
SMARCB1 Deficiency as a Driver of the Hallmarks of Cancer in Rhabdoid Tumours: Novel Insights into Dysregulated Energy Metabolism, Emerging Targets, and Ongoing Clinical Trials


Abstract
1. Introduction
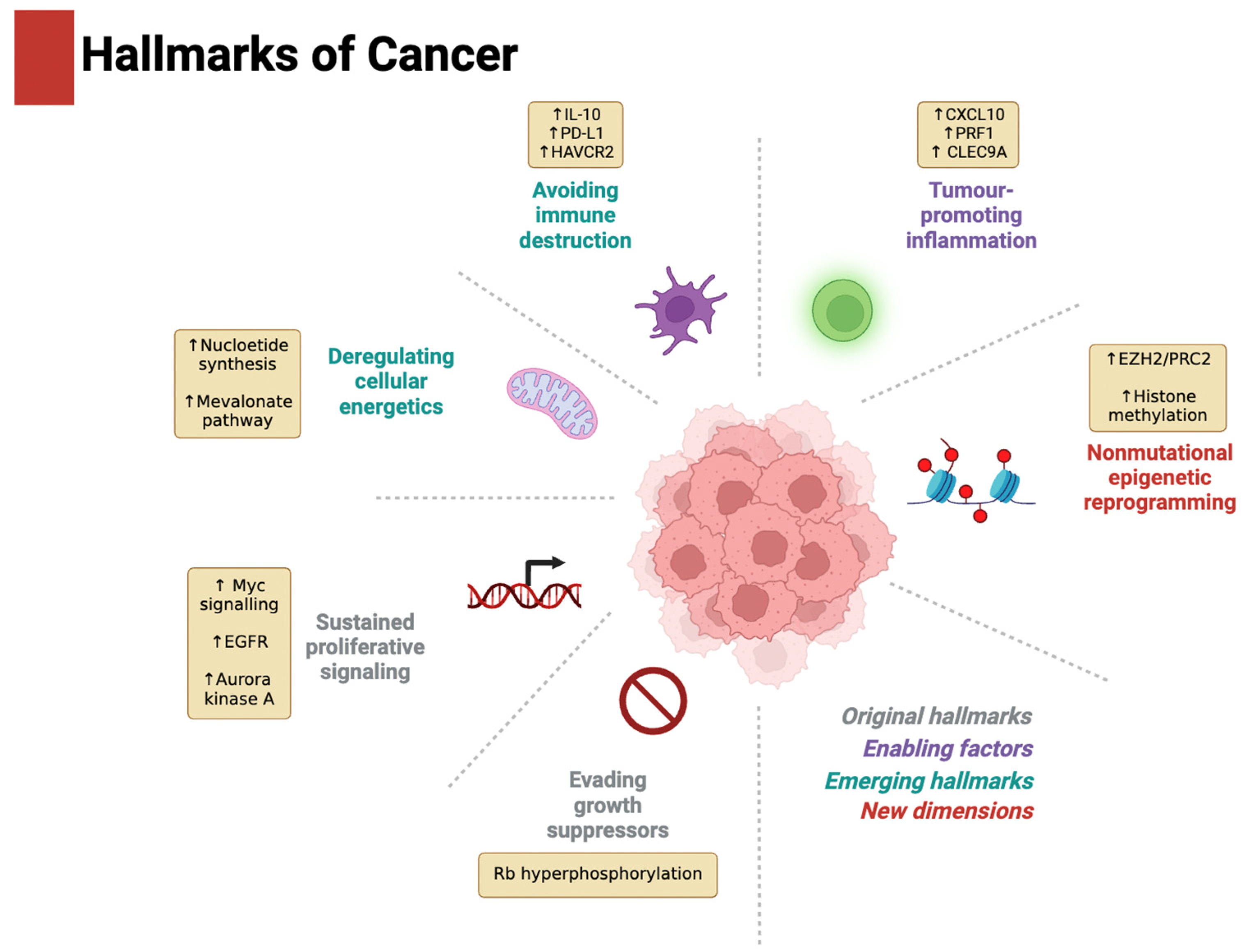
2. Driver of the Hallmarks of Cancer
2.1. Evading Growth Suppressors and Sustaining Proliferative Signalling
2.2. Tumour-Promoting Inflammation and Avoiding Immune Destruction
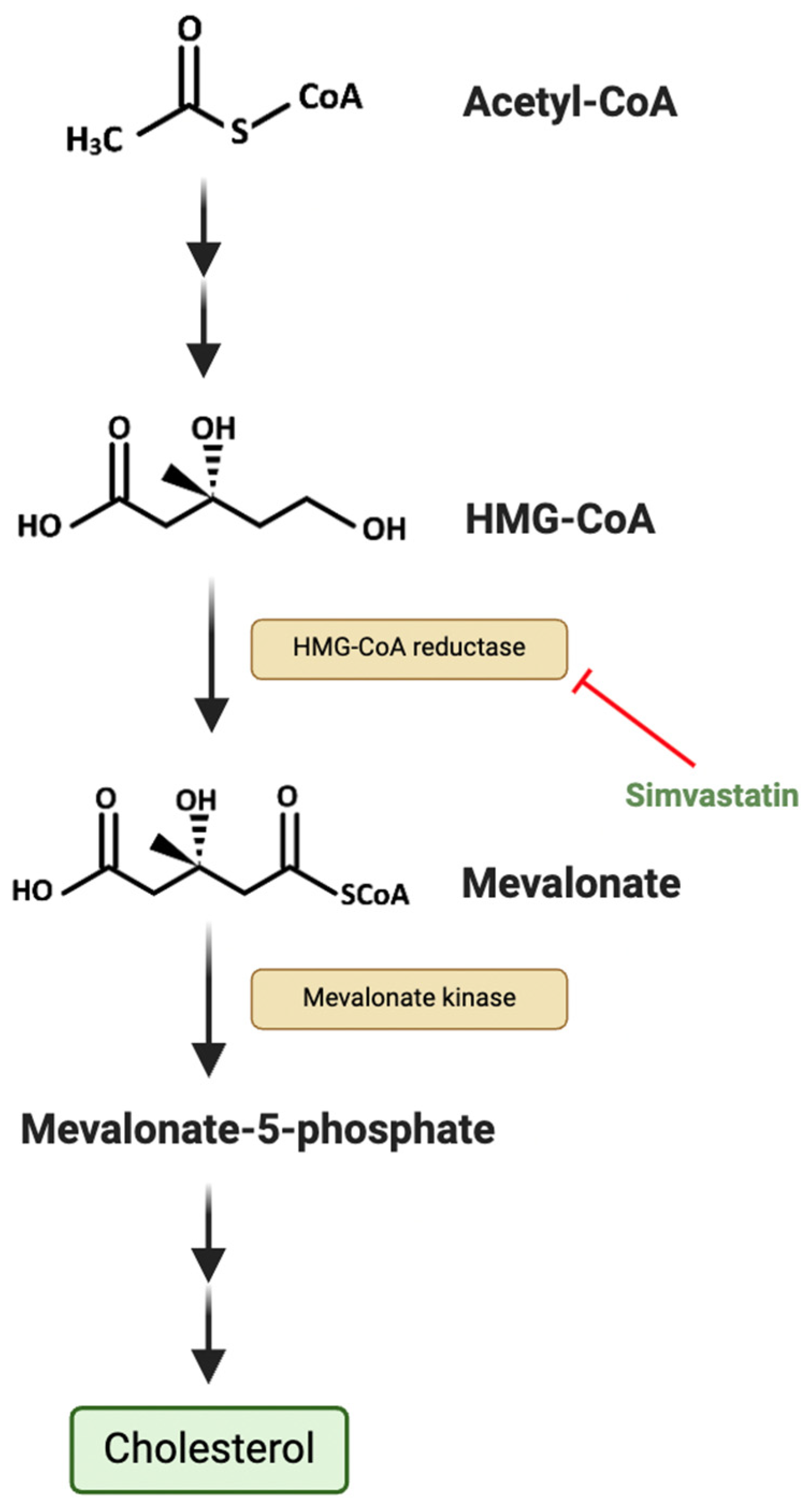
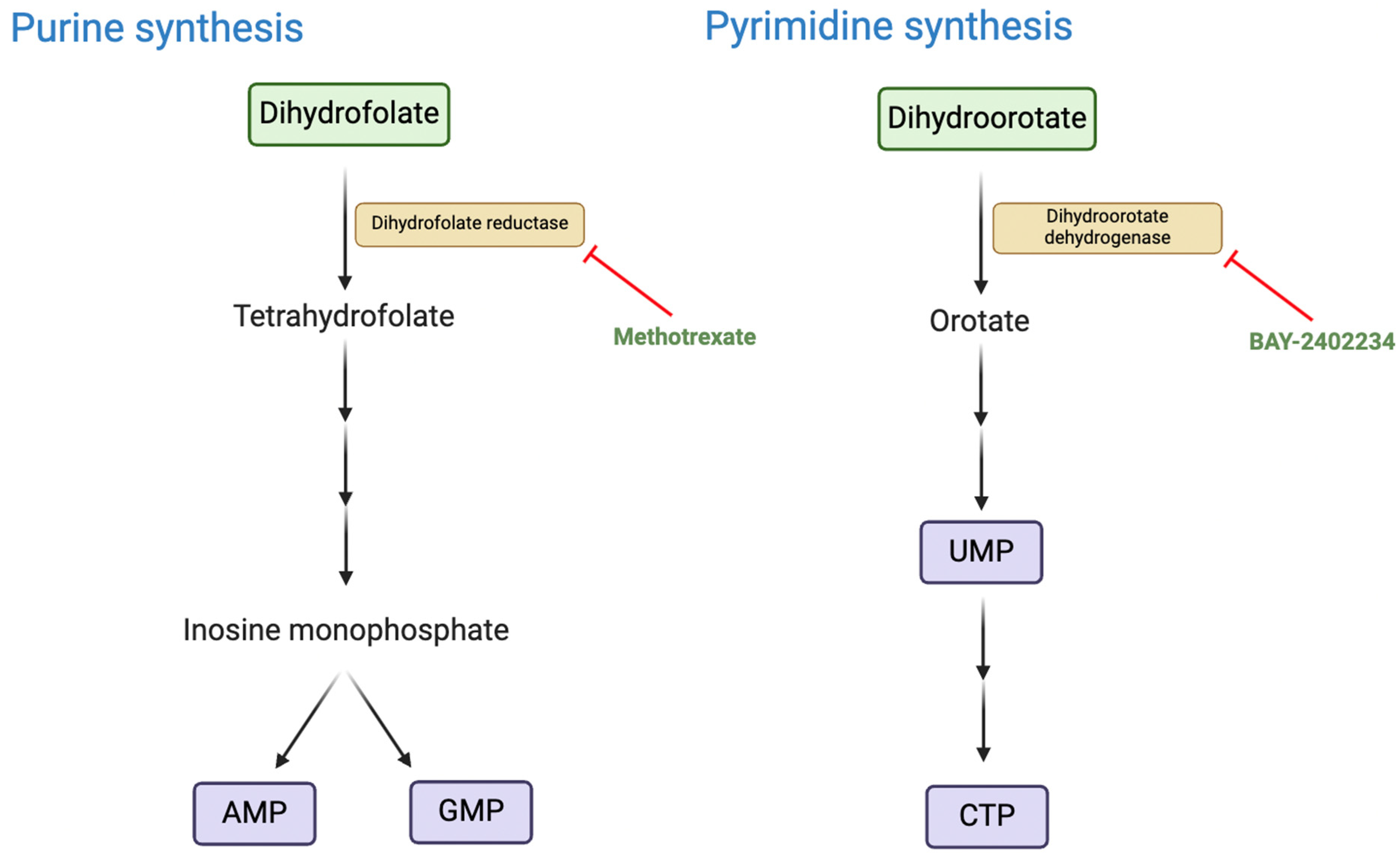
2.3. Deregulating Cellular Energetics
2.4. Non-Mutational Epigenetic Reprogramming
2.5. Activating Invasion and Metastasis
3. Ongoing Trials and Future Directions
3.1. Immunotherapies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial Number | Phase | SMARCB-1 Targeting Therapies | Disorder | Target | Status | Sites | Primary Outcome Measures |
|---|---|---|---|---|---|---|---|
| NCT04416568 [58] | II | Nivolumab and Ipilimumab | SMARCB1-negative tumours | PD-1, CTLA-4 | Recruiting | Texas, USA | Objective overall response rate |
| NCT05286801 [60] | I/II | Tiragolumab and Atezolizumab | SMARCB1- or SMARCA4-deficient tumours, including RTs | TIGIT, PD-L1 | Recruiting | USA, Canada, and Australia | Objective response rate and dose-limiting toxicities |
| NCT05407441 [62] | I/II | Tazemetostat, nivolumab, ipilimumab | SMARCB1-negative or SMARCA4-deficient tumours, including RTs | EZH2, PD-1, CTLA-4 | Recruiting | Boston, MA, USA | Toxicity and dosing parameters |
| NCT06622941 [68] | II | Nivolumab (ONO-4538) | RTs | PD-1 | Not yet recruiting | Osaka and Tokyo, Japan | Objective response rate |
| Trial Number | Phase | SMARCB-1-Targeting Therapies | Disorder | Target | Status | Sites | Primary Outcome Measures |
|---|---|---|---|---|---|---|---|
| NCT06193759 [69] | I | Cytotoxic T lymphocytes directed against proteogenomically determined tumour-specific antigens | Paediatric brain tumours, including RTs | Various tumour-specific antigens | Recruiting | Washington, USA | Various adverse events and toxicity parameters |
| NCT05835687 [70] | I | Locoregional autologous B7-H3-CAR T cells | Primary CNS neoplasms, including RTs | B7-H3-positive tumours | Recruiting | Tennessee, USA | Maximum tolerated dose |
| NCT05103631 [65] | I | GPC3-CART cells and IL-15 | Solid tumours, including RTs | GPC3-positive tumours | Recruiting | Texas, USA | Dose-limiting toxicities |
| NCT04897321 [71] | I | Autologous B7-H3-CAR T cells | Solid tumours, including RTs | B7-H3-positive tumours | Recruiting | Tennessee, USA | Maximum tolerated dose |
| NCT04715191 [66] | I | GPC3-CART cells and IL-15/21 | Paediatric solid tumours, including RTs | GPC3-positive tumours | Recruiting | Texas, USA | Dose-limiting toxicities |
| NCT04377932 [67] | I | GPC3-CART cells and IL-15 | Paediatric solid tumours, including RTs | GPC3-positive tumours | Recruiting | Texas, USA | Dose-limiting toxicities |
| NCT04185038 [72] | I | Locoregional autologous B7-H3-CAR T cells | Paediatric CNS tumours, including RTs | B7-H3-positive tumours | Recruiting | Washington, USA | Feasibility and adverse event parameters |
| NCT03618381 [73] | I | EGFR806 CAR T cell immunotherapy | Recurrent/refractory solid tumours in children and young adults, including RTs | EGFR | Recruiting | Washington, USA | Maximum tolerated dose, feasibility, and adverse event parameters |
| NCT04483778 [74] | I | B7-H3-CAR T cells | Recurrent/refractory solid tumours in children and young adults, including RTs | B7-H3-positive tumours | Active, not recruiting | Washington, USA | Various safety, tolerability, toxicity, and feasibility parameters |
3.2. Targeting Epigenetic Aberrations
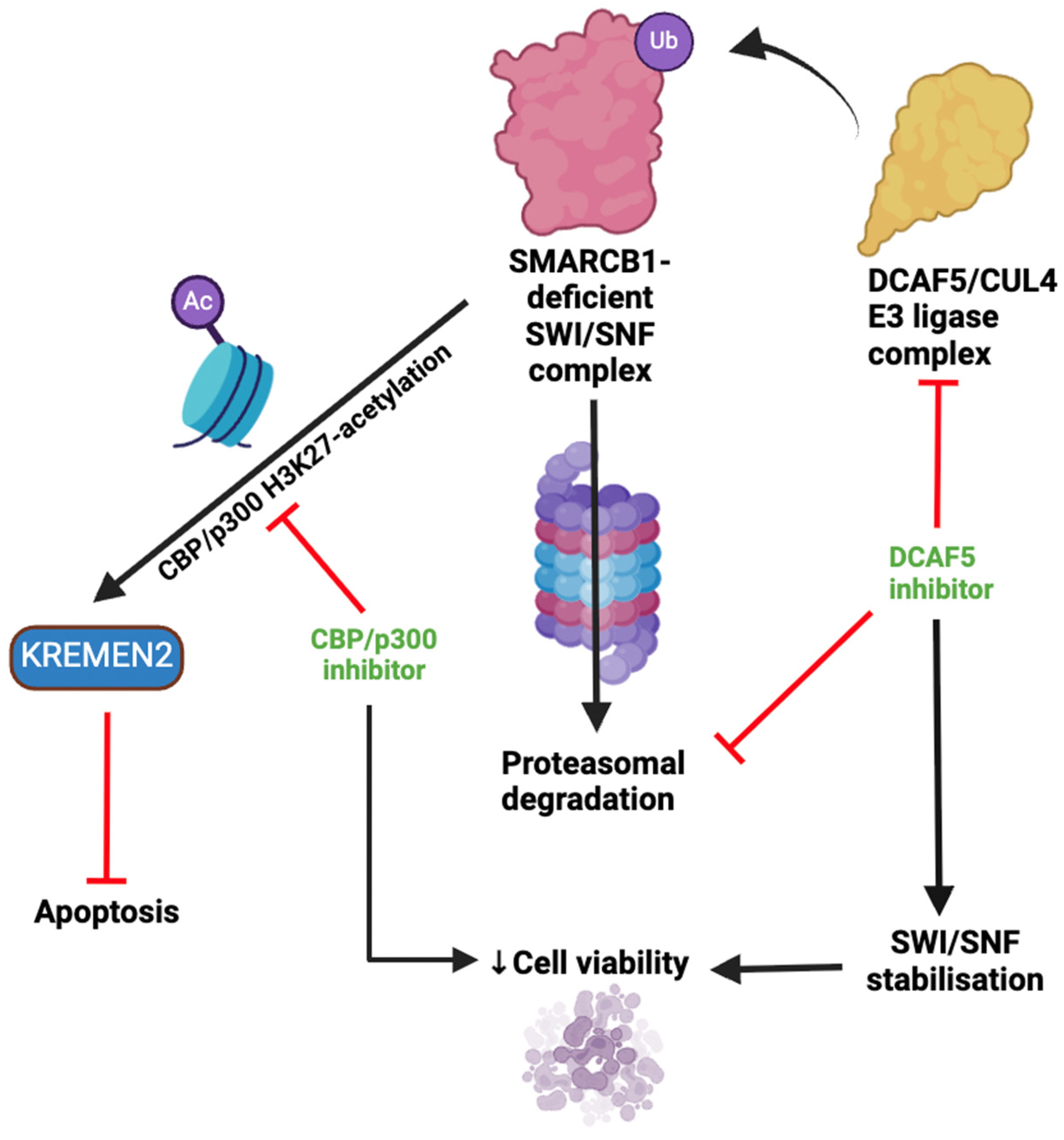
3.3. Targeting Auxiliary Oncogenic Aberrations
3.4. Next Frontiers in Rhabdoid Tumour Research and Treatment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pawel, B.R. SMARCB1-deficient Tumors of Childhood: A Practical Guide. Pediatr. Dev. Pathol. 2017, 21, 6–28. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.W.; Hong, A.L. SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers 2022, 14, 3645. [Google Scholar] [CrossRef]
- Gupta, N.K.; Godbole, N.; Sanmugananthan, P.; Gunda, S.; Kasula, V.; Baggett, M.; Gajjar, A.; Kouam, R.W.; D’Amico, R.; Rodgers, S. Management of Atypical Teratoid/Rhabdoid Tumors in the Pediatric Population: A Systematic Review and Meta-Analysis. World Neurosurg. 2024, 181, e504–e515. [Google Scholar] [CrossRef]
- Nemes, K.; Bens, S.; Bourdeaut, F.; Johann, P.; Kordes, U.; Siebert, R.; Frühwald, M.C. Rhabdoid Tumor Predisposition Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Sultan, I.; Qaddoumi, I.; Rodríguez-Galindo, C.; Nassan, A.A.; Ghandour, K.; Al-Hussaini, M. Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr. Blood Cancer 2010, 54, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.S.; Stewart, C.; Carter, S.L.; Ambrogio, L.; Cibulskis, K.; Sougnez, C.; Lawrence, M.S.; Auclair, D.; Mora, J.; Golub, T.R.; et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J. Clin. Investig. 2012, 122, 2983–2988. [Google Scholar] [CrossRef]
- Nemes, K.; Fruhwald, M.C. Emerging therapeutic targets for the treatment of malignant rhabdoid tumors. Expert Opin. Ther. Targets 2018, 22, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, S.N.; Chetty, R. Gene of the month: SMARCB1. J. Clin. Pathol. 2016, 69, 484–489. [Google Scholar] [CrossRef]
- Roberts, C.W.; Leroux, M.M.; Fleming, M.D.; Orkin, S.H. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell 2002, 2, 415–425. [Google Scholar] [CrossRef]
- Han, Z.Y.; Richer, W.; Freneaux, P.; Chauvin, C.; Lucchesi, C.; Guillemot, D.; Grison, C.; Lequin, D.; Pierron, G.; Masliah-Planchon, J.; et al. The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat. Commun. 2016, 7, 10421. [Google Scholar] [CrossRef]
- Pathak, R.; Zin, F.; Thomas, C.; Bens, S.; Gayden, T.; Karamchandani, J.; Dudley, R.W.; Nemes, K.; Johann, P.D.; Oyen, F.; et al. Inhibition of nuclear export restores nuclear localization and residual tumor suppressor function of truncated SMARCB1/INI1 protein in a molecular subset of atypical teratoid/rhabdoid tumors. Acta Neuropathol. 2021, 142, 361–374. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Betz, B.L.; Strobeck, M.W.; Reisman, D.N.; Knudsen, E.S.; Weissman, B.E. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene 2002, 21, 5193–5203. [Google Scholar] [CrossRef]
- García-Gutiérrez, L.; Delgado, M.D.; León, J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef]
- Weissmiller, A.M.; Wang, J.; Lorey, S.L.; Howard, G.C.; Martinez, E.; Liu, Q.; Tansey, W.P. Inhibition of MYC by the SMARCB1 tumor suppressor. Nat. Commun. 2019, 10, 2014. [Google Scholar] [CrossRef]
- Amati, B.; Land, H. Myc—Max—Mad: A transcription factor network controlling cell cycle progression, differentiation and death. Curr. Opin. Genet. Dev. 1994, 4, 102–108. [Google Scholar] [CrossRef]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Darr, J.; Klochendler, A.; Isaac, S.; Geiger, T.; Eden, A. Phosphoproteomic analysis reveals Smarcb1 dependent EGFR signaling in Malignant Rhabdoid tumor cells. Mol. Cancer 2015, 14, 167. [Google Scholar] [CrossRef]
- Wong, J.P.; Todd, J.R.; Finetti, M.A.; McCarthy, F.; Broncel, M.; Vyse, S.; Luczynski, M.T.; Crosier, S.; Ryall, K.A.; Holmes, K.; et al. Dual Targeting of PDGFRα and FGFR1 Displays Synergistic Efficacy in Malignant Rhabdoid Tumors. Cell Rep. 2016, 17, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Gregory, G.L.; Copple, I.M. Modulating the expression of tumor suppressor genes using activating oligonucleotide technologies as a therapeutic approach in cancer. Mol. Ther. Nucleic Acids 2023, 31, 211–223. [Google Scholar] [CrossRef]
- Ge, M.; Luo, J.; Wu, Y.; Shen, G.; Kuang, X. The biological essence of synthetic lethality: Bringing new opportunities for cancer therapy. MedComm—Oncology 2024, 3, e70. [Google Scholar] [CrossRef]
- Radko-Juettner, S.; Yue, H.; Myers, J.A.; Carter, R.D.; Robertson, A.N.; Mittal, P.; Zhu, Z.; Hansen, B.S.; Donovan, K.A.; Hunkeler, M.; et al. Targeting DCAF5 suppresses SMARCB1-mutant cancer by stabilizing SWI/SNF. Nature 2024, 628, 442–449. [Google Scholar] [CrossRef]
- Dedes, K.J.; Wilkerson, P.M.; Wetterskog, D.; Weigelt, B.; Ashworth, A.; Reis-Filho, J.S. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle 2011, 10, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.E.; Johann, P.D.; Milne, K.; Zapatka, M.; Buellesbach, A.; Ishaque, N.; Iskar, M.; Erkek, S.; Wei, L.; Tessier-Cloutier, B.; et al. Identification and Analyses of Extra-Cranial and Cranial Rhabdoid Tumor Molecular Subgroups Reveal Tumors with Cytotoxic T Cell Infiltration. Cell Rep. 2019, 29, 2338–2354.e7. [Google Scholar] [CrossRef]
- Forrest, S.J.; Al-Ibraheemi, A.; Doan, D.; Ward, A.; Clinton, C.M.; Putra, J.; Pinches, R.S.; Kadoch, C.; Chi, S.N.; DuBois, S.G.; et al. Genomic and Immunologic Characterization of INI1-Deficient Pediatric Cancers. Clin. Cancer Res. 2020, 26, 2882–2890. [Google Scholar] [CrossRef]
- Tran, S.; Plant-Fox, A.S.; Chi, S.N.; Narendran, A. Current advances in immunotherapy for atypical teratoid rhabdoid tumor (ATRT). Neurooncol. Pract. 2023, 10, 322–334. [Google Scholar] [CrossRef]
- Pecci, F.; Cognigni, V.; Giudice, G.C.; Paoloni, F.; Cantini, L.; Saini, K.S.; Abushukair, H.M.; Naqash, A.R.; Cortellini, A.; Mazzaschi, G.; et al. Unraveling the link between cholesterol and immune system in cancer: From biological mechanistic insights to clinical evidence. A narrative review. Crit. Rev. Oncol./Hematol. 2025, 209, 104654. [Google Scholar] [CrossRef]
- Ye, L.; Wen, X.; Qin, J.; Zhang, X.; Wang, Y.; Wang, Z.; Zhou, T.; Di, Y.; He, W. Metabolism-regulated ferroptosis in cancer progression and therapy. Cell Death Dis. 2024, 15, 196. [Google Scholar] [CrossRef]
- Xiao, M.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Li, J.; Xu, H.; Zhao, Y.; Yu, X.; Shi, S. Functional significance of cholesterol metabolism in cancer: From threat to treatment. Exp. Mol. Med. 2023, 55, 1982–1995. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, F.; Yokogami, K.; Yamada, A.; Moritake, H.; Watanabe, T.; Yamashita, S.; Sato, Y.; Takeshima, H. Targeting cholesterol biosynthesis for AT/RT: Comprehensive expression analysis and validation in newly established AT/RT cell line. Hum. Cell 2024, 37, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Cash, T.; Jonus, H.C.; Tsvetkova, M.; Beumer, J.H.; Sadanand, A.; Lee, J.Y.; Henry, C.J.; Aguilera, D.; Harvey, R.D.; Goldsmith, K.C. A phase 1 study of simvastatin in combination with topotecan and cyclophosphamide in pediatric patients with relapsed and/or refractory solid and CNS tumors. Pediatr Blood Cancer 2023, 70, e30405. [Google Scholar] [CrossRef]
- Dorsch, M.; Kowalczyk, M.; Planque, M.; Heilmann, G.; Urban, S.; Dujardin, P.; Forster, J.; Ueffing, K.; Nothdurft, S.; Oeck, S.; et al. Statins affect cancer cell plasticity with distinct consequences for tumor progression and metastasis. Cell Rep. 2021, 37, 110056. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Fiorentino, R.; Chiarelli, F. Statins in Children, an Update. Int. J. Mol. Sci. 2023, 24, 1366. [Google Scholar] [CrossRef]
- Kes, M.M.G.; Morales-Rodriguez, F.; Zaal, E.A.; de Souza, T.; Proost, N.; van de Ven, M.; van den Heuvel-Eibrink, M.M.; Jansen, J.W.A.; Berkers, C.R.; Drost, J. Metabolic profiling of patient-derived organoids reveals nucleotide synthesis as a metabolic vulnerability in malignant rhabdoid tumors. Cell Rep. Med. 2025, 6, 101878. [Google Scholar] [CrossRef]
- Kenny, C.; O’Meara, E.; Ulas, M.; Hokamp, K.; O’Sullivan, M.J. Global Chromatin Changes Resulting from Single-Gene Inactivation-The Role of SMARCB1 in Malignant Rhabdoid Tumor. Cancers 2021, 13, 2561. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lee, R.S.; Alver, B.H.; Haswell, J.R.; Wang, S.; Mieczkowski, J.; Drier, Y.; Gillespie, S.M.; Archer, T.C.; Wu, J.N.; et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 2017, 49, 289–295. [Google Scholar] [CrossRef]
- Hoy, S.M. Tazemetostat: First Approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.; Troisi, E.C.; Howard, T.P.; Haswell, J.R.; Wolf, B.K.; Hawk, W.H.; Ramos, P.; Oberlick, E.M.; Tzvetkov, E.P.; et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 2019, 10, 1881. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Ogiwara, H. Efficacy of glutathione inhibitor eprenetapopt against the vulnerability of glutathione metabolism in SMARCA4-, SMARCB1- and PBRM1-deficient cancer cells. Sci. Rep. 2024, 14, 31321. [Google Scholar] [CrossRef]
- Sasaki, M.; Kato, D.; Murakami, K.; Yoshida, H.; Takase, S.; Otsubo, T.; Ogiwara, H. Targeting dependency on a paralog pair of CBP/p300 against de-repression of KREMEN2 in SMARCB1-deficient cancers. Nat. Commun. 2024, 15, 4770. [Google Scholar] [CrossRef]
- Mao, B.; Wu, W.; Davidson, G.; Marhold, J.; Li, M.; Mechler, B.M.; Delius, H.; Hoppe, D.; Stannek, P.; Walter, C.; et al. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature 2002, 417, 664–667. [Google Scholar] [CrossRef] [PubMed]
- Sumia, I.; Pierani, A.; Causeret, F. Kremen1-induced cell death is regulated by homo- and heterodimerization. Cell Death Discov. 2019, 5, 91. [Google Scholar] [CrossRef]
- Amara, C.S.; Kami Reddy, K.R.; Yuntao, Y.; Chan, Y.S.; Piyarathna, D.W.B.; Dobrolecki, L.E.; Shih, D.J.H.; Shi, Z.; Xu, J.; Huang, S.; et al. The IL6/JAK/STAT3 signaling axis is a therapeutic vulnerability in SMARCB1-deficient bladder cancer. Nat. Commun. 2024, 15, 1373. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Balanis, N.; Carlin, C.R.; Schiemann, W.P. STAT3 and epithelial-mesenchymal transitions in carcinomas. Jakstat 2014, 3, e28975. [Google Scholar] [CrossRef]
- Liu, W.H.; Chen, M.T.; Wang, M.L.; Lee, Y.Y.; Chiou, G.Y.; Chien, C.S.; Huang, P.I.; Chen, Y.W.; Huang, M.C.; Chiou, S.H.; et al. Cisplatin-selected resistance is associated with increased motility and stem-like properties via activation of STAT3/Snail axis in atypical teratoid/rhabdoid tumor cells. Oncotarget 2015, 6, 1750–1768. [Google Scholar] [CrossRef]
- Murzabdillaeva, A.; Elzamly, S.; Brown, R.; Buryanek, J.; Jafri, S.; Rowe, J. Prometastatic CXCR4 and Histone Methyltransferase EZH2 are Upregulated in SMARCB1/INI1-deficient and TP53-mutated Metastatic Poorly Differentiated Chordoma to the Liver. Am. J. Clin. Pathol. 2020, 154 (Suppl. S1), S151. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Y.; Liu, W.; Zhang, W.; Xu, J. EZH2-mediated loss of miR-622 determines CXCR4 activation in hepatocellular carcinoma. Nat. Commun. 2015, 6, 8494. [Google Scholar] [CrossRef]
- Chien, Y.C.; Chen, J.N.; Chen, Y.H.; Chou, R.H.; Lee, H.C.; Yu, Y.L. Epigenetic Silencing of miR-9 Promotes Migration and Invasion by EZH2 in Glioblastoma Cells. Cancers 2020, 12, 1781. [Google Scholar] [CrossRef] [PubMed]
- Marhelava, K.; Pilch, Z.; Bajor, M.; Graczyk-Jarzynka, A.; Zagozdzon, R. Targeting Negative and Positive Immune Checkpoints with Monoclonal Antibodies in Therapy of Cancer. Cancers 2019, 11, 1756. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccines Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef]
- Liu, P.C.; Ssu, C.T.; Tsao, Y.P.; Liou, T.L.; Tsai, C.Y.; Chou, C.T.; Chen, M.H.; Leu, C.M. Cytotoxic T lymphocyte-associated antigen-4-Ig (CTLA-4-Ig) suppresses Staphylococcus aureus-induced CD80, CD86, and pro-inflammatory cytokine expression in human B cells. Arthritis Res. Ther. 2020, 22, 64. [Google Scholar] [CrossRef] [PubMed]
- Bolm, L.; Petruch, N.; Sivakumar, S.; Annels, N.E.; Frampton, A.E. Gene of the month: T-cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT). J. Clin. Pathol. 2022, 75, 217–221. [Google Scholar] [CrossRef]
- Davis, K.L.; Fox, E.; Isikwei, E.; Reid, J.M.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; Mackall, C.L. A Phase I/II Trial of Nivolumab plus Ipilimumab in Children and Young Adults with Relapsed/Refractory Solid Tumors: A Children’s Oncology Group Study ADVL1412. Clin. Cancer Res. 2022, 28, 5088–5097. [Google Scholar] [CrossRef]
- Forrest, S.J.; Yi, J.; Kline, C.; Cash, T.; Reddy, A.T.; Cote, G.M.; Merriam, P.; Czaplinski, J.; Bhushan, K.; DuBois, S.G.; et al. Phase II study of nivolumab and ipilimumab in children and young adults with INI1-negative cancers. J. Clin. Oncol. 2021, 39 (Suppl. S15), TPS10055-TPS10055. [Google Scholar] [CrossRef]
- Nutsch, K.; Banta, K.L.; Wu, T.D.; Tran, C.W.; Mittman, S.; Duong, E.; Nabet, B.Y.; Qu, Y.; Williams, K.; Muller, S.; et al. TIGIT and PD-L1 co-blockade promotes clonal expansion of multipotent, non-exhausted antitumor T cells by facilitating co-stimulation. Nat. Cancer. 2024, 5, 1834–1851. [Google Scholar] [CrossRef]
- A Phase 1/2 Study of Tiragolumab (NSC# 827799) and Atezolizumab (NSC# 783608) in Patients with Relapsed or Refractory SMARCB1 or SMARCA4 Deficient Tumors. 2022. Available online: https://www.dana-farber.org/clinical-trials/22-528 (accessed on 28 April 2025).
- Kang, N.; Eccleston, M.; Clermont, P.L.; Latarani, M.; Male, D.K.; Wang, Y.; Crea, F. EZH2 inhibition: A promising strategy to prevent cancer immune editing. Epigenomics 2020, 12, 1457–1476. [Google Scholar] [CrossRef]
- Yi, J.S.; Czaplinski, J.; Bhushan, K.; Forrest, S.J.; Shukla, N.; Mack, S.; Hong, A.; Strachan, M.; DuBois, S.G.; London, W.B.; et al. TRLS-15. “TAZNI”: A phase I/II combination trial of tazemetostat with nivolumab and ipilimumab for children with INI1-negative or SMARCA4-deficient tumors (NCT05407441). Neuro-Oncology 2024, 26 (Suppl. S4). [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Dabas, P.; Danda, A. Revolutionizing cancer treatment: A comprehensive review of CAR-T cell therapy. Med. Oncol. 2023, 40, 275. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Interleukin-15 Armored Glypican 3-Specific Chimeric Antigen Receptor Expressed in Autologous T Cells for Solid Tumors. ClinicalTrials.gov Identifier: NCT05103631. Available online: https://clinicaltrials.gov/study/NCT05103631 (accessed on 28 April 2025).
- ClinicalTrials.gov. Interleukin-15 and -21 Armored Glypican-3-Specific Chimeric Antigen Receptor Expressed in T Cells for Pediatric Solid Tumors. ClinicalTrials.gov Identifier: NCT04715191. Available online: https://clinicaltrials.gov/study/NCT04715191 (accessed on 28 April 2025).
- ClinicalTrials.gov. Interleukin-15 Armored Glypican 3-Specific Chimeric Antigen Receptor Expressed in T Cells for Pediatric Solid Tumors. ClinicalTrials.gov Identifier: NCT04377932. Available online: https://clinicaltrials.gov/study/NCT04377932 (accessed on 28 April 2025).
- ClinicalTrials.gov. Study to ONO-4538 in Patients with Rhabdoid Tumor. ClinicalTrials.gov Identifier: NCT06622941. Available online: https://clinicaltrials.gov/study/NCT06622941 (accessed on 28 April 2025).
- ClinicalTrials.gov. Immunotherapy for Malignant Pediatric Brain Tumors Employing Adoptive Cellular Therapy (IMPACT). ClinicalTrials.gov Identifier: NCT06193759. Available online: https://clinicaltrials.gov/study/NCT06193759 (accessed on 28 April 2025).
- ClinicalTrials.gov. Loc3CAR: Locoregional Delivery of B7-H3-CAR T Cells for Pediatric Patients With Primary CNS Tumors. ClinicalTrials.gov Identifier: NCT05835687. Available online: https://clinicaltrials.gov/study/NCT05835687 (accessed on 28 April 2025).
- ClinicalTrials.gov. B7-H3-Specific Chimeric Antigen Receptor Autologous T-Cell Therapy for Pediatric Patients with Solid Tumors (3CAR). ClinicalTrials.gov Identifier: NCT04897321. Available online: https://clinicaltrials.gov/study/NCT04897321 (accessed on 28 April 2025).
- ClinicalTrials.gov. Study of B7-H3-Specific CAR T Cell Locoregional Immunotherapy for Diffuse Intrinsic Pontine Glioma/Diffuse Midline Glioma and Recurrent or Refractory Pediatric Central Nervous System Tumors. ClinicalTrials.gov Identifier: NCT04185038. Available online: https://clinicaltrials.gov/study/NCT04185038 (accessed on 28 April 2025).
- ClinicalTrials.gov. EGFR806 CAR T Cell Immunotherapy for Recurrent/Refractory Solid Tumors in Children and Young Adults. ClinicalTrials.gov Identifier: NCT03618381. Available online: https://clinicaltrials.gov/study/NCT03618381 (accessed on 28 April 2025).
- ClinicalTrials.gov. B7H3 CAR T Cell Immunotherapy for Recurrent/Refractory Solid Tumors in Children and Young Adults. ClinicalTrials.gov Identifier: NCT04483778. Available online: https://clinicaltrials.gov/study/NCT04483778 (accessed on 28 April 2025).
- Esteller, M.; Dawson, M.A.; Kadoch, C.; Rassool, F.V.; Jones, P.A.; Baylin, S.B. The Epigenetic Hallmarks of Cancer. Cancer Discov. 2024, 14, 1783–1809. [Google Scholar] [CrossRef] [PubMed]
- Sadida, H.Q.; Abdulla, A.; Marzooqi, S.A.; Hashem, S.; Macha, M.A.; Akil, A.S.A.; Bhat, A.A. Epigenetic modifications: Key players in cancer heterogeneity and drug resistance. Transl. Oncol. 2024, 39, 101821. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. EZH2 Inhibitor Tazemetostat in Pediatric Subjects with Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma. ClinicalTrials.gov Identifier: NCT02601937. Available online: https://clinicaltrials.gov/study/NCT02601937 (accessed on 28 April 2025).
- Chi, S.N.; Bourdeaut, F.; Casanova, M.; Kilburn, L.B.; Hargrave, D.R.; McCowage, G.B.; Pinto, N.R.; Yang, J.; Chadha, R.; Kahali, B.; et al. Update on phase 1 study of tazemetostat, an enhancer of zeste homolog 2 inhibitor, in pediatric patients with relapsed or refractory integrase interactor 1–negative tumors. J. Clin. Oncol. 2022, 40 (Suppl. 16), 10040. [Google Scholar] [CrossRef]
- Ge, Z.; Da, Y.; Xue, Z.; Zhang, K.; Zhuang, H.; Peng, M.; Li, Y.; Li, W.; Simard, A.; Hao, J.; et al. Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp. Neurol. 2013, 241, 56–66. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Vorinostat and Temozolomide in Treating Young Patients with Relapsed or Refractory Primary Brain Tumors or Spinal Cord Tumors. ClinicalTrials.gov Identifier: NCT01076530. Available online: https://clinicaltrials.gov/study/NCT01076530 (accessed on 28 April 2025).
- Hummel, T.R.; Wagner, L.; Ahern, C.; Fouladi, M.; Reid, J.M.; McGovern, R.M.; Ames, M.M.; Gilbertson, R.J.; Horton, T.; Ingle, A.M.; et al. A pediatric phase 1 trial of vorinostat and temozolomide in relapsed or refractory primary brain or spinal cord tumors: A Children’s Oncology Group phase 1 consortium study. Pediatr. Blood Cancer 2013, 60, 1452–1457. [Google Scholar] [CrossRef]
- Nemes, K.; Johann, P.D.; Tüchert, S.; Melchior, P.; Vokuhl, C.; Siebert, R.; Furtwängler, R.; Frühwald, M.C. Current and Emerging Therapeutic Approaches for Extracranial Malignant Rhabdoid Tumors. Cancer Manag. Res. 2022, 14, 479–498. [Google Scholar] [CrossRef]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef]
- Upadhyaya, S.; Campagne, O.; Robinson, G.W.; Onar-Thomas, A.; Orr, B.; Billups, C.A.; Tatevossian, R.G.; Broniscer, A.; Kilburn, L.B.; Baxter, P.A.; et al. Phase II study of alisertib as a single agent in recurrent or progressive atypical teratoid rhabdoid tumors. J. Clin. Oncol. 2020, 38 (Suppl. 15), 10542. [Google Scholar] [CrossRef]
- Green, A.L.; Minard, C.G.; Liu, X.; Safgren, S.L.; Pinkney, K.; Harris, L.; Link, G.; DeSisto, J.; Voss, S.; Nelson, M.D.; et al. Phase 1 trial of selinexor in pediatric recurrent/refractory solid and CNS tumors (ADVL1414): A Children’s Oncology Group Phase 1 Consortium Trial. Clin. Cancer Res. 2025, 31, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Bourdeaut, F.; DuBois, S.G.; Fischer, M.; Geller, J.I.; Gottardo, N.G.; Marabelle, A.; Pearson, A.D.J.; Modak, S.; Cash, T.; et al. A Phase I Study of the CDK4/6 Inhibitor Ribociclib (LEE011) in Pediatric Patients with Malignant Rhabdoid Tumors, Neuroblastoma, and Other Solid Tumors. Clin. Cancer Res. 2017, 23, 2433–2441. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Study of Efficacy and Safety of Ribociclib (LEE011) in Combination with Topotecan and Temozolomide (TOTEM) in Pediatric Patients With Relapsed or Refractory Neuroblastoma and Other Solid Tumors. ClinicalTrials.gov Identifier: NCT05429502. Available online: https://clinicaltrials.gov/study/NCT05429502 (accessed on 28 April 2025).
- ClinicalTrials.gov. A Study of Selinexor in People with Wilms Tumors and Other Solid Tumors. ClinicalTrials.gov Identifier: NCT05985161. Available online: https://clinicaltrials.gov/study/NCT05985161 (accessed on 28 April 2025).
- ClinicalTrials.gov. Study of Palbociclib Combined with Chemotherapy in Pediatric Patients with Recurrent/Refractory Solid Tumors. ClinicalTrials.gov Identifier: NCT03709680. Available online: https://clinicaltrials.gov/study/NCT03709680 (accessed on 28 April 2025).
- Cesur-Ergün, B.; Demir-Dora, D. Gene therapy in cancer. J. Gene Med. 2023, 25, e3550. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Moghe, M.; Rait, A.; Donaldson, K.; Harford, J.B.; Chang, E.H. SMARCB1 Gene Therapy Using a Novel Tumor-Targeted Nanomedicine Enhances Anti-Cancer Efficacy in a Mouse Model of Atypical Teratoid Rhabdoid Tumors. Int. J. Nanomed. 2024, 19, 5973–5993. [Google Scholar] [CrossRef]
- McDermott, J.; Sturtevant, D.; Kathad, U.; Varma, S.; Zhou, J.; Kulkarni, A.; Biyani, N.; Schimke, C.; Reinhold, W.C.; Elloumi, F.; et al. Artificial intelligence platform, RADR®, aids in the discovery of DNA damaging agent for the ultra-rare cancer Atypical Teratoid Rhabdoid Tumors. Front. Drug Discov. 2022, 2. [Google Scholar] [CrossRef]

| Trial Number | Phase | SMARCB-1-Targeting Therapies | Disorder | Target | Status | Sites | Primary Outcome Measures |
|---|---|---|---|---|---|---|---|
| NCT05429502 [87] | I/II | Ribociclib | Solid tumours, including RTs | CDK4/6 | Recruiting | USA, UK, Spain, Singapore, Italy, Germany, France, and Australia | Overall response rate and dose-limiting toxicities |
| NCT05985161 [88] | II | Selinexor | Solid tumours, including RTs | Exportin-1 | Recruiting | Various USA sites | Complete and partial response |
| NCT03709680 [89] | I/II | Palbociclib | Recurrent/refractory solid tumours, including RTs | CDK4/6 | Active, not recruiting | >10 countries, including USA, UK, Brazil, and Korea | Event-free survival, first-cycle dose-limiting toxicities, frequency of adverse events, complete response or partial response |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shakerdi, A.L.; Pidgeon, G.P. SMARCB1 Deficiency as a Driver of the Hallmarks of Cancer in Rhabdoid Tumours: Novel Insights into Dysregulated Energy Metabolism, Emerging Targets, and Ongoing Clinical Trials. Metabolites 2025, 15, 304. https://doi.org/10.3390/metabo15050304
Shakerdi AL, Pidgeon GP. SMARCB1 Deficiency as a Driver of the Hallmarks of Cancer in Rhabdoid Tumours: Novel Insights into Dysregulated Energy Metabolism, Emerging Targets, and Ongoing Clinical Trials. Metabolites. 2025; 15(5):304. https://doi.org/10.3390/metabo15050304
Chicago/Turabian StyleShakerdi, Abdul L., and Graham P. Pidgeon. 2025. "SMARCB1 Deficiency as a Driver of the Hallmarks of Cancer in Rhabdoid Tumours: Novel Insights into Dysregulated Energy Metabolism, Emerging Targets, and Ongoing Clinical Trials" Metabolites 15, no. 5: 304. https://doi.org/10.3390/metabo15050304
APA StyleShakerdi, A. L., & Pidgeon, G. P. (2025). SMARCB1 Deficiency as a Driver of the Hallmarks of Cancer in Rhabdoid Tumours: Novel Insights into Dysregulated Energy Metabolism, Emerging Targets, and Ongoing Clinical Trials. Metabolites, 15(5), 304. https://doi.org/10.3390/metabo15050304