
Exploring the Mechanism of Luteolin in Protecting Chickens from Ammonia Poisoning Based on Proteomic Technology



Abstract

1. Introduction
2. Experimental Methods
2.1. Ethical Considerations
2.2. Primary Cell Culture and Construction of the Lut-Protected Ammonia Poisoning Injury Model
2.3. Protein Isolation and Proteolytic Processing
2.4. Utilizing Liquid Chromatography and Tandem Mass Spectrometry for Quantitative Analysis
2.5. Database Searching
2.6. Data Quality Control and Quantitative Analysis
2.7. Bioinformatics Analysis
2.7.1. Functional Annotation of Differentially Expressed Proteins (DEPs)
2.7.2. Functional Classification and Enrichment Analysis
2.7.3. Enrichment-Based Clustering
2.7.4. Protein–Protein Interaction (PPI) Network
2.8. Comparative Analysis of mRNA Expression Levels
2.9. Analytical Statistics
3. Results and Analysis
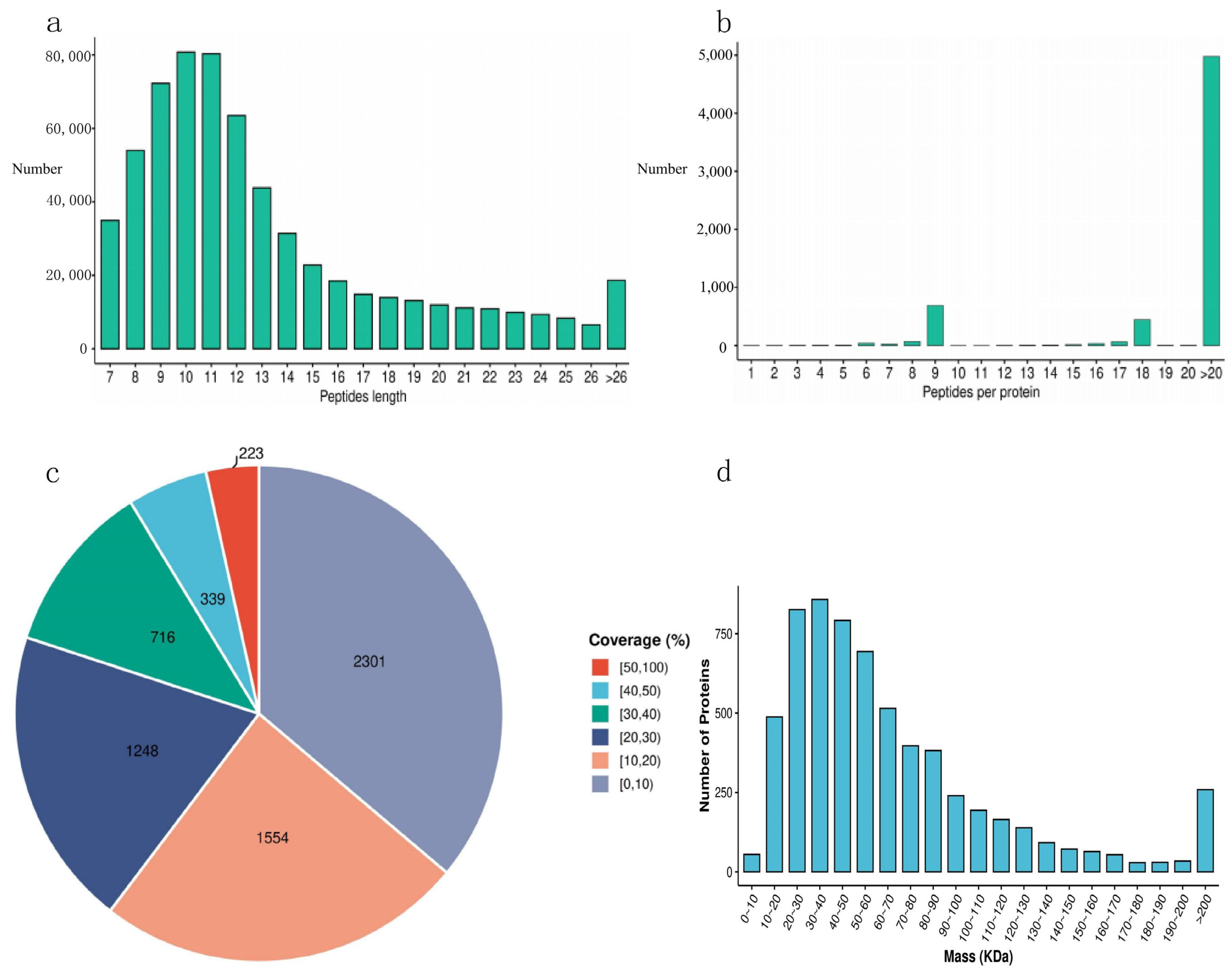
3.1. Analysis of Data Quality Assurance
3.2. Protein Quantitative Analysis
3.3. Protein Functional Annotation
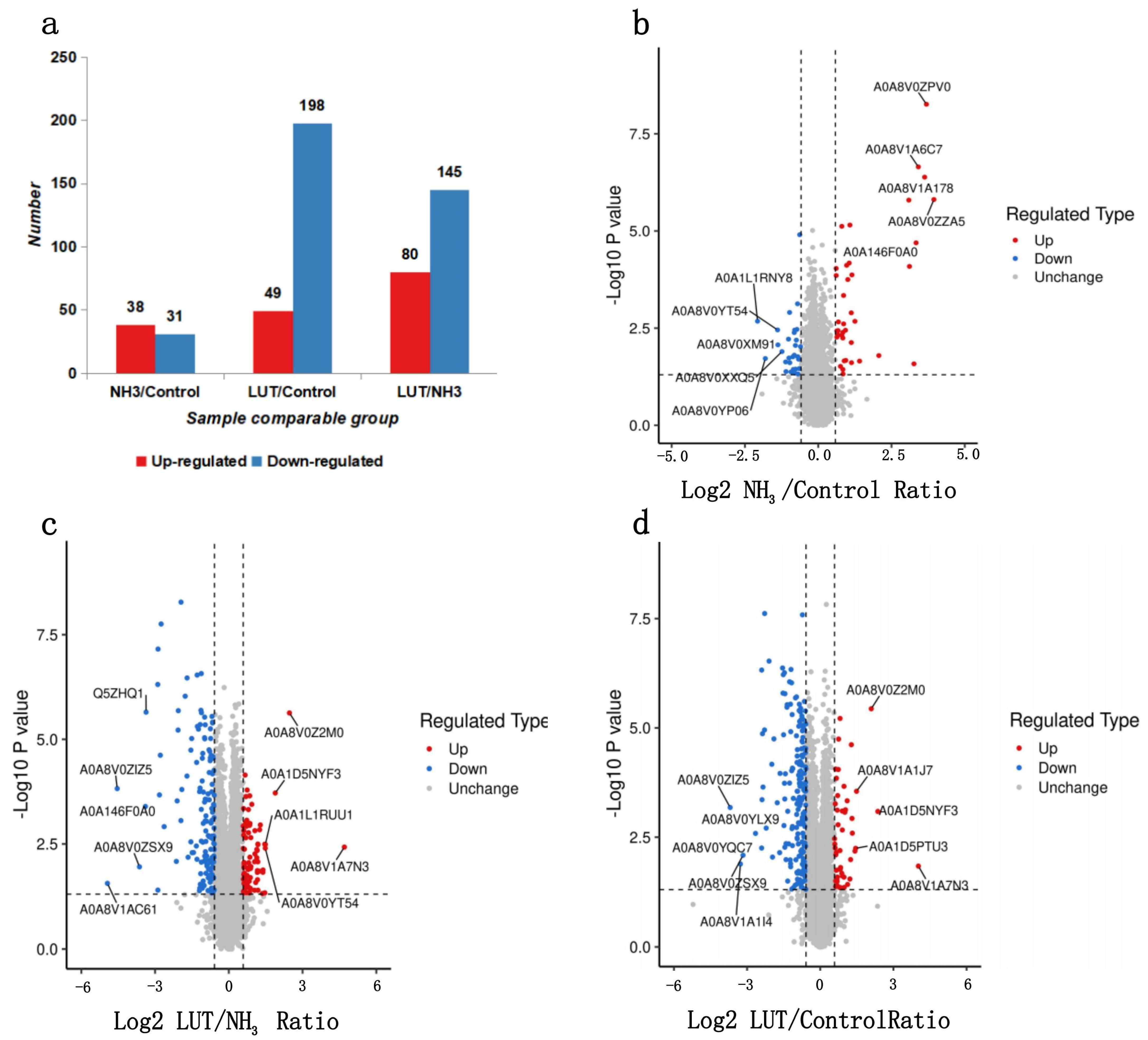
3.4. The Specific Number of DEPs
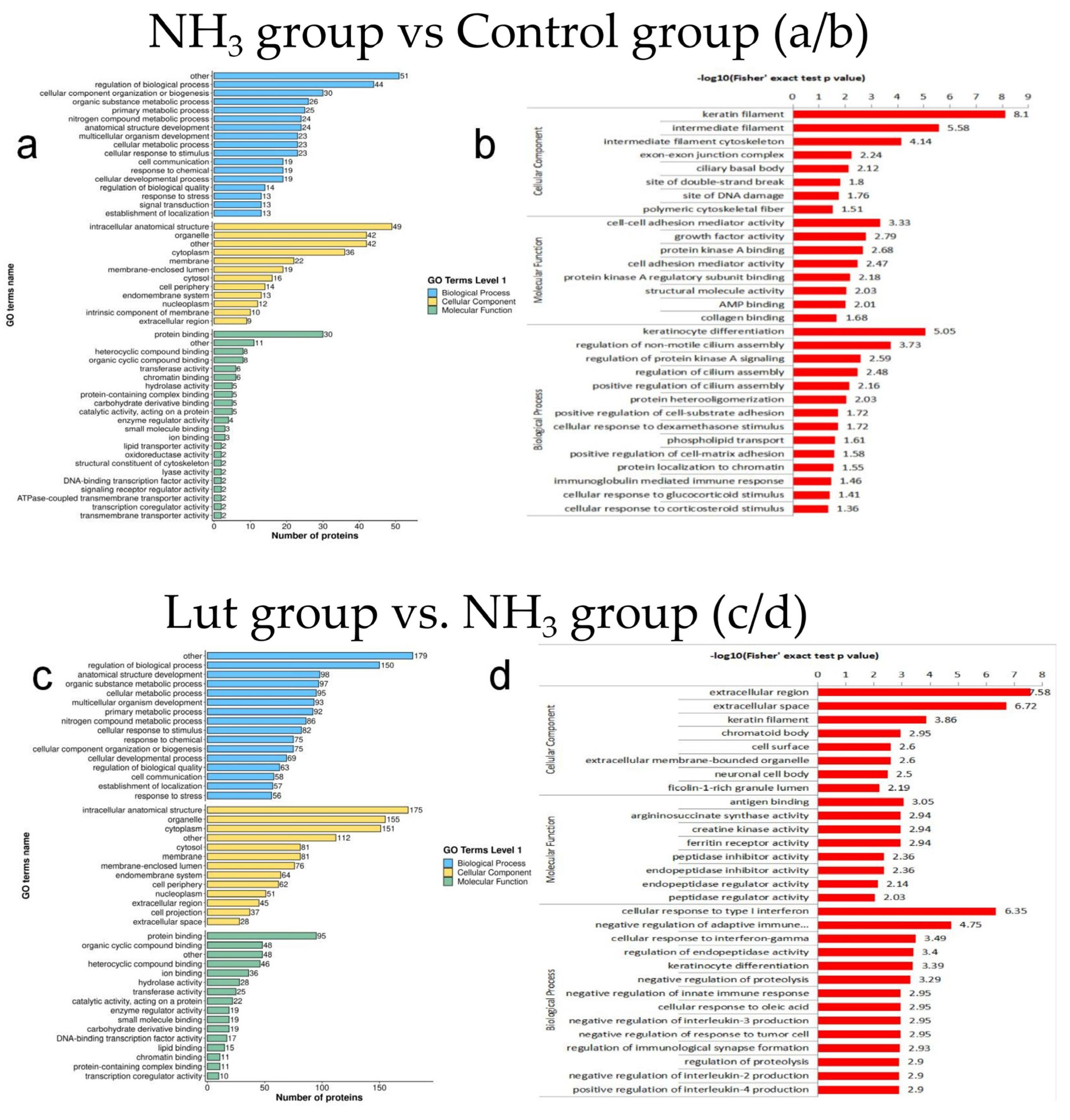
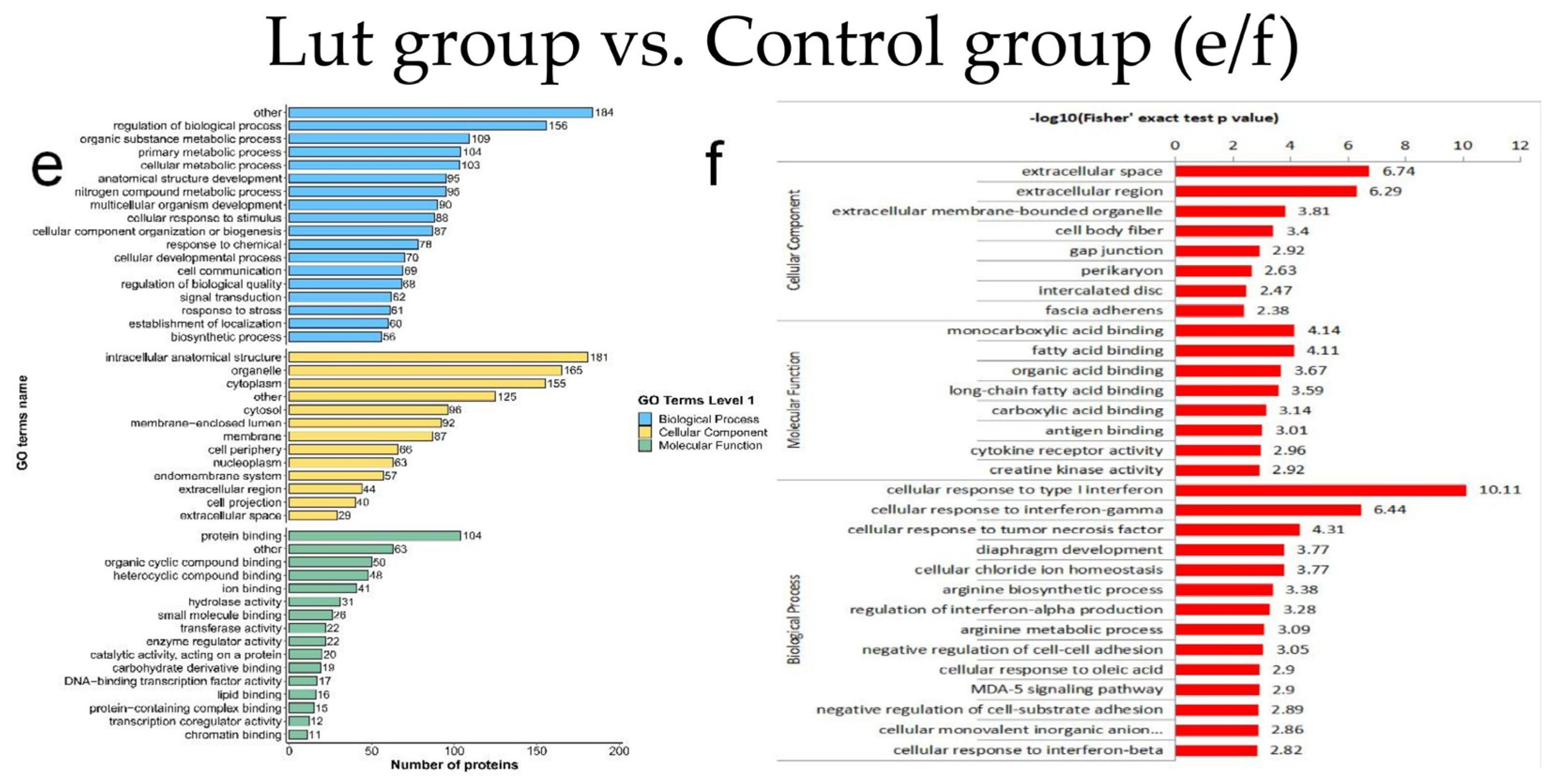
3.5. Results of GO Functional Classification and Enrichment Analysis of DEPs
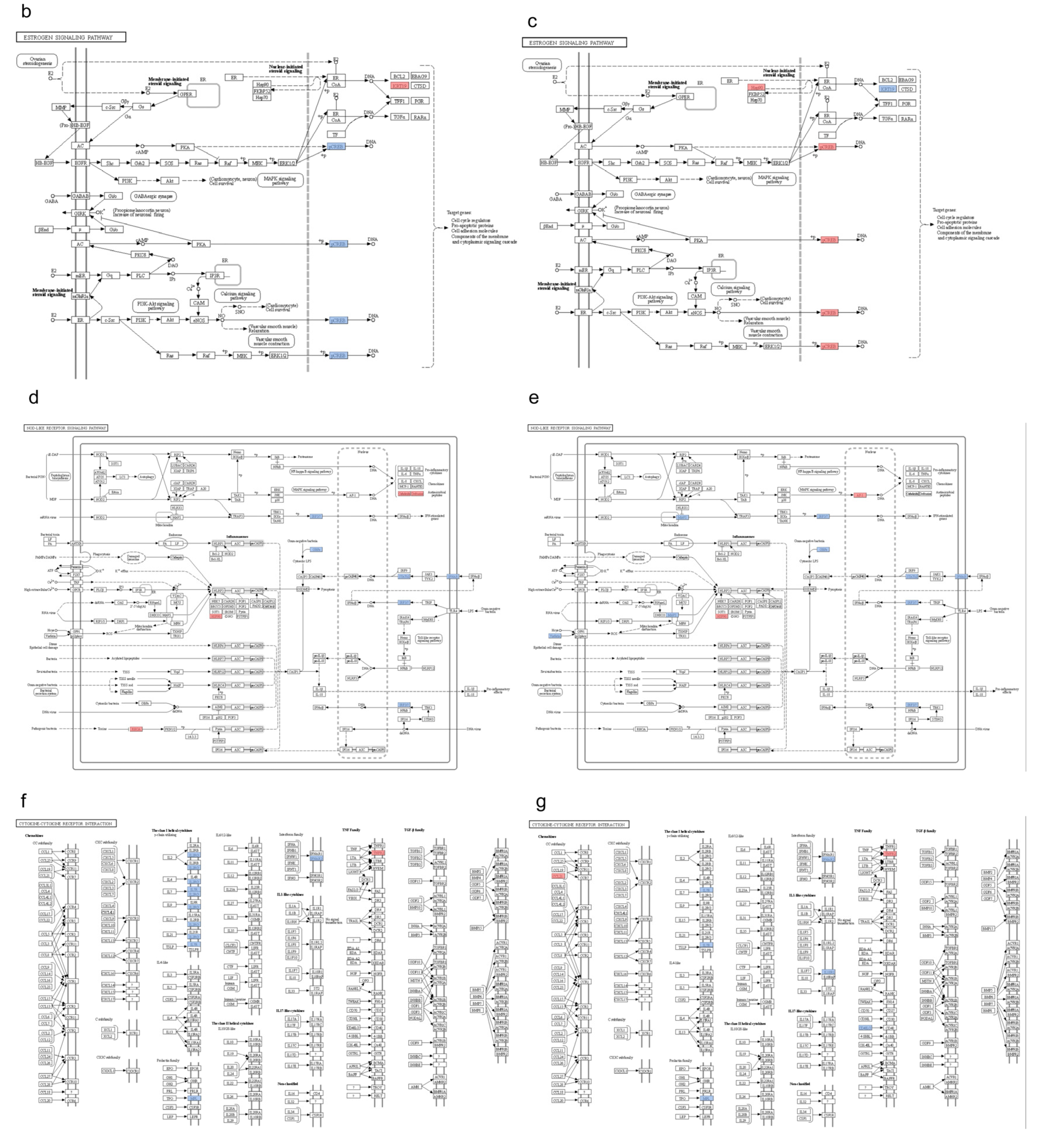
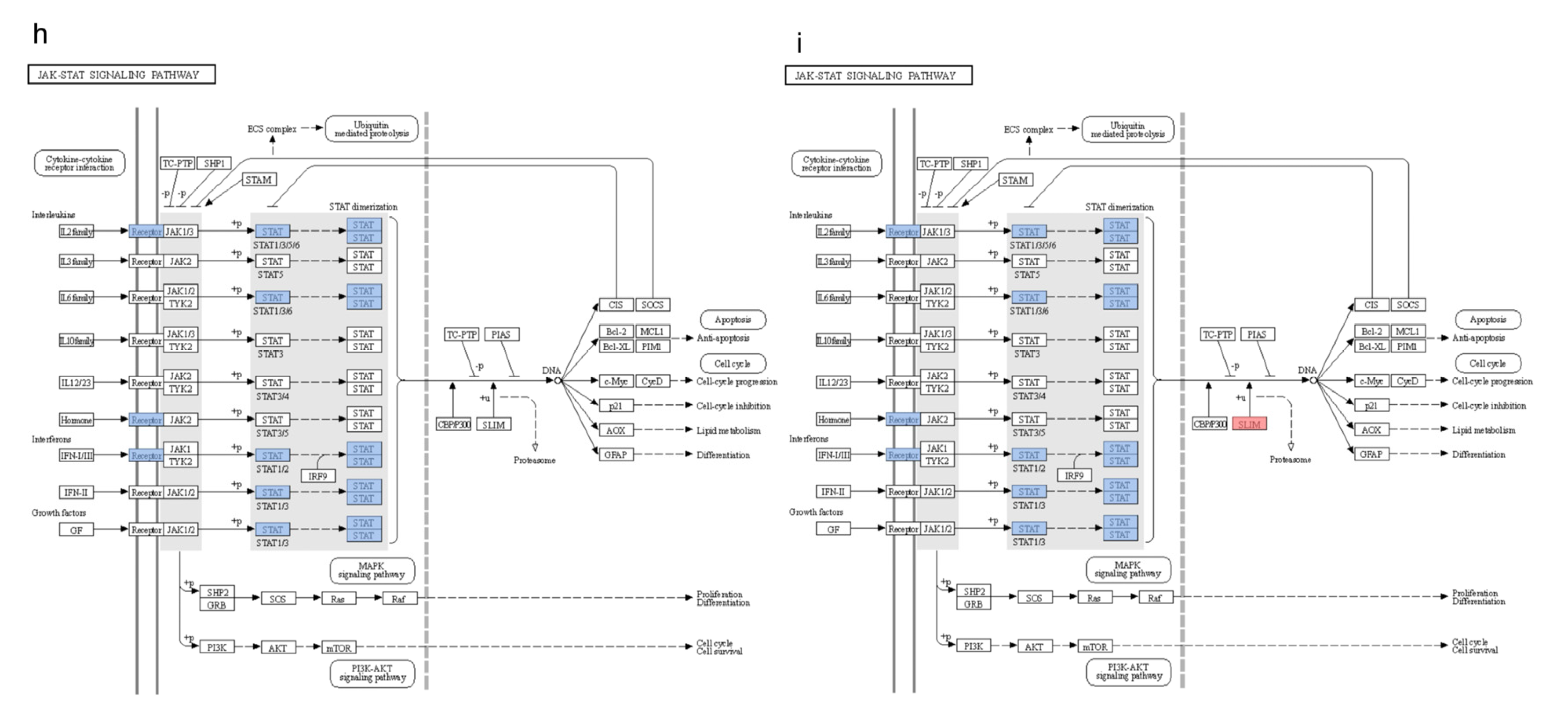
3.6. KEGG Pathway Clustering Analysis
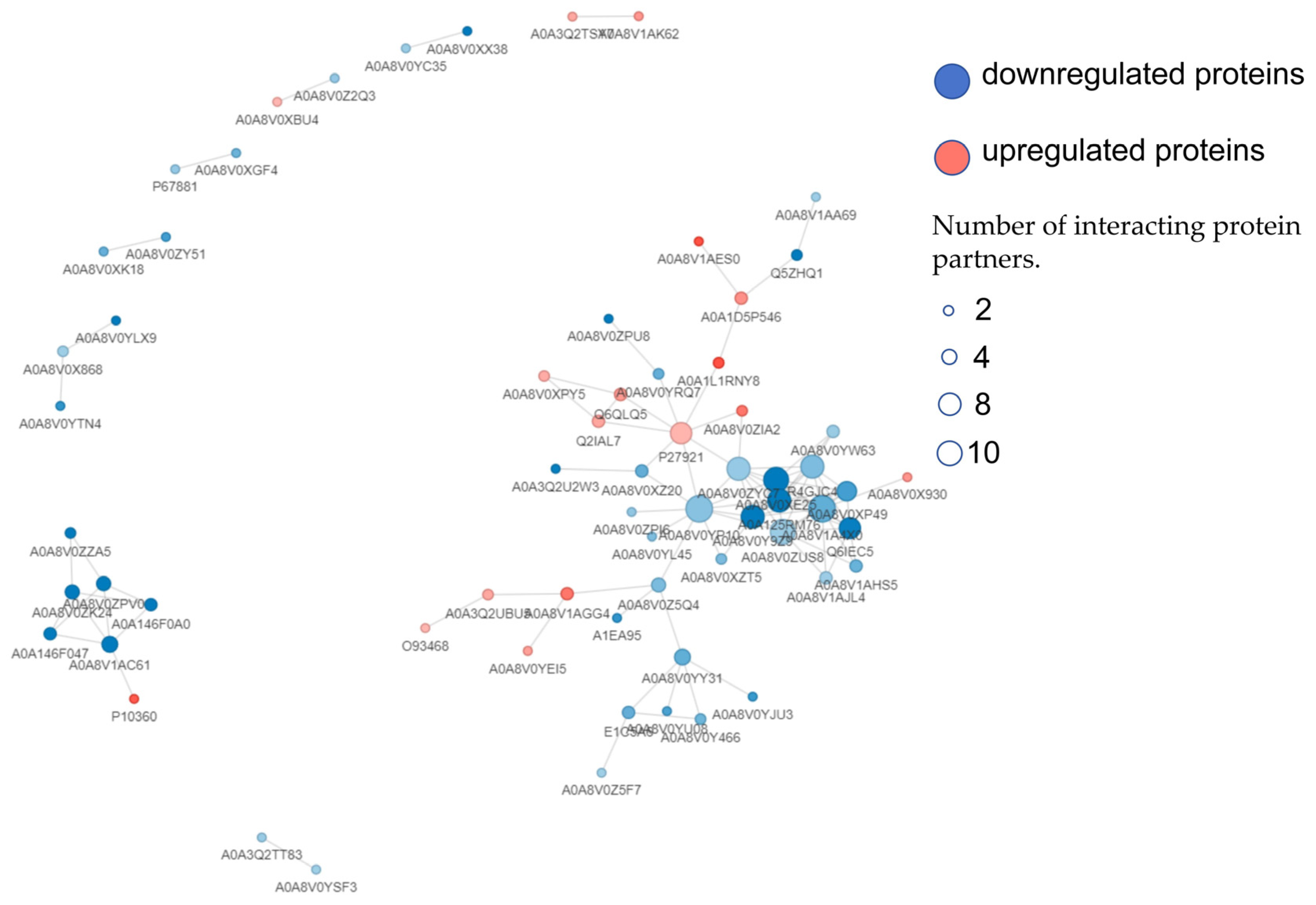
3.7. PPI Analysis of DEPs
3.8. Lut-Induced Gene Expression Associated with Apoptosis of Chicken Spleen Cells Exposed to Ammonia
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NH3 | Ammonia |
| Lut | Luteolin |
| 4D-FastDIA | Four-Dimensional Fast Data-Independent Acquisition |
| DEPs | Differentially Expressed Proteins |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| qRT-PCR | quantitative real-time polymerase chain reaction |
| IFNAR2 | Interferon Alpha and Beta Receptor subunit 2 |
| STAT1 | Signal Transducer and Activator of Transcription 1 |
| HPLC | High-Performance Liquid Chromatography |
| MS | Mass Spectrometry |
| DIA | Data-Independent Acquisition |
| FDR | False Discovery Rate |
| COGs | Clusters of Orthologous Groups |
| PPI | Protein–Protein Interaction |
| IRF-7 | Interferon Regulatory Factor 7 |
| IFN-α | Interferon-alpha |
| CRE | Cyclic AMP-responsive Element |
| Caspase-9 | Cysteinyl Aspartate Specific Protease 9 |
| API-5 | Apoptosis Inhibitor 5 |
| SOCS-3 | Suppressor of Cytokine Signaling 3 |
| IL | Interleukin |
| GSDMD | Gasdermin D |
| NF-κB | Nuclear Factor-kappa B |
References
- Shah, S.W.A.; Chen, J.; Han, Q.; Xu, Y.; Ishfaq, M.; Teng, X. Ammonia inhalation impaired immune function and mitochondrial integrity in the broilers bursa of fabricius: Implication of oxidative stress and apoptosis. Ecotoxicol. Environ. Saf. 2020, 190, 110078. [Google Scholar] [CrossRef] [PubMed]
- Naseem, S.; King, A.J. Ammonia production in poultry houses can affect health of humans, birds, and the environment-techniques for its reduction during poultry production. Environ. Sci. Pollut. Res. Int. 2018, 25, 15269–15293. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.M.; Rowe, D.E.; Cathcart, T.C. Litter ammonia generation: Moisture content and organic versus inorganic bedding materials. Poult. Sci. 2011, 90, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, H.; Tang, H.; Ran, L.; Wang, D.; Yang, F.; Zhang, H.; Teng, X.; Chen, D. Golgi apparatus regulated pyroptosis through the miR-32-5p/Golga7/NLRP3 axis in chicken splenic lymphocytes exposure to ammonia. Environ. Pollut. 2024, 362, 124923. [Google Scholar] [CrossRef]
- Chen, D.; Ning, F.; Zhang, J.; Tang, Y.; Teng, X. NF-κB pathway took part in the development of apoptosis mediated by miR-15a and oxidative stress via mitochondrial pathway in ammonia-treated chicken splenic lymphocytes. Sci. Total Environ. 2020, 729, 139017. [Google Scholar] [CrossRef]
- Miles, D.M.; Branton, S.L.; Lott, B.D. Atmospheric ammonia is detrimental to the performance of modern commercial broilers. Poult. Sci. 2004, 83, 1650–1654. [Google Scholar] [CrossRef]
- Harborne, J.B.; Williams, C.A. Advances in flavonoid research since 1992. Phytochemistry 2000, 55, 481–504. [Google Scholar] [CrossRef]
- Chagas, M.; Behrens, M.D.; Moragas-Tellis, C.J.; Penedo, G.X.M.; Silva, A.R.; Gonçalves-de-Albuquerque, C.F. Flavonols and Flavones as Potential anti-Inflammatory, Antioxidant, and Antibacterial Compounds. Oxid. Med. Cell. Longev. 2022, 2022, 9966750. [Google Scholar] [CrossRef]
- Boeing, T.; de Souza, P.; Speca, S.; Somensi, L.B.; Mariano, L.N.B.; Cury, B.J.; Anjos, M.F.D.; Quintão, N.L.M.; Dubuqoy, L.; Desreumax, P.; et al. Luteolin prevents irinotecan-induced intestinal mucositis in mice through antioxidant and anti-inflammatory properties. Br. J. Pharmacol. 2020, 177, 2393–2408. [Google Scholar] [CrossRef]
- Shen, R.; Ma, L.; Zheng, Y. Anti-inflammatory effects of luteolin on acute gouty arthritis rats via TLR/MyD88/NF-κB pathway. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2020, 45, 115–122. [Google Scholar]
- Lee, M.N.; Lee, Y.; Wu, D.; Pae, M. Luteolin inhibits NLRP3 inflammasome activation via blocking ASC oligomerization. J. Nutr. Biochem. 2021, 92, 108614. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shen, F.; Liu, J.; Tang, H.; Zhang, K.; Teng, X.; Yang, F. The protective effect of Luteolin on chicken spleen lymphocytes from ammonia poisoning through mitochondria and balancing energy metabolism disorders. Poult. Sci. 2023, 102, 103093. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shen, F.; Liu, J.; Tang, H.; Teng, X.; Yang, F.; Liu, H. Luteolin enhanced antioxidant capability and induced pyroptosis through NF-κB/NLRP3/Caspase-1 in splenic lymphocytes exposure to ammonia. Sci. Total Environ. 2024, 919, 170699. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Sharma, N.; Kour, S.; Sodhi, S.S.; Gupta, M.K.; Lee, S.J.; Son, Y.O. Applications of Omics Technology for Livestock Selection and Improvement. Front. Genet. 2022, 13, 774113. [Google Scholar] [CrossRef]
- Sun, C.; Zhu, T.; Zhu, Y.; Li, B.; Zhang, J.; Liu, Y.; Juan, C.; Yang, S.; Zhao, Z.; Wan, R.; et al. Hepatotoxic mechanism of diclofenac sodium on broiler chicken revealed by iTRAQ-based proteomics analysis. J. Vet. Sci. 2022, 23, e56. [Google Scholar] [CrossRef]
- Chen, D.; Hu, G.; Zhang, S.; Zhang, H.; Teng, X. Ammonia-triggered apoptosis via immune function and metabolic process in the thymuses of chickens by proteomics analysis. Ecotoxicol. Environ. Saf. 2020, 198, 110619. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, K.; Zhao, X.; Liu, S.; Wang, L.; Yang, X.; Jiang, Z. Fermented Feed Modulates Meat Quality and Promotes the Growth of Longissimus Thoracis of Late-Finishing Pigs. Animals 2020, 10, 1682. [Google Scholar] [CrossRef]
- Kearney, G.D.; Shaw, R.; Prentice, M.; Tutor-Marcom, R. Evaluation of respiratory symptoms and respiratory protection behavior among poultry workers in small farming operations. J. Agromed. 2014, 19, 162–170. [Google Scholar] [CrossRef]
- Han, Q.; Tong, J.; Sun, Q.; Teng, X.; Zhang, H.; Teng, X. The involvement of miR-6615-5p/Smad7 axis and immune imbalance in ammonia-caused inflammatory injury via NF-κB pathway in broiler kidneys. Poult. Sci. 2020, 99, 5378–5388. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, M.; Zhao, X.; Feng, J. Ammonia exposure induced intestinal inflammation injury mediated by intestinal microbiota in broiler chickens via TLR4/TNF-α signaling pathway. Ecotoxicol. Environ. Saf. 2021, 226, 112832. [Google Scholar] [CrossRef]
- Zhu, P.; Wu, X.; Ni, L.; Chen, K.; Dong, Z.; Du, J.; Kong, F.; Mao, Y.; Tao, H.; Chu, M.; et al. Inhibition of PP2A ameliorates intervertebral disc degeneration by reducing annulus fibrosus cells apoptosis via p38/MAPK signal pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 166888. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mo, S.; Jin, Y.; Zheng, Z.; Wang, H.; Wu, S.; Ren, Z.; Wu, J. Ammonia-induced excess ROS causes impairment and apoptosis in porcine IPEC-J2 intestinal epithelial cells. Ecotoxicol. Environ. Saf. 2022, 243, 114006. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a Citrus Flavonoid, Attenuates LPS-Induced Neuroinflammation, Apoptosis and Memory Impairments by Modulating TLR4/NF-κB Signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Chapter Three—Estrogen receptor signaling mechanisms. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 135–170. [Google Scholar]
- Knowlton, A.A.; Lee, A.R. Estrogen and the cardiovascular system. Pharmacol. Ther. 2012, 135, 54–70. [Google Scholar] [CrossRef]
- Cheng, C.; Wu, H.; Wang, M.; Wang, L.; Zou, H.; Li, S.; Liu, R. Estrogen ameliorates allergic airway inflammation by regulating activation of NLRP3 in mice. Biosci. Rep. 2019, 39, 101042. [Google Scholar] [CrossRef]
- Shi, L.; Feng, Y.; Lin, H.; Ma, R.; Cai, X. Role of estrogen in hepatocellular carcinoma: Is inflammation the key? J. Transl. Med. 2014, 12, 93. [Google Scholar] [CrossRef]
- Chen, D.; Tang, H.; Liu, J.; Zhang, H.; Rao, K.; Teng, X.; Yang, F.; Liu, H. Luteolin-mediated phosphoproteomic changes in chicken splenic lymphocytes: Unraveling the detoxification mechanisms against ammonia-induced stress. J. Hazard. Mater. 2025, 485, 136931. [Google Scholar] [CrossRef]
- Titlow, J.S.; Kiourlappou, M.; Palanca, A.; Lee, J.Y.; Gala, D.S.; Ennis, D.; Yu, J.J.S.; Young, F.L.; Pinto, D.M.S.; Garforth, S.; et al. Systematic analysis of YFP traps reveals common mRNA/protein discordance in neural tissues. J. Cell Biol. 2023, 222, 101083. [Google Scholar] [CrossRef]
- Yosef, N.; Shalek, A.K.; Gaublomme, J.T.; Jin, H.; Lee, Y.; Awasthi, A.; Wu, C.; Karwacz, K.; Xiao, S.; Jorgolli, M.; et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature 2013, 496, 461–468. [Google Scholar] [CrossRef]
- Platnich, J.M.; Muruve, D.A. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch. Biochem. Biophys. 2019, 670, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yu, S.; Zhang, W. NOD-like Receptor Signaling Pathway in Gastrointestinal Inflammatory Diseases and Cancers. Int. J. Mol. Sci. 2023, 24, 14511. [Google Scholar] [CrossRef] [PubMed]
- Imani, D.; Azimi, A.; Salehi, Z.; Rezaei, N.; Emamnejad, R.; Sadr, M.; Izad, M. Association of nod-like receptor protein-3 single nucleotide gene polymorphisms and expression with the susceptibility to relapsing-remitting multiple sclerosis. Int. J. Immunogenet. 2018, 45, 329–336. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Dai, Z.; Liu, W.C.; Chen, X.Y.; Wang, X.; Li, J.L.; Zhang, X. Gasdermin D-mediated pyroptosis: Mechanisms, diseases, and inhibitors. Front. Immunol. 2023, 14, 1178662. [Google Scholar] [CrossRef]
- Zhang, L.; Zeng, X.X.; Li, Y.M.; Chen, S.K.; Tang, L.Y.; Wang, N.; Yang, X.; Lin, M.J. Keratin 1 attenuates hypoxic pulmonary artery hypertension by suppressing pulmonary artery media smooth muscle expansion. Acta Physiol. 2021, 231, e13558. [Google Scholar] [CrossRef]
- Zhang, T.; Sun, H.-C.; Zhou, H.-Y.; Luo, J.-T.; Zhang, B.-L.; Wang, P.; Wang, L.; Qin, L.-X.; Ren, N.; Ye, S.-L.; et al. Interferon alpha inhibits hepatocellular carcinoma growth through inducing apoptosis and interfering with adhesion of tumor endothelial cells. Cancer Lett. 2010, 290, 204–210. [Google Scholar] [CrossRef]
- Li, Y.; Tian, Y.Y.; Wang, J.; Lin, R.; Zhang, Y.; Zhang, M.M.; Dong, T.W.; Li, M.; Xie, Y.H.; Zheng, X.H.; et al. Main active components of Ilex rotunda Thunb. protect against ulcerative colitis by restoring the intestinal mucosal barrier and modulating the cytokine-cytokine interaction pathways. J. Ethnopharmacol. 2024, 318 Pt B, 116961. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, D.; Chen, Z.; Lin, L.; Shao, Q.; Zhang, H.; Li, P.; Lv, H. Predicting biomarkers in laryngeal squamous cell carcinoma based on the cytokine-cytokine receptor interaction pathway. Heliyon 2024, 10, e37738. [Google Scholar] [CrossRef]
- Olbei, M.; Thomas, J.P.; Hautefort, I.; Treveil, A.; Bohar, B.; Madgwick, M.; Gul, L.; Csabai, L.; Modos, D.; Korcsmaros, T. CytokineLink: A Cytokine Communication Map to Analyse Immune Responses—Case Studies in Inflammatory Bowel Disease and COVID-19. Cells 2021, 10, 2242. [Google Scholar] [CrossRef]
- Sheng, Y.; Lin, Y.; Qiang, Z.; Shen, X.; He, Y.; Li, L.; Li, S.; Zhang, G.; Wang, F. Protein kinase a suppresses antiproliferative effect of interferon-α in hepatocellular carcinoma by activation of protein tyrosine phosphatase SHP2. J. Biol. Chem. 2025, 301, 108195. [Google Scholar] [CrossRef] [PubMed]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Nordmann, T.M.; Anderton, H.; Hasegawa, A.; Schweizer, L.; Zhang, P.; Stadler, P.C.; Sinha, A.; Metousis, A.; Rosenberger, F.A.; Zwiebel, M.; et al. Spatial proteomics identifies JAKi as treatment for a lethal skin disease. Nature 2024, 635, 1001–1009. [Google Scholar] [CrossRef]
- Nunes, C.; Almeida, L.; Barbosa, R.M.; Laranjinha, J. Luteolin suppresses the JAK/STAT pathway in a cellular model of intestinal inflammation. Food Funct. 2017, 8, 387–396. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer (5′-3′) | Size (bp) |
|---|---|---|
| β-actin | F: CCCAGCCATGTATGTAGCCATCC R: AACACCATCACCAGAGTCCATCAC | 91 |
| IRF7 | F: CAGCACAAAGCCCAAGGAGTC R: TTGCCACTGTTGAGGGAGGAG | 114 |
| IFN-α | F: ACCACCCACGACATCCTTCAG R: GGCTTTGGCGTTGGCTGTC | 88 |
| IFNAR2 | F: TTCCCAAGAAGATGCTGTTGACTG R: TGTGGTTTCTGCGTGCTTTCTG | 87 |
| CRE | F: GTGGCGATGGATGCGTTGTC R: CTGCTGTTGCTGGGAAGTTGTC | 81 |
| Caspase-9 | F: AAGGTGAGTGGCTCGTGGTAC R: GTAGCATGGTTAGCAGGTCTTCAG | 85 |
| STAT1 | F: CTCTGGAACGATGGCTGTATCATG R: GCTCCCTCTTTACTGCTTTCACTG | 119 |
| API-5 | F: TGCAGTTCAGTTATGTCGAGTGTC R: TTTGGCTGTGAGGAAATCTGGAAG | 80 |
| SOCS-3 | F: GCCTCAAGACGTTCAGCTCTAAG R: CTCCAGTAGAAGCCGCTCTCC | 85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Shama, A.; Tang, H.; Zhao, T.; Zhang, X.; Yang, F.; Chen, D. Exploring the Mechanism of Luteolin in Protecting Chickens from Ammonia Poisoning Based on Proteomic Technology. Metabolites 2025, 15, 326. https://doi.org/10.3390/metabo15050326
Jin Y, Shama A, Tang H, Zhao T, Zhang X, Yang F, Chen D. Exploring the Mechanism of Luteolin in Protecting Chickens from Ammonia Poisoning Based on Proteomic Technology. Metabolites. 2025; 15(5):326. https://doi.org/10.3390/metabo15050326
Chicago/Turabian StyleJin, Yu, Azi Shama, Haojinming Tang, Ting Zhao, Xinyu Zhang, Falong Yang, and Dechun Chen. 2025. "Exploring the Mechanism of Luteolin in Protecting Chickens from Ammonia Poisoning Based on Proteomic Technology" Metabolites 15, no. 5: 326. https://doi.org/10.3390/metabo15050326
APA StyleJin, Y., Shama, A., Tang, H., Zhao, T., Zhang, X., Yang, F., & Chen, D. (2025). Exploring the Mechanism of Luteolin in Protecting Chickens from Ammonia Poisoning Based on Proteomic Technology. Metabolites, 15(5), 326. https://doi.org/10.3390/metabo15050326