMed]
- Vera-Ramirez, L.; Ramirez-Tortosa, M.; Perez-Lopez, P.; Granados-Principal, S.; Battino, M.; Quiles, J.L. Long-term effects of systemic cancer treatment on DNA oxidative damage: The potential for targeted therapies. Cancer Lett. 2012, 327, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Dziegielewska-Gesiak, S. Metabolic Syndrome in an Aging Society—Role of Oxidant-Antioxidant Imbalance and Inflammation Markers in Disentangling Atherosclerosis. Clin. Interv. Aging 2021, 16, 1057–1070. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Bartosz, G. Effect of Antioxidants Supplementation on Aging and Longevity. BioMed Res. Int. 2014, 2014, 404680. [Google Scholar] [CrossRef]
- Lauro, D.; Pastore, D.; Capuani, B.; Pacifici, F.; Palmirotta, R.; Abete, P.; Roselli, M.; Bellia, A.; Federici, M.; Di Daniele, N.; et al. Role of Serum and Glucocorticoid-Inducible Kinase (SGK)-1 in Senescence: A Novel Molecular Target Against Age-Related Diseases. Curr. Med. Chem. 2015, 22, 3765–3788. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-Y.; Wu, Y.H.; Liu, L.K.; Lee, W.J.; Hwang, A.C.; Peng, L.N.; Lin, M.H.; Chen, L.K. Association Among Serum Insulin-Like Growth Factor-1, Frailty, Muscle Mass, Bone Mineral Density, and Physical Performance Among Community-Dwelling Middle-Aged and Older Adults in Taiwan. Rejuvenation Res. 2018, 21, 270–277. [Google Scholar] [CrossRef]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef]
- Bale, B.F.; Doneen, A.L.; Leimgruber, P.P.; Vigerust, D.J. The critical issue linking lipids and inflammation: Clinical utility of stopping oxidative stress. Front. Cardiovasc. Med. 2022, 9, 1042729. [Google Scholar] [CrossRef]
- Kang, E.; Li, Y.; Kim, B.; Huh, K.Y.; Han, M.; Ahn, J.H.; Sung, H.Y.; Park, Y.S.; Lee, S.E.; Lee, S.; et al. Identification of Serum Metabolites for Predicting Chronic Kidney Disease Progression according to Chronic Kidney Disease Cause. Metabolites 2022, 12, 1125. [Google Scholar] [CrossRef]
- Karakousis, N.D.; Biliou, S.; Pyrgioti, E.E.; Georgakopoulos, P.N.; Liakopoulos, V.; Papanas, N. Frailty, sarcopenia and diabetic kidney disease: Where do we stand? Int. Urol. Nephrol. 2023, 55, 1173–1181. [Google Scholar] [CrossRef]
- Trim, W.; Turner, J.E.; Thompson, D. Parallels in Immunometabolic Adipose Tissue Dysfunction with Ageing and Obesity. Front. Immunol. 2018, 9, 330241. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Catena, G.; Gaudio, G.; D’Angelo, A.; Maffioli, P. Adipose tissue dysfunction and metabolic disorders: Is it possible to predict who will develop type 2 diabetes mellitus? Role of markErs in the progreSsion of dIabeteS in obese paTIeNts (The RESISTIN trial). Cytokine 2020, 127, 154947. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; Zeeshan, H.M.A.; Kim, H.-R.; Chae, H.-J. Nox4 regulates the eNOS uncoupling process in aging endothelial cells. Free Radic. Biol. Med. 2017, 113, 26–35. [Google Scholar] [CrossRef]
- Lassègue, B.; Martín, A.S.; Griendling, K.K. Biochemistry, Physiology and Pathophysiology of NADPH Oxidases in the Cardiovascular System. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef]
- Silva, J.F.; Diniz, T.F.; Lima, P.M.; Santos, R.L.; Cortes, S.F.; Coimbra, C.C.; Lemos, V.S. Obesity, Inflammation, and Exercise Training: Relative Contribution of iNOS and eNOS in the Modulation of Vascular Function in the Mouse Aorta. Front. Physiol. 2016, 7, 386. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.J.P.; Shin, D.S.; Getzoff, E.D.; Tainer, J.A. The structural biochemistry of the superoxide dismutases. Biochim. Biophys. Acta 2010, 1804, 245–262. [Google Scholar] [CrossRef]
- VCulotta, C.; Yang, M.; O’Halloran, T.V. Activation of superoxide dismutases: Putting the metal to the pedal. Biochim. Biophys. Acta 2006, 1763, 747–758. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef]
- Valentine, J.S.; Doucette, P.A.; Potter, S.Z. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 2005, 74, 563–593. [Google Scholar] [CrossRef]
- Miao, L.; Clair, D.K.S. Regulation of superoxide dismutase genes: Implications in disease. Free Radic. Biol. Med. 2009, 47, 344–356. [Google Scholar] [CrossRef]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef] [PubMed]
- Minc, E.; De Coppet, P.; Masson, P.; Thiery, L.; Dutertre, S.; Amor-Guéret, M.; Jaulin, C. The human copper-zinc superoxide dismutase gene (SOD1) proximal promoter is regulated by Sp1, Egr-1, and WT1 via non-canonical binding sites. J. Biol. Chem. 1999, 274, 503–509. [Google Scholar] [CrossRef]
- Baldelli, S.; Aquilano, K.; Rotilio, G.; Ciriolo, M.R. Glutathione and copper, zinc superoxide dismutase are modulated by overexpression of neuronal nitric oxide synthase. Int. J. Biochem. Cell Biol. 2008, 40, 2660–2670. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, K.H.; Rho, H.M. Transcriptional activation of the Cu,Zn-superoxide dismutase gene through the AP2 site by ginsenoside Rb2 extracted from a medicinal plant, Panax ginseng. J. Biol. Chem. 1996, 271, 24539–24543. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.J.; Kim, H.T.; Cho, G.; Rho, H.M.; Jung, G. Sp1 and C/EBP-related factor regulate the transcription of human Cu/Zn SOD gene. Gene 1996, 178, 177–185. [Google Scholar] [CrossRef]
- Chang, M.S.; Yoo, H.Y.; Rho, H.M. Positive and negative regulatory elements in the upstream region of the rat Cu/Zn-superoxide dismutase gene. Biochem. J. 1999, 339 Pt 2, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.Y.; Chang, M.S.; Rho, H.M. Induction of the rat Cu/Zn superoxide dismutase gene through the peroxisome proliferator-responsive element by arachidonic acid. Gene 1999, 234, 87–91. [Google Scholar] [CrossRef]
- Cho, G.; Kang, S.; Seo, S.J.; Kim, Y.; Jung, G. The transcriptional repression of the human Cu/Zn superoxide dismutase(sod1) gene by the anticancer drug, mitomycin C (MMC). Biochem. Mol. Biol. Int. 1997, 42, 949–956. [Google Scholar] [CrossRef]
- Park, E.Y.; Rho, H.M. The transcriptional activation of the human copper/zinc superoxide dismutase gene by 2,3,7,8-tetrachlorodibenzo-p-dioxin through two different regulator sites, the antioxidant responsive element and xenobiotic responsive element. Mol. Cell. Biochem. 2002, 240, 47–55. [Google Scholar] [CrossRef]
- Oates, N.; Pamphlett, R. An epigenetic analysis of SOD1 and VEGF in ALS. Amyotroph. Lateral Scler. 2007, 8, 83–86. [Google Scholar] [CrossRef]
- Ozata, M.; Oktenli, C.; Aydin, A.; Sanisoglu, S.Y.; Bolu, E.; Yilmaz, M.I.; Sayal, A.; Isimer, A.; Ozdemir, I.C. Increased oxidative stress and hypozincemia in male obesity. Clin. Biochem. 2002, 35, 627–631. [Google Scholar] [CrossRef]
- Abou-Seif, M.A.; Youssef, A.-A. Evaluation of some biochemical changes in diabetic patients. Clin. Chim. Acta Int. J. Clin. Chem. 2004, 346, 161–170. [Google Scholar] [CrossRef]
- Colak, E.; Majkić-Singh, N.; Stanković, S.; Srecković-Dimitrijević, V.; Djordjević, P.B.; Lalić, K.; Lalić, N. Parameters of antioxidative defense in type 2 diabetic patients with cardiovascular complications. Ann. Med. 2005, 37, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Yuan, J.Q.; Lv, Y.B.; Gao, X.; Yin, Z.X.; Kraus, V.B.; Luo, J.S.; Chei, C.L.; Matchar, D.B.; Zeng, Y.; et al. Associations between superoxide dismutase, malondialdehyde and all-cause mortality in older adults: A community-based cohort study. BMC Geriatr. 2019, 19, 104. [Google Scholar] [CrossRef]
- Masle, A.M.; Kibel, A.; Jukić, I.; Čičak, P.; Selthofer-Relatić, K.; Stupin, A.; Mihaljević, Z.; Šušnjara, P.; Breškić Ćurić, Ž.; Bačun, T.; et al. Enhancing Endothelial Function with Nutrient-Enriched Table Hen Eggs: A Randomized Study in Patients Recovering from Acute Coronary Syndrome. Clin. Interv. Aging 2024, 19, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Quetglas-Llabrés, M.M.; Monserrat-Mesquida, M.; Bouzas, C.; García, S.; Mateos, D.; Ugarriza, L.; Gómez, C.; Sureda, A.; Tur, J.A. Long-Term Impact of Nutritional Intervention with Increased Polyphenol Intake and Physical Activity Promotion on Oxidative and Inflammatory Profiles in Patients with Metabolic Syndrome. Nutrients 2024, 16, 2121. [Google Scholar] [CrossRef] [PubMed]
- Tavakoli, A.; Akhgarjand, C.; Ansar, H.; Houjaghani, H.; Khormani, A.; Djafarian, K.; Rostamian, A.; Ranjbar, M.; Farsani, G.M. The effects of intermittent fasting on antioxidant and inflammatory markers and liver enzymes in postmenopausal, overweight and obese women with rheumatoid arthritis: A randomized controlled trial. Sci. Rep. 2025, 15, 2357. [Google Scholar] [CrossRef]
- Bednarska-Makaruk, M.; Rodo, M.; Szirkowiec, W.; Mossakowska, M.; Puzianowska-Kuźnicka, M.; Skalska, A.; Zdrojewski, T.; Ryglewicz, D.; Wehr, H. Paraoxonase 1 activity and level of antibodies directed against oxidized low density lipoproteins in a group of an elderly population in Poland—PolSenior study. Arch. Gerontol. Geriatr. 2015, 60, 153–161. [Google Scholar] [CrossRef]
- Meisinger, C.; Freuer, D.; Bub, A.; Linseisen, J. Association between inflammatory markers and serum paraoxonase and arylesterase activities in the general population: A cross-sectional study. Lipids Health Dis. 2021, 20, 81. [Google Scholar] [CrossRef]
- Erdman, V.; Tuktarova, I.; Nasibullin, T.; Timasheva, Y.; Petintseva, A.; Korytina, G. Polygenic markers of survival and longevity in the antioxidant genes PON1, PON2, MTHFR, MSRA, SOD1, NQO1, and CAT in a 20-year follow-up study in the population from the Volga-Ural region. Gene 2024, 919, 148510. [Google Scholar] [CrossRef]
- Krishnamurthy, H.K.; Rajavelu, I.; Pereira, M.; Jayaraman, V.; Krishna, K.; Wang, T.; Bei, K.; Rajasekaran, J.J. Inside the genome: Understanding genetic influences on oxidative stress. Front. Genet. 2024, 15, 1397352. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://string-db.org (accessed on 20 April 2025).
- Gusti, A.M.T.; Qusti, S.Y.; Alshammari, E.M.; Toraih, E.A.; Fawzy, M.S. Antioxidants-Related Superoxide Dismutase (SOD), Catalase (CAT), Glutathione Peroxidase (GPX), Glutathione-S-Transferase (GST), and Nitric Oxide Synthase (NOS) Gene Variants Analysis in an Obese Population: A Preliminary Case-Control Study. Antioxidants 2021, 10, 595. [Google Scholar] [CrossRef]
- Kopp, T.I.; Vogel, U.; Dragsted, L.O.; Tjonneland, A.; Ravn-Haren, G. Association between single nucleotide polymorphisms in the antioxidant genes CAT, GR and SOD1, erythrocyte enzyme activities, dietary and life style factors and breast cancer risk in a Danish, prospective cohort study. Oncotarget 2017, 8, 62984–62997. [Google Scholar] [CrossRef]
- Huang, J.-Q.; Zhou, J.-C.; Wu, Y.-Y.; Ren, F.-Z.; Lei, X.G. Role of glutathione peroxidase 1 in glucose and lipid metabolism-related diseases. Free Radic. Biol. Med. 2018, 127, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.M.; Veeramachaneni, R.; Deng, D.; Putluri, N.; Putluri, V.; Cardenas, M.F.; Wheeler, D.A.; Decker, W.K.; Frederick, A.I.; Kazi, S.; et al. Glutathione peroxidase 2 is a metabolic driver of the tumor immune microenvironment and immune checkpoint inhibitor response. J. Immunother. Cancer 2022, 10, e004752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Liao, Y.; Zhu, C.; Zou, Z. GPX4, ferroptosis, and diseases. Biomed. Pharmacother. 2024, 174, 116512. [Google Scholar] [CrossRef]
- Montazeri-Najafabady, N.; Dabbaghmanesh, M.H.; Jahromi, B.N.; Chatrabnous, N.; Chatrsimin, F. The impact of GSTM1 and GSTT1 polymorphisms on susceptibility to gestational diabetes in Iranian population. J. Matern.-Fetal Neonatal Med. 2022, 35, 1451–1456. [Google Scholar] [CrossRef]
- Shnayder, N.A.; Petrova, M.M.; Moskaleva, P.V.; Shesternya, P.A.; Pozhilenkova, E.A.; Nasyrova, R.F. The Role of Single-Nucleotide Variants of NOS1, NOS2, and NOS3 Genes in the Comorbidity of Arterial Hypertension and Tension-Type Headache. Molecules 2021, 26, 1556. [Google Scholar] [CrossRef]
- Mercado, N.; Kizawa, Y.; Ueda, K.; Xiong, Y.; Kimura, G.; Moses, A.; Curtis, J.M.; Ito, K.; Barnes, P.J. Activation of transcription factor Nrf2 signalling by the sphingosine kinase inhibitor SKI-II is mediated by the formation of Keap1 dimers. PLoS ONE 2014, 9, e88168. [Google Scholar] [CrossRef]
- Arab, Z.N.; Khayatan, D.; Razavi, S.M.; Zare, K.; Kheradkhah, E.; Momtaz, S.; Ferretti, G.; Bacchetti, T.; Sathyapalan, T.; Emami, S.A. Phytochemicals as Modulators of Paraoxonase-1 in Health and Diseases. Antioxidants 2022, 11, 1273. [Google Scholar] [CrossRef]
- Witte, I.; Foerstermann, U.; Devarajan, A.; Reddy, S.T.; Horke, S. Protectors or Traitors: The Roles of PON2 and PON3 in Atherosclerosis and Cancer. J. Lipids 2012, 2012, 342806. [Google Scholar] [CrossRef]
- Zhang, X.; Lv, S.; Guo, C.; Shi, C.; Chi, Y.; Zhao, L.; Wang, G.; Wang, Z. Gene-gene interaction between PPARG and CYP1A1 gene on coronary artery disease in the Chinese Han Population. Oncotarget 2017, 8, 34398–34404. [Google Scholar] [CrossRef]
- Song, Y.; Raheel, T.M.; Jia, A.; Dai, G.; Liu, L.; Long, X.; He, C. rs10865710 polymorphism in PPARG promoter is associated with the severity of type 2 diabetes mellitus and coronary artery disease in a Chinese population. Postgrad. Med. J. 2022, 98, 778–787. [Google Scholar] [CrossRef]
- Torsdottir, G.; Kristinsson, J.; Snaedal, J.; Sveinbjörnsdóttir, S.; Gudmundsson, G.; Hreidarsson, S.; Jóhannesson, T. Case-control studies on ceruloplasmin and superoxide dismutase (SOD1) in neurodegenerative diseases: A short review. J. Neurol. Sci. 2010, 299, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Tran, G.-B.; Nguyen, C.T. Anti-oxidative effects of superoxide dismutase 3 on inflammatory diseases. J. Mol. Med. Berl. Ger. 2020, 98, 59–69. [Google Scholar] [CrossRef]
- Burdon, K.P.; Rudock, M.E.; Lehtinen, A.B.; Langefeld, C.D.; Bowden, D.W.; Register, T.C.; Liu, Y.; Freedman, B.I.; Carr, J.J.; Hedrick, C.C.; et al. Human lipoxygenase pathway gene variation and association with markers of subclinical atherosclerosis in the diabetes heart study. Mediators Inflamm. 2010, 2010, 170153. [Google Scholar] [CrossRef] [PubMed]
- Racis, M.; Stanisławska-Sachadyn, A.; Sobiczewski, W.; Wirtwein, M.; Krzemiński, M.; Krawczyńska, N.; Limon, J.; Rynkiewicz, A.; Gruchała, M. Association of Genes Related to Oxidative Stress with the Extent of Coronary Atherosclerosis. Life 2020, 10, 210. [Google Scholar] [CrossRef]
- Akhtar, S.; Mahjabeen, I.; Akram, Z.; Kayani, M.A. CYP1A1 and GSTP1 gene variations in breast cancer: A systematic review and case-control study. Fam. Cancer 2016, 15, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Regensburger, M.; Ip, C.W.; Kohl, Z.; Schrader, C.; Urban, P.P.; Kassubek, J.; Jost, W.H. Clinical benefit of MAO-B and COMT inhibition in Parkinson’s disease: Practical considerations. J. Neural Transm. 2023, 130, 847–861. [Google Scholar] [CrossRef]
- Miranda, K.M.; Ridnour, L.A.; Cheng, R.Y.S.; Wink, D.A.; Thomas, D.D. The Chemical Biology of NO that Regulates Oncogenic Signaling and Metabolism: NOS2 and Its Role in Inflammatory Disease. Crit. Rev. Oncog. 2023, 28, 27–45. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef]
- Mousavi, S.; Tabari, M.A.K.; Bagheri, A.; Samieefar, N.; Shaterian, N.; Kelishadi, R. The Role of p66Shc in Diabetes: A Comprehensive Review from Bench to Bedside. J. Diabetes Res. 2022, 2022, 7703520. [Google Scholar] [CrossRef]
- Martín-Vázquez, E.; Cobo-Vuilleumier, N.; López-Noriega, L.; Lorenzo, P.I.; Gauthier, B.R. The PTGS2/COX2-PGE(2) signaling cascade in inflammation: Pro or anti? A case study with type 1 diabetes mellitus. Int. J. Biol. Sci. 2023, 19, 4157–4165. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Bian, Y.; Liu, L.; Liu, L.; Liu, X.; Ma, S. Molecular pathways associated with oxidative stress and their potential applications in radiotherapy (Review). Int. J. Mol. Med. 2022, 49, 65. [Google Scholar] [CrossRef]
- Pignatelli, P.; Menichelli, D.; Pastori, D.; Violi, F. Oxidative stress and cardiovascular disease: New insights. Pol. Heart J. Kardiologia Pol. 2018, 76, 713–722. [Google Scholar] [CrossRef]
- Kresanov, P.; Vasankari, T.; Ahotupa, M.; Kaikkonen, J.; Hutri-Kähönen, N.; Juonala, M.; Kähönen, M.; Lehtimäki, T.; Viikari, J.; Raitakari, O.T. Paraoxonase-1 and oxidized lipoprotein lipids. The Cardiovascular Risk in Young Finns Study. Atherosclerosis 2015, 241, 502–506. [Google Scholar] [CrossRef]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar]
- Gusti, A.M.T.; Qusti, S.Y.; Bahijri, S.M.; Toraih, E.A.; Bokhari, S.; Attallah, S.M.; Alzahrani, A.; Alshehri, W.M.; Alotaibi, H.; Fawzy, M.S. Glutathione S-Transferase (GSTT1 rs17856199) and Nitric Oxide Synthase (NOS2 rs2297518) Genotype Combination as Potential Oxidative Stress-Related Molecular Markers for Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 1385–1403. [Google Scholar] [CrossRef]
- Sarhangi, N.; Sharifi, F.; Hashemian, L.; Hassani Doabsari, M.; Heshmatzad, K.; Rahbaran, M.; Jamaldini, S.H.; Aghaei Meybodi, H.R.; Hasanzad, M. PPARG (Pro12Ala) genetic variant and risk of T2DM: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 12764. [Google Scholar] [CrossRef] [PubMed]
- Polvani, S.; Tarocchi, M.; Galli, A. PPAR and Oxidative Stress: Con() Catenating NRF2 and FOXO. PPAR Res. 2021, 2012, 641087. [Google Scholar] [CrossRef]
- Zimta, A.-A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.J.; Kabeer, A.; Abbas, Z.; Siddiqui, H.A.; Calina, D.; Sharifi-Rad, J.; Cho, W.C. Interplay of oxidative stress, cellular communication and signaling pathways in cancer. Cell Commun. Signal. 2024, 22, 7. [Google Scholar] [CrossRef]
- Wang, X.; Guo, Z.; Wang, J.; Lu, C.; Wang, J.; Jin, R.; Mo, Z. SNRPB promotes the progression of hepatocellular carcinoma via regulating cell cycle, oxidative stress, and ferroptosis. Aging 2024, 16, 348. [Google Scholar] [CrossRef]
- Milosic, F.; Hengstschläger, M.; Osmanagic-Myers, S. Premature aging in genetic diseases: What conclusions can be drawn for physiological aging. Front. Aging 2024, 4, 1327833. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.J.; Gordon, L.B. Hutchinson–Gilford progeria syndrome. Handb. Clin. Neurol. 2015, 66, 249–264. [Google Scholar] [CrossRef]
- Mozzini, C.; Setti, A.; Cicco, S.; Pagani, M. The Most Severe Paradigm of Early Cardiovascular Disease: Hutchinson-Gilford Progeria. Focus on the Role of Oxidative Stress. Curr. Probl. Cardiol. 2022, 47, 100900. [Google Scholar] [CrossRef]
- Young, S.G.; Meta, M.; Yang, S.H.; Fong, L.G. Prelamin A farnesylation and progeroid syndromes. J. Biol. Chem. 2006, 281, 39741–39745. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Lautrup, S.; Jensen, M.B.; Yang, B.; SenGupta, T.; Caponio, D.; Khezri, R.; Demarest, T.G.; Aman, Y. NAD+ augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019, 10, 5284. [Google Scholar] [CrossRef]
- Kyng, K.J.; May, A.; Brosh, R.M., Jr.; Cheng, W.H.; Chen, C.; Becker, K.G.; Bohr, V.A. The transcriptional response after oxidative stress is defective in Cockayne syndrome group B cells. Oncogene 2003, 22, 1135–1149. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Human Body System | Age-Related Changes in Laboratory Tests |
|---|---|
| Digestive system | Liver dysfunction: increase in globulin, VII and VIII factors, alkaline phosphatase [13] Variable (increase, decrease, no change) changes in aminotransferases [13] Gastric pH increase [18] Decrease absorption of Ca2+ and Fe2+ [18] |
| Endocrine system | Glucose level increase (1–2 mg/dl each decade from 30 y.o.) Postprandial glucose concentration increase (4 mg/dl each decade from 30 y.o.) [12] Decrease in thyroid hormones, renin, aldosterone, growth hormone, testosterone, estrogens, vitamin D, and calciferol absorption [10,11,14,19,20] Increase in antidiuretic hormone |
| Immune system | Decreases in immunoglobulin G, immunoglobulin M, and bone marrow reserve (changes within the normal range) [15,16] Increases in antibody levels, immunoglobulin A, and erythrocyte sedimentation Rate [15,16] Rheumatoid factor false positive presence [17] Changes in lymphocyte number and function (e.g., tuberculin test false negative) [16] |
| Urinary tract | Decrease in glomerular filtration rate and creatine clearance (10 mL/min/1.73m2 each decade from 40 y.o.) [9] |
| Enzyme of Interest | Study Population | Age Group | Disease/Condition of Interest | Results/Sample Material | Interpretation/Conclusion | Study No |
|---|---|---|---|---|---|---|
| CAT | Patients with MetS—long-term follow-up | 55–75 y.o. | Metabolic syndrome | ↑ CAT activity (no BMI reduction)/plasma | Compensatory response to persistent oxidative stress | [80] |
| CAT | Postmenopausal women with RA | 48–64 y.o. | Rheumatoid arthritis | ↑ CAT activity/serum | Anti-inflammatory effect of intermittent fasting | [81] |
| GPX | ACS patients—nutritional intervention | 48–66 y.o. | Post-acute coronary syndrome | ↑ GPX activity/serum | Reversible changes under redox-optimized diet | [79] |
| GPX, SOD | Patients with type 2 diabetes and CVD | ~60 y.o. | Metabolic and cardiovascular comorbidities | ↓ GPX/plasma and ↓SOD/RBC | Indicative of increased oxidative burden | [77] |
| PON1 | General elderly population (PolSenior study) | ≥65 y.o. | Age, metabolic dysfunction, inflammation | ↓ Arylesterase activity/serum | Decline with age and inflammatory markers | [82] |
| PON1 | Patients with CAD and type 2 diabetes | 56–72 y.o. | Coronary artery disease | ↓ PON1 activity/serum | Low PON1 linked to increased cardiovascular risk | [83] |
| SOD | General elderly cohort | ≥65 y.o. (median of 86 years) | Mortality follow-up | ↑ SOD activity in women/plasma | Associated with lower all-cause mortality | [78] |
| Gene | MIM Number | Type | Gene Function Result | Age-Related Disease | References |
|---|---|---|---|---|---|
| CAT | 115500 | Antioxidant | Decomposes hydrogen peroxide into water and oxygen | Cardiovascular disease, diabetes, cancer | [79,88] |
| GPX1 | 138320 | Antioxidant | Reduces hydrogen peroxide to water | Atherosclerosis, diabetes | [89] |
| GPX2 | 603749 | Antioxidant | Reduces hydrogen peroxide and lipid hydroperoxides | Cancer | [90] |
| GPX4 | 138322 | Antioxidant | Reduces lipid peroxides, crucial for ferroptosis regulation | Cardiovascular disease, neurogenerative diseases, cancer | [91] |
| GSTT1 | 600436 | Antioxidant | Detoxifies xenobiotics | Diabetes, cancer | [92] |
| NOS1 | 163731 | Antioxidant | Produces nitric oxide, regulates neurotransmission | Neurogenerative diseases, hypertension | [93] |
| NOS3 | 163729 | Antioxidant | Endothelial nitric oxide production, regulates vascular tone | Cardiovascular disease, hypertension | [93] |
| NRF2 | 600492 | Antioxidant | Regulates antioxidant response elements (AREs) | Neurogenerative diseases, chronic obstructive pulmonary disease | [94] |
| PON1 | 168820 | Antioxidant | Hydrolyzes lipid peroxides, anti-atherosclerotic | Cardiovascular disease, neurogenerative diseases | [95] |
| PON2 | 602447 | Antioxidant | Cellular antioxidant, protects against oxidative stress | Cancer, neurogenerative diseases | [96] |
| PON3 | 602448 | Antioxidant | Prevents LDL oxidation, anti-atherosclerotic | Atherosclerosis, metabolic syndrome | [96] |
| PPARG | 601487 | Antioxidant | Regulates lipid metabolism, inflammation control | Cardiovascular disease, diabetes | [97,98] |
| SOD1 | 147450 | Antioxidant | Converts superoxide radicals into oxygen and hydrogen peroxide | Amyotrophic lateral sclerosis, Alzheimer’s disease, cardiovascular diseases | [43,99] |
| SOD2 | 147460 | Antioxidant | Mitochondrial superoxide scavenging | Parkinson’s disease, cardiovascular diseases, cancer | [43,99] |
| SOD3 | 185490 | Antioxidant | Extracellular superoxide scavenging | Inflammatory diseases, atherosclerosis | [43,100] |
| ALOX15 | 603693 | Pro-oxidant | Produces lipid peroxidation products | Atherosclerosis, diabetes | [101] |
| CYBA | 608508 | Pro-oxidant | Component of NADPH oxidase, generates reactive oxygen species | Atherosclerosis, hypertension | [102] |
| CYP1A1 | 108330 | Pro-oxidant | Metabolizes xenobiotics, generates oxidative metabolites | Cardiovascular disease, cancer | [97,103] |
| MAO | 309850 | Pro-oxidant | Catalyzes oxidation of neurotransmitters, produces hydrogen peroxide | Neurodegenerative diseases, Parkinson’s disease | [104] |
| NOS2 | 163730 | Pro-oxidant | Produces nitric oxide in immune response | Inflammatory diseases, cancer | [105] |
| NOX1 | 300763 | Pro-oxidant | Produces reactive oxygen species | Atherosclerosis, cancer | [106] |
| NOX2 | 300481 | Pro-oxidant | Mediates oxidative burst in immune cells | Alzheimer’s disease, stroke | [107] |
| NOX4 | 605261 | Pro-oxidant | Regulates reactive oxygen species in mitochondria | Cardiovascular disease, hypertension | [108] |
| P66Shc | 600619 | Pro-oxidant | Regulates mitochondrial reactive oxygen species production | Diabetes | [109] |
| PTGS2 | 600262 | Pro-oxidant | Involved in prostaglandin synthesis, inflammation | Inflammatory diseases, cancer | [110] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Płóciniczak, A.; Bukowska-Olech, E.; Wysocka, E. The Complexity of Oxidative Stress in Human Age-Related Diseases—A Review. Metabolites 2025, 15, 479. https://doi.org/10.3390/metabo15070479
Płóciniczak A, Bukowska-Olech E, Wysocka E. The Complexity of Oxidative Stress in Human Age-Related Diseases—A Review. Metabolites. 2025; 15(7):479. https://doi.org/10.3390/metabo15070479
Chicago/Turabian StylePłóciniczak, Alicja, Ewelina Bukowska-Olech, and Ewa Wysocka. 2025. "The Complexity of Oxidative Stress in Human Age-Related Diseases—A Review" Metabolites 15, no. 7: 479. https://doi.org/10.3390/metabo15070479
APA StylePłóciniczak, A., Bukowska-Olech, E., & Wysocka, E. (2025). The Complexity of Oxidative Stress in Human Age-Related Diseases—A Review. Metabolites, 15(7), 479. https://doi.org/10.3390/metabo15070479