
The Effect of Dexamethasone-Mediated Atrophy on Mitochondrial Function and BCAA Metabolism During Insulin Resistance in C2C12 Myotubes

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Methods
2.1. Cell Culture
2.2. Quantitative Real Time Polymerase Chain Reaction (qRT-PCR)
2.3. Immunoblotting
2.4. Seahorse Metabolic Assays
2.5. Fluorescent Staining and Fluorescent Microscopy
2.6. Liquid Chromatography–Mass Spectrometry (LC–MS)
2.7. Statistical Analyses
2.8. Additional Materials, Methods, and Experimental Procedures
3. Results
3.1. Dexamethasone-Induced Atrophy Reduces pAKT Expression in Cultured Myotubes
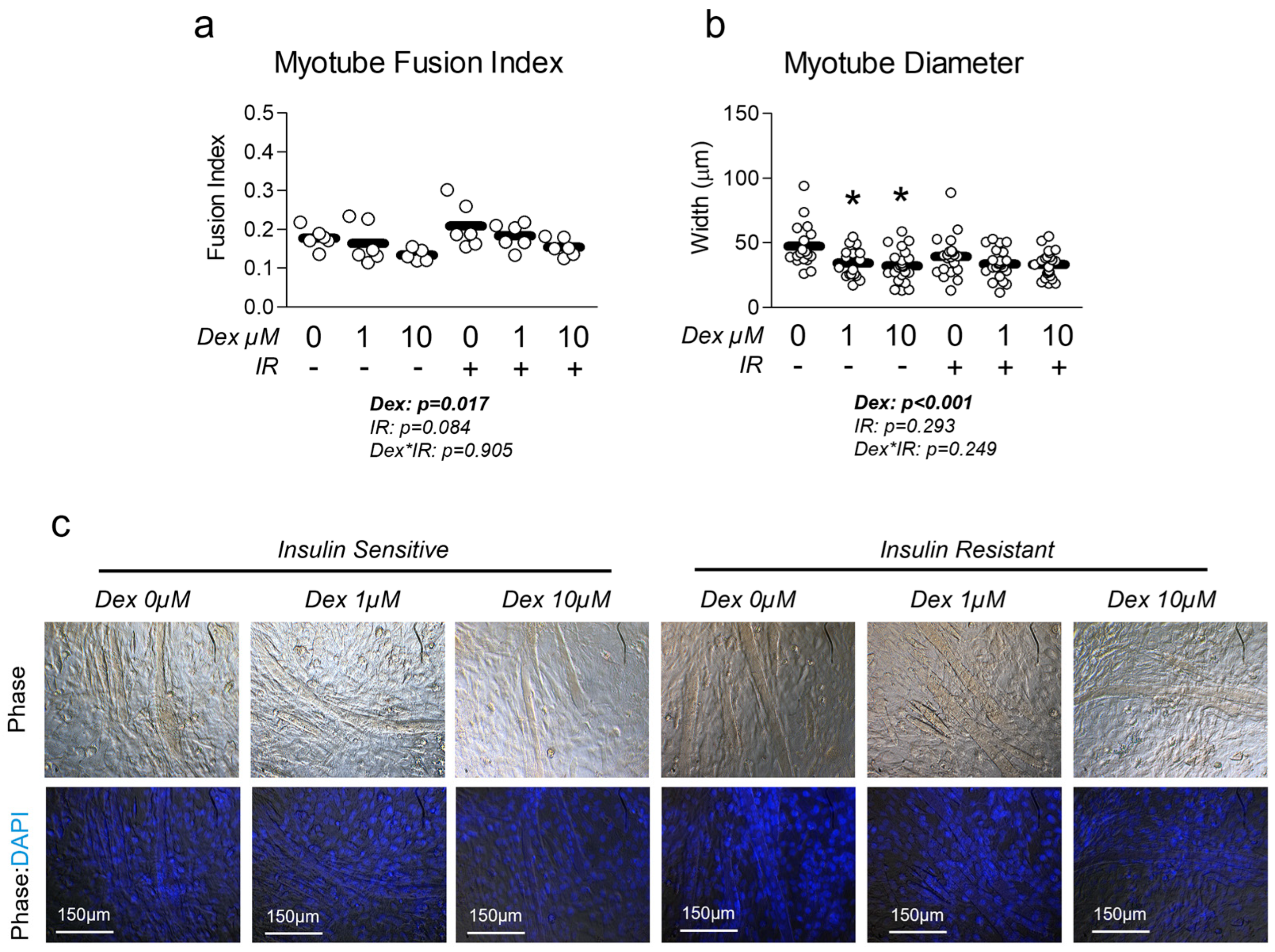
3.2. Dexamethasone Reduces Myotube Diameter and Fusion Index, Which Is Unaffected by Insulin Resistance
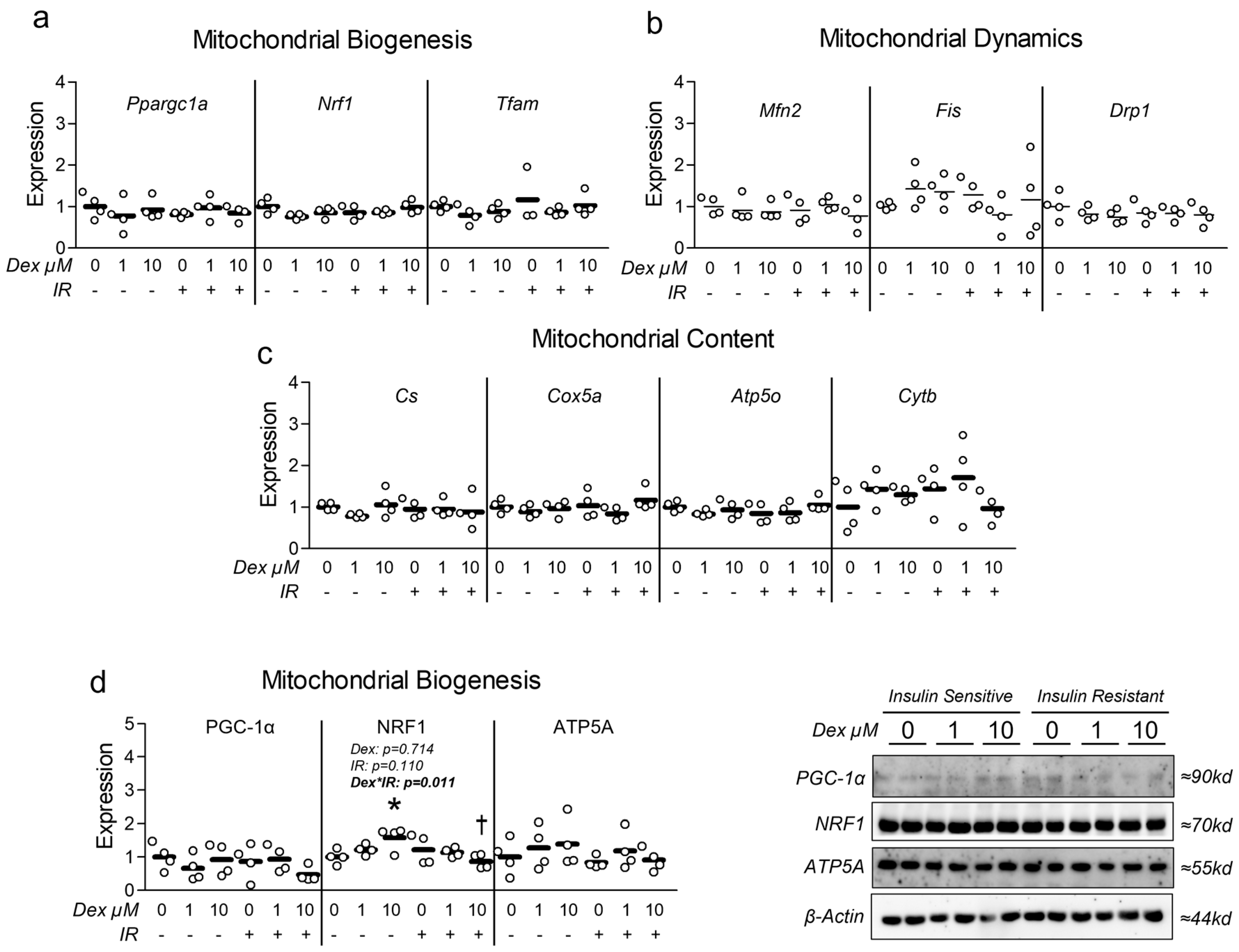
3.3. Dexamethasone-Induced Atrophy Reduces Myotube Mitochondrial Content and Function, Which Is Worsened by Insulin Resistance
3.4. Dexamethasone Does Not Alter BCAA Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Z.J.; Zhu, C.F. Causal relationship between insulin resistance and sarcopenia. Diabetol. Metab. Syndr. 2023, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Neinast, M. Branched Chain Amino Acids in Metabolic Disease. Curr. Diabetes Rep. 2018, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Gannon, N.P.; Schnuck, J.K.; Vaughan, R.A. BCAA Metabolism and Insulin Sensitivity–Dysregulated by Metabolic Status? Mol. Nutr. Food Res. 2018, 62, 1700756. [Google Scholar] [CrossRef] [PubMed]
- Holeček, M. Why Are Branched-Chain Amino Acids Increased in Starvation and Diabetes? Nutrients 2020, 12, 3087. [Google Scholar] [CrossRef]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef]
- Newgard, C.B. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012, 15, 606–614. [Google Scholar] [CrossRef]
- White, P.J.; McGarrah, R.W.; Herman, M.A.; Bain, J.R.; Shah, S.H.; Newgard, C.B. Insulin action, type 2 diabetes, and branched-chain amino acids: A two-way street. Mol. Metab. 2021, 52, 101261. [Google Scholar] [CrossRef]
- Mann, G.; Mora, S.; Madu, G.; Adegoke, O.A.J. Branched-chain Amino Acids: Catabolism in Skeletal Muscle and Implications for Muscle and Whole-body Metabolism. Front. Physiol. 2021, 12, 702826. [Google Scholar] [CrossRef]
- Vanweert, F.; Schrauwen, P.; Phielix, E. Role of branched-chain amino acid metabolism in the pathogenesis of obesity and type 2 diabetes-related metabolic disturbances BCAA metabolism in type 2 diabetes. Nutr. Diabetes 2022, 12, 35. [Google Scholar] [CrossRef]
- Haufe, S.; Engeli, S.; Kaminski, J.; Witt, H.; Rein, D.; Kamlage, B.; Utz, W.; Fuhrmann, J.C.; Haas, V.; Mahler, A.; et al. Branched-chain amino acid catabolism rather than amino acids plasma concentrations is associated with diet-induced changes in insulin resistance in overweight to obese individuals. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 858–864. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Gogg, S.; Lotta, L.A.; Stancakova, A.; Nerstedt, A.; Boren, J.; Bluher, M.; Ferrannini, E.; Langenberg, C.; Wareham, N.J.; et al. Elevated Plasma Levels of 3-Hydroxyisobutyric Acid Are Associated With Incident Type 2 Diabetes. eBioMedicine 2018, 27, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, M.S.; Jersin, R.; Ulvik, A.; Madsen, A.; McCann, A.; Svensson, P.A.; Svensson, M.K.; Nedrebø, B.G.; Gudbrandsen, O.A.; Tell, G.S.; et al. 3-Hydroxyisobutyrate, A Strong Marker of Insulin Resistance in Type 2 Diabetes and Obesity That Modulates White and Brown Adipocyte Metabolism. Diabetes 2020, 69, 1903–1916. [Google Scholar] [CrossRef]
- Boulet, M.M.; Chevrier, G.; Grenier-Larouche, T.; Pelletier, M.; Nadeau, M.; Scarpa, J.; Prehn, C.; Marette, A.; Adamski, J.; Tchernof, A. Alterations of plasma metabolite profiles related to adipose tissue distribution and cardiometabolic risk. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E736–E746. [Google Scholar] [CrossRef] [PubMed]
- Gannon, N.P.; Vaughan, R.A. Leucine-induced anabolic-catabolism: Two sides of the same coin. Amino Acids 2016, 48, 321–336. [Google Scholar] [CrossRef]
- Zhang, L.; Li, F.; Guo, Q.; Duan, Y.; Wang, W.; Zhong, Y.; Yang, Y.; Yin, Y. Leucine Supplementation: A Novel Strategy for Modulating Lipid Metabolism and Energy Homeostasis. Nutrients 2020, 12, 1299. [Google Scholar] [CrossRef]
- Ruocco, C.; Segala, A.; Valerio, A.; Nisoli, E. Essential amino acid formulations to prevent mitochondrial dysfunction and oxidative stress. Curr. Opin. Clin. Nutr. Metab. Care 2021, 24, 88–95. [Google Scholar] [CrossRef]
- Hinkle, J.S.; Rivera, C.N.; Vaughan, R.A. Branched-Chain Amino Acids and Mitochondrial Biogenesis: An Overview and Mechanistic Summary. Mol. Nutr. Food Res. 2022, 66, e2200109. [Google Scholar] [CrossRef] [PubMed]
- Le Couteur, D.G.; Solon-Biet, S.M.; Cogger, V.C.; Ribeiro, R.; de Cabo, R.; Raubenheimer, D.; Cooney, G.J.; Simpson, S.J. Branched chain amino acids, aging and age-related health. Ageing Res. Rev. 2020, 64, 101198. [Google Scholar] [CrossRef]
- Petersen, M.C.; Smith, G.I.; Palacios, H.H.; Farabi, S.S.; Yoshino, M.; Yoshino, J.; Cho, K.; Davila-Roman, V.G.; Shankaran, M.; Barve, R.A.; et al. Cardiometabolic characteristics of people with metabolically healthy and unhealthy obesity. Cell Metab. 2024, 36, 745–761.e745. [Google Scholar] [CrossRef]
- Zhiyin, L.; Jinliang, C.; Qiunan, C.; Yunfei, Y.; Qian, X. Fucoxanthin rescues dexamethasone induced C2C12 myotubes atrophy. Biomed. Pharmacother. 2021, 139, 111590. [Google Scholar] [CrossRef]
- Shen, S.; Liao, Q.; Liu, J.; Pan, R.; Lee, S.M.; Lin, L. Myricanol rescues dexamethasone-induced muscle dysfunction via a sirtuin 1-dependent mechanism. J. Cachexia Sarcopenia Muscle 2019, 10, 429–444. [Google Scholar] [CrossRef]
- Ko, H.; Bekele, T.T.; Le, T.T.; Cha, K.H.; Kim, S.; Youn, H.-Y.; Jung, S.H.; Myungsuk, K. Identification of components from Aralia elata and their effects on muscle health and gut microbiota. J. Funct. Foods 2024, 121, 106384. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, S.; Zhang, Z.; Tang, M.; Meng, Z.; Peng, Z.; Liao, Y.; Yang, X.; Nüssler, A.K.; Liu, L.; et al. Gouqi-derived nanovesicles (GqDNVs) inhibited dexamethasone-induced muscle atrophy associating with AMPK/SIRT1/PGC1α signaling pathway. J. Nanobiotechnol. 2024, 22, 276. [Google Scholar] [CrossRef]
- Kim, R.; Kim, H.; Im, M.; Park, S.K.; Han, H.J.; An, S.; Kang, J.S.; Lee, S.J.; Bae, G.U. BST204 Protects Dexamethasone-Induced Myotube Atrophy through the Upregulation of Myotube Formation and Mitochondrial Function. Int. J. Environ. Res. Public Health 2021, 18, 2367. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, S.W.; Lee, S.H.; Jung, D.W.; Williams, D.R. Inhibiting 5-lipoxygenase prevents skeletal muscle atrophy by targeting organogenesis signalling and insulin-like growth factor-1. J. Cachexia Sarcopenia Muscle 2022, 13, 3062–3077. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, H.M.; Kim, J.H.; Lee, J.H.; Zhang, K.; Guo, S.; Lee, D.H.; Gao, E.M.; Son, R.H.; Kim, S.M.; et al. Preventive effects of the butanol fraction of. Heliyon 2022, 8, e11597. [Google Scholar] [CrossRef]
- Xie, Y.; Perry, B.D.; Espinoza, D.; Zhang, P.; Price, S.R. Glucocorticoid-induced CREB activation and myostatin expression in C2C12 myotubes involves phosphodiesterase-3/4 signaling. Biochem. Biophys. Res. Commun. 2018, 503, 1409–1414. [Google Scholar] [CrossRef]
- Collao, N.; Akohene-Mensah, P.; Nallabelli, J.; Binet, E.R.; Askarian, A.; Lloyd, J.; Niemiro, G.M.; Beals, J.W.; van Vliet, S.; Rajgara, R.; et al. The role of L-type amino acid transporter 1. Am. J. Physiol. Cell Physiol. 2022, 323, C595–C605. [Google Scholar] [CrossRef]
- Bröer, A.; Gauthier-Coles, G.; Rahimi, F.; van Geldermalsen, M.; Dorsch, D.; Wegener, A.; Holst, J.; Bröer, S. Ablation of the ASCT2 (SLC1A5) gene encoding a neutral amino acid transporter reveals transporter plasticity and redundancy in cancer cells. J. Biol. Chem. 2019, 294, 4012–4026. [Google Scholar] [CrossRef]
- Cormerais, Y.; Massard, P.A.; Vucetic, M.; Giuliano, S.; Tambutté, E.; Durivault, J.; Vial, V.; Endou, H.; Wempe, M.F.; Parks, S.K.; et al. The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J. Biol. Chem. 2018, 293, 2877–2887. [Google Scholar] [CrossRef]
- Poncet, N.; Mitchell, F.E.; Ibrahim, A.F.; McGuire, V.A.; English, G.; Arthur, J.S.; Shi, Y.B.; Taylor, P.M. The catalytic subunit of the system L1 amino acid transporter (slc7a5) facilitates nutrient signalling in mouse skeletal muscle. PLoS ONE 2014, 9, e89547. [Google Scholar] [CrossRef] [PubMed]
- VanDerStad, L.R.; Wyatt, E.C.; Vaughan, R.A. The antidiabetic SGLT2 inhibitor canagliflozin reduces mitochondrial metabolism in a model of skeletal muscle insulin resistance. Diabet. Med. 2024, 41, e15271. [Google Scholar] [CrossRef] [PubMed]
- Lyon, E.S.; Rivera, M.E.; Johnson, M.A.; Sunderland, K.L.; Vaughan, R.A. Actions of chronic physiological 3-hydroxyisobuterate treatment on mitochondrial metabolism and insulin signaling in myotubes. Nutr. Res. 2019, 66, 22–31. [Google Scholar] [CrossRef]
- Rivera, M.E.; Lyon, E.S.; Johnson, M.A.; Sunderland, K.L.; Vaughan, R.A. Effect of valine on myotube insulin sensitivity and metabolism with and without insulin resistance. Mol. Cell. Biochem. 2020, 468, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Rivera, M.E.; Lyon, E.S.; Johnson, M.A.; Vaughan, R.A. Leucine increases mitochondrial metabolism and lipid content without altering insulin signaling in myotubes. Biochimie 2020, 168, 124–133. [Google Scholar] [CrossRef]
- Kumar, N.; Dey, C.S. Metformin enhances insulin signalling in insulin-dependent and-independent pathways in insulin resistant muscle cells. Br. J. Pharmacol. 2002, 137, 329–336. [Google Scholar] [CrossRef]
- Kumar, N.; Dey, C.S. Development of insulin resistance and reversal by thiazolidinediones in C2C12 skeletal muscle cells. Biochem. Pharmacol. 2003, 65, 249–257. [Google Scholar] [CrossRef]
- Chicco, A.G.; Adibi, S.A.; Liu, W.Q.; Morris, S.M.; Paul, H.S. Regulation of gene expression of branched-chain keto acid dehydrogenase complex in primary cultured hepatocytes by dexamethasone and a cAMP analog. J. Biol. Chem. 1994, 269, 19427–19434. [Google Scholar] [CrossRef]
- Rivera, C.N.; Kamer, M.M.; Rivera, M.E.; Watne, R.M.; Macgowan, T.C.; Wommack, A.J.; Vaughan, R.A. Insulin resistance promotes extracellular BCAA accumulation without altering LAT1 content, independent of prior BCAA treatment in a myotube model of skeletal muscle. Mol. Cell. Endocrinol. 2023, 559, 111800. [Google Scholar] [CrossRef]
- Xiao, Q.; Sun, C.C.; Tang, C.F. Heme oxygenase-1: A potential therapeutic target for improving skeletal muscle atrophy. Exp. Gerontol. 2023, 184, 112335. [Google Scholar] [CrossRef]
- Liu, J.; Peng, Y.; Wang, X.; Fan, Y.; Qin, C.; Shi, L.; Tang, Y.; Cao, K.; Li, H.; Long, J. Mitochondrial Dysfunction Launches Dexamethasone-Induced Skeletal Muscle Atrophy via AMPK/FOXO3 Signaling. Mol. Pharm. 2016, 13, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.J.; Carter, S.; Nicholson, T.; Allen, S.L.; Morgan, P.T.; Jones, S.W.; Rendeiro, C.; Breen, L. (-)-Epicatechin and its colonic metabolite hippuric acid protect against dexamethasone-induced atrophy in skeletal muscle cells. J. Nutr. Biochem. 2022, 110, 109150. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Chen, Y.C.; Chan, Y.C.; Liu, C.; Chang, S.J. Oligonol. Int. J. Mol. Sci. 2024, 25, 1549. [Google Scholar] [CrossRef]
- She, P.; Reid, T.M.; Bronson, S.K.; Vary, T.C.; Hajnal, A.; Lynch, C.J.; Hutson, S.M. Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab. 2007, 6, 181–194. [Google Scholar] [CrossRef]
- Bai, G.H.; Tsai, M.C.; Tsai, H.W.; Chang, C.C.; Hou, W.H. Effects of branched-chain amino acid-rich supplementation on EWGSOP2 criteria for sarcopenia in older adults: A systematic review and meta-analysis. Eur. J. Nutr. 2022, 61, 637–651. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ragland, K.J.; Travis, K.B.; Spry, E.R.; Zaman, T.; Lundin, P.M.; Vaughan, R.A. The Effect of Dexamethasone-Mediated Atrophy on Mitochondrial Function and BCAA Metabolism During Insulin Resistance in C2C12 Myotubes. Metabolites 2025, 15, 322. https://doi.org/10.3390/metabo15050322
Ragland KJ, Travis KB, Spry ER, Zaman T, Lundin PM, Vaughan RA. The Effect of Dexamethasone-Mediated Atrophy on Mitochondrial Function and BCAA Metabolism During Insulin Resistance in C2C12 Myotubes. Metabolites. 2025; 15(5):322. https://doi.org/10.3390/metabo15050322
Chicago/Turabian StyleRagland, Kayla J., Kipton B. Travis, Emmalie R. Spry, Toheed Zaman, Pamela M. Lundin, and Roger A. Vaughan. 2025. "The Effect of Dexamethasone-Mediated Atrophy on Mitochondrial Function and BCAA Metabolism During Insulin Resistance in C2C12 Myotubes" Metabolites 15, no. 5: 322. https://doi.org/10.3390/metabo15050322
APA StyleRagland, K. J., Travis, K. B., Spry, E. R., Zaman, T., Lundin, P. M., & Vaughan, R. A. (2025). The Effect of Dexamethasone-Mediated Atrophy on Mitochondrial Function and BCAA Metabolism During Insulin Resistance in C2C12 Myotubes. Metabolites, 15(5), 322. https://doi.org/10.3390/metabo15050322