
Double-Weighted Bayesian Model Combination for Metabolomics Data Description and Prediction

 ,
,  , , , , ,
, , , , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Data Pretreatment
2.3. Ensemble Machine Learning
2.4. Hyperparameter Optimization and Feature Selection
3. Results
3.1. Study 1: NMR-Based Metabolic Profiling for Diagnostic and Prognostic Purposes in Critically Ill Children (Grauslys et al. [12])
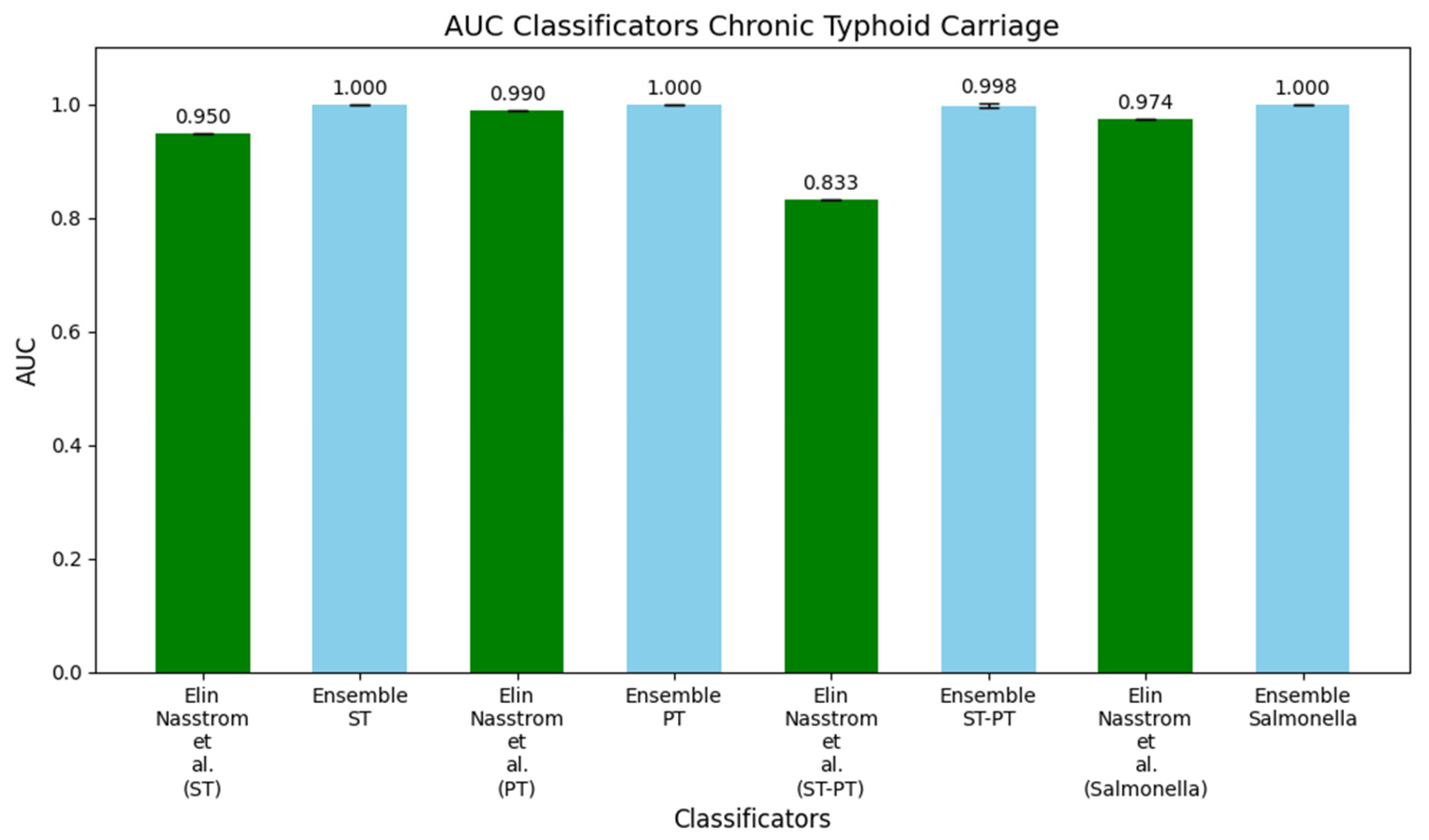
3.2. Study 2: Diagnostic Metabolite Biomarkers of Chronic Typhoid Carriage (Näsström et al. [13])
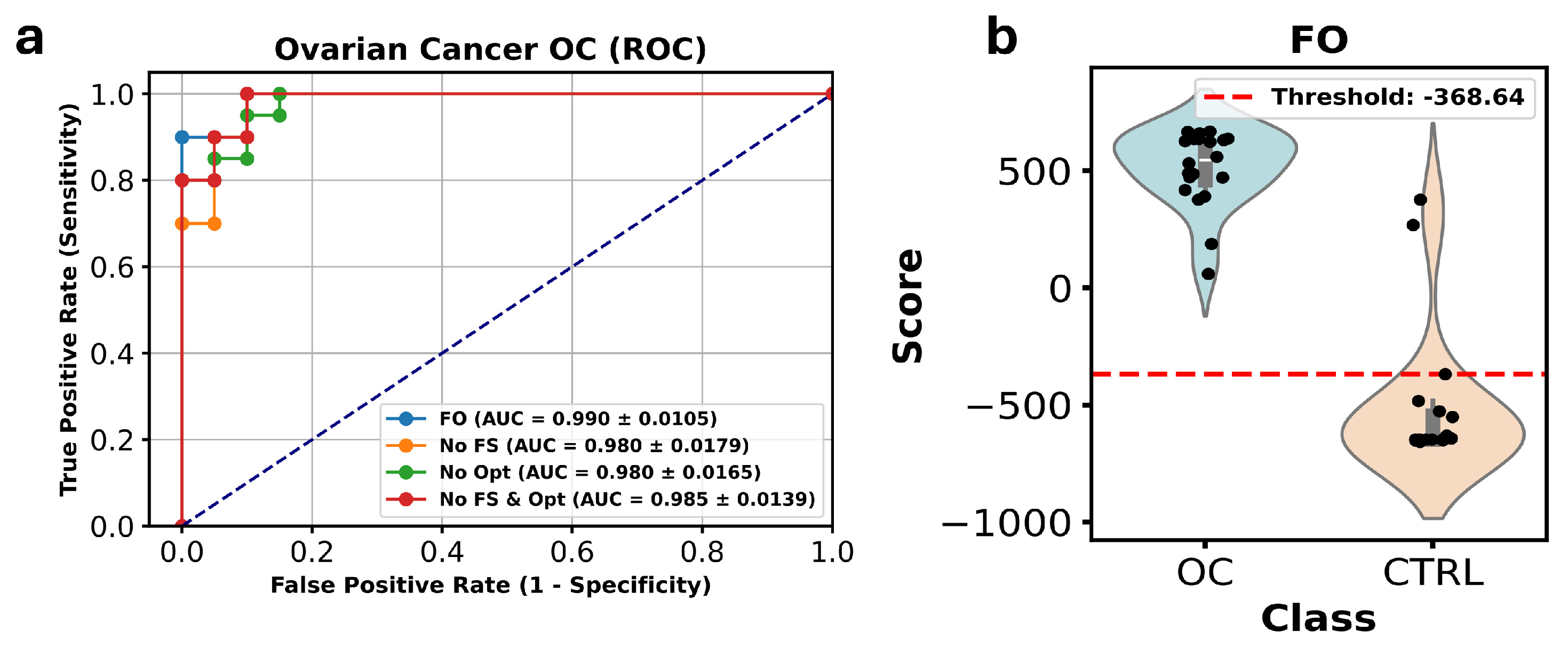
3.3. Study 3: Profiling the Metabolome of Uterine Fluid for Early Detection of Ovarian Cancer (Pan Wang et al. [14])
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bruggeman, F.J.; Westerhoff, H.V. The Nature of Systems Biology. Trends Microbiol. 2007, 15, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J. Systems Biology of Metabolism. Annu. Rev. Biochem. 2017, 86, 245–275. [Google Scholar] [CrossRef]
- Kitano, H. Systems Biology: A Brief Overview. Science 2002, 295, 1662–1664. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, A.; Foroozan, E.; Tahmasebi, A.; Taghizadeh, M.S.; Bolhassani, M.; Jafari, M. System Network Analysis of Rosmarinus Officinalis Transcriptome and Metabolome—Key Genes in Biosynthesis of Secondary Metabolites. PLoS ONE 2023, 18, e0282316. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond Biomarkers and towards Mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef]
- Troisi, J.; EBSCOhost (Eds.) Metabolomics Perspectives: From Theory to Practical Application; Academic Press: London, UK, 2022; ISBN 978-0-323-85062-9. [Google Scholar]
- Jacob, M.; Lopata, A.L.; Dasouki, M.; Abdel Rahman, A.M. Metabolomics toward Personalized Medicine. Mass Spectrom. Rev. 2019, 38, 221–238. [Google Scholar] [CrossRef]
- Troisi, J.; Richards, S.M.; Scala, G.; Landolfi, A. Chapter 7—Approaches in Untargeted Metabolomics. In Metabolomics Perspectives; Troisi, J., Ed.; Academic Press: Cambridge, MA, USA, 2022; pp. 237–262. ISBN 978-0-323-85062-9. [Google Scholar]
- Szymańska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-Check: Validation of Diagnostic Statistics for PLS-DA Models in Metabolomics Studies. Metabolomics 2012, 8, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Gromski, P.S.; Muhamadali, H.; Ellis, D.I.; Xu, Y.; Correa, E.; Turner, M.L.; Goodacre, R. A Tutorial Review: Metabolomics and Partial Least Squares-Discriminant Analysis—A Marriage of Convenience or a Shotgun Wedding. Anal. Chim. Acta 2015, 879, 10–23. [Google Scholar] [CrossRef]
- Habehh, H.; Gohel, S. Machine Learning in Healthcare. Curr. Genom. 2021, 22, 291–300. [Google Scholar] [CrossRef]
- Grauslys, A.; Phelan, M.M.; Broughton, C.; Baines, P.B.; Jennings, R.; Siner, S.; Paulus, S.C.; Carrol, E.D. NMR-Based Metabolic Profiling Provides Diagnostic and Prognostic Information in Critically Ill Children with Suspected Infection. Sci. Rep. 2020, 10, 20198. [Google Scholar] [CrossRef]
- Näsström, E.; Jonsson, P.; Johansson, A.; Dongol, S.; Karkey, A.; Basnyat, B.; Tran Vu Thieu, N.; Trinh Van, T.; Thwaites, G.E.; Antti, H.; et al. Diagnostic Metabolite Biomarkers of Chronic Typhoid Carriage. PLoS Negl. Trop. Dis. 2018, 12, e0006215. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Ma, J.; Li, W.; Wang, Q.; Xiao, Y.; Jiang, Y.; Gu, X.; Wu, Y.; Dong, S.; Guo, H.; et al. Profiling the Metabolome of Uterine Fluid for Early Detection of Ovarian Cancer. Cell Rep. Med. 2023, 4, 101061. [Google Scholar] [CrossRef]
- Yi, X.; Xu, Y.; Hu, Q.; Krishnamoorthy, S.; Li, W.; Tang, Z. ASN-SMOTE: A Synthetic Minority Oversampling Method with Adaptive Qualified Synthesizer Selection. Complex. Intell. Syst. 2022, 8, 2247–2272. [Google Scholar] [CrossRef]
- Fluss, R.; Faraggi, D.; Reiser, B. Estimation of the Youden Index and Its Associated Cutoff Point. Biom. J. J. Math. Methods Biosci. 2005, 47, 458–472. [Google Scholar] [CrossRef]
- Kennedy, A.D.; Wittmann, B.M.; Evans, A.M.; Miller, L.A.D.; Toal, D.R.; Lonergan, S.; Elsea, S.H.; Pappan, K.L. Metabolomics in the Clinic: A Review of the Shared and Unique Features of Untargeted Metabolomics for Clinical Research and Clinical Testing. J. Mass Spectrom. 2018, 53, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Singla, J.; Nkenyereye, L.; Jha, S.; Prashar, D.; Joshi, G.P.; El-Sappagh, S.; Islam, M.S.; Islam, S.M.R. Medical Diagnostic Systems Using Artificial Intelligence (AI) Algorithms: Principles and Perspectives. IEEE Access 2020, 8, 228049–228069. [Google Scholar] [CrossRef]
- Fridman, L.; Ding, L.; Jenik, B.; Reimer, B. Arguing Machines: Human Supervision of Black Box AI Systems That Make Life-Critical Decisions. In Proceedings of the 2019 IEEE/CVF Conference on Computer Vision and Pattern Recognition Workshops (CVPRW), Long Beach, CA, USA, 16–17 June 2019; IEEE: Piscataway, NJ, USA, 2019; pp. 1335–1343. [Google Scholar]
- Asakura, T.; Date, Y.; Kikuchi, J. Application of Ensemble Deep Neural Network to Metabolomics Studies. Anal. Chim. Acta 2018, 1037, 230–236. [Google Scholar] [CrossRef]
- Edeh, M.O.; Dalal, S.; Dhaou, I.B.; Agubosim, C.C.; Umoke, C.C.; Richard-Nnabu, N.E.; Dahiya, N. Artificial Intelligence-Based Ensemble Learning Model for Prediction of Hepatitis C Disease. Front. Public. Health 2022, 10, 892371. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, P.; Uddin, S.; Hajati, F.; Moni, M.A. Ensemble Learning for Disease Prediction: A Review. Healthcare 2023, 11, 1808. [Google Scholar] [CrossRef]
- ShahrjooiHaghighi, A.; Frigui, H.; Zhang, X.; Wei, X.; Shi, B.; McClain, C.J. Ensemble Feature Selection for Biomarker Discovery in Mass Spectrometry-Based Metabolomics. In Proceedings of the 34th ACM/SIGAPP Symposium on Applied Computing, Limassol, Cyprus, 8–12 April 2019; ACM: New York, NY, USA, 2019; pp. 19–24. [Google Scholar]
- Shahrjooihaghighi, A.; Frigui, H.; Zhang, X.; Wei, X.; Shi, B.; Trabelsi, A. An Ensemble Feature Selection Method for Biomarker Discovery. In Proceedings of the 2017 IEEE International Symposium on Signal Processing and Information Technology (ISSPIT), Bilbao, Spain, 18–20 December 2017; IEEE: Piscataway, NJ, USA, 2017; pp. 416–421. [Google Scholar]
- Han, S.; Huang, J.; Foppiano, F.; Prehn, C.; Adamski, J.; Suhre, K.; Li, Y.; Matullo, G.; Schliess, F.; Gieger, C.; et al. TIGER: Technical Variation Elimination for Metabolomics Data Using Ensemble Learning Architecture. Brief. Bioinform. 2022, 23, bbab535. [Google Scholar] [CrossRef]
- Netzer, M.; Hanser, F.; Breit, M.; Weinberger, K.M.; Baumgartner, C.; Baumgarten, D. Ensemble Based Approach for Time Series Classification in Metabolomics. Stud. Health Technol. Inf. 2019, 260, 89–96. [Google Scholar]
- Cao, Y.; Geddes, T.A.; Yang, J.Y.H.; Yang, P. Ensemble Deep Learning in Bioinformatics. Nat. Mach. Intell. 2020, 2, 500–508. [Google Scholar] [CrossRef]
- Yang, P.; Yee, H.Y.; Zhou, B.; Zomaya, A.Y. A Review of Ensemble Methods in Bioinformatics. Curr. Bioinform. 2010, 5, 296–308. [Google Scholar] [CrossRef]
- Troisi, J.; Colucci, A.; Cavallo, P.; Richards, S.; Symes, S.; Landolfi, A.; Scala, G.; Maiorino, F.; Califano, A.; Fabiano, M.; et al. A Serum Metabolomic Signature for the Detection and Grading of Bladder Cancer. Appl. Sci. 2021, 11, 2835. [Google Scholar] [CrossRef]
- Troisi, J.; Tafuro, M.; Lombardi, M.; Scala, G.; Richards, S.M.; Symes, S.J.K.; Ascierto, P.A.; Delrio, P.; Tatangelo, F.; Buonerba, C.; et al. A Metabolomics-Based Screening Proposal for Colorectal Cancer. Metabolites 2022, 12, 110. [Google Scholar] [CrossRef]
- Troisi, J.; Raffone, A.; Travaglino, A.; Belli, G.; Belli, C.; Anand, S.; Giugliano, L.; Cavallo, P.; Scala, G.; Symes, S.; et al. Development and Validation of a Serum Metabolomic Signature for Endometrial Cancer Screening in Postmenopausal Women. JAMA Netw. Open 2020, 3, e2018327. [Google Scholar] [CrossRef]
- Troisi, J.; Sarno, L.; Landolfi, A.; Scala, G.; Martinelli, P.; Venturella, R.; Di Cello, A.; Zullo, F.; Guida, M. Metabolomic Signature of Endometrial Cancer. J. Proteome Res. 2018, 17, 804–812. [Google Scholar] [CrossRef]
- Troisi, J.; Mollo, A.; Lombardi, M.; Scala, G.; Richards, S.M.; Symes, S.J.K.; Travaglino, A.; Neola, D.; de Laurentiis, U.; Insabato, L.; et al. The Metabolomic Approach for the Screening of Endometrial Cancer: Validation from a Large Cohort of Women Scheduled for Gynecological Surgery. Biomolecules 2022, 12, 1229. [Google Scholar] [CrossRef]
- Troisi, J.; Cavallo, P.; Richards, S.; Symes, S.; Colucci, A.; Sarno, L.; Landolfi, A.; Scala, G.; Adair, D.; Ciccone, C. Noninvasive Screening for Congenital Heart Defects Using a Serum Metabolomics Approach. Prenat. Diagn. 2021, 41, 743–753. [Google Scholar] [CrossRef]
- Troisi, J.; Landolfi, A.; Sarno, L.; Richards, S.; Symes, S.; Adair, D.; Ciccone, C.; Scala, G.; Martinelli, P.; Guida, M. A Metabolomics-Based Approach for Non-Invasive Screening of Fetal Central Nervous System Anomalies. Metabolomics 2018, 14, 77. [Google Scholar] [CrossRef]
- Troisi, J.; Sarno, L.; Martinelli, P.; Di Carlo, C.; Landolfi, A.; Scala, G.; Rinaldi, M.; D’Alessandro, P.; Ciccone, C.; Guida, M. A Metabolomics-Based Approach for Non-Invasive Diagnosis of Chromosomal Anomalies. Metabolomics 2017, 13, 140. [Google Scholar] [CrossRef]
- Troisi, J.; Lombardi, M.; Scala, G.; Cavallo, P.; Tayler, R.S.; Symes, S.J.K.; Richards, S.M.; Adair, D.C.; Fasano, A.; McCowan, L.M.; et al. A Screening Test Proposal for Congenital Defects Based on Maternal Serum Metabolomics Profile. Am. J. Obstet. Gynecol. 2022, 228, 342.e1–342.e12. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Configuration | Ensemble | NB | GLM | LR | FLM | DL | DT | RF | GBT | SVM | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy CB Model | FO | 92.9% | - | 96.7% | - | 73.3% | 83.3% | 70.0% | 60.0% | 86.7% | - |
| No FS | 90.5% | - | 93.3% | 60.0% | 80.0% | 80.0% | - | 63.3% | 60.0% | 80.0% | |
| No Opt | 88.1% | - | 83.3% | - | 73.3% | 83.3% | - | - | 76.7% | 73.3% | |
| No FS and Opt | 95.2% | - | 93.3% | 60.0% | 70.0% | 80.0% | 63.3% | 90.0% | 73.3% | 90.0% | |
| Accuracy CV Model | FO | 88.1% | 73.3% | 86.7% | - | 86.7% | 76.7% | 70.0% | 63.3% | 60.0% | 73.3% |
| No FS | 81% | 70% | 73.3% | - | 73.3% | 86.7% | 70.0% | 73.3% | 63.3% | 73.3% | |
| No Opt | 85.7% | 73.3% | 73.3% | - | 66.7% | 76.7% | 73.3% | 80.0% | 70.0% | - | |
| No FS and Opt | 83.3% | 70% | 73.3% | - | 76.7% | 86.7% | 73.3% | 70.0% | 63.3% | 80.0% | |
| Accuracy CB-CV Model | FO | 92.5% | 86.7% | 78.7% | - | 96.7% | 79.3% | 72.7% | 72.7% | 78.7% | 86.0% |
| No FS | 90% | 68% | 93.3% | - | 90.0% | 90.0% | 83.3% | 68.7% | 78.7% | 82.7% | |
| No Opt | 90% | 86.7% | 86.7% | - | 90.0% | 83.3% | - | - | 79.3% | 79.3% | |
| No FS and Opt | 92.5% | 68% | 93.3% | - | 86.7% | 90.0% | 76.7% | 75.3% | 78.7% | 93.3% |
| Configuration | Ensemble | NB | GLM | LR | FLM | DL | DT | RF | GBT | SVM | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy ST Model | FO | 100.0% | 90.0% | 96.7% | - | 93.3% | 89.3% | 90.0% | 100.0% | 93.3% | 100.0% |
| No FS | 100.0% | 100.0% | 100.0% | 96.7% | 100.0% | 96.7% | 93.3% | 96.7% | 90.0% | 100.0% | |
| No Opt | 97.5% | 90.0% | 96.7% | 89.3% | - | 100.0% | 93.3% | 93.3% | 90.0% | - | |
| No FS and Opt | 97.6% | 100.0% | 100.0% | 96.7% | 90.0% | 96.7% | 93.3% | 93.3% | 90.0% | - | |
| Accuracy PT Model | FO | 92.9% | - | 96.7% | - | 73.3% | 83.3% | 70.0% | 60.0% | 86.7% | - |
| No FS | 90.5% | - | 93.3% | 60.0% | 80.0% | 80.0% | - | 63.3% | 60.0% | 80.0% | |
| No Opt | 88.1% | - | 83.3% | - | 73.3% | 83.3% | - | - | 76.7% | 73.3% | |
| No FS and Opt | 95.2% | - | 93.3% | 60.0% | 70.0% | 80.0% | 63.3% | 90.0% | 73.3% | 90.0% | |
| Accuracy ST-PT Model | FO | 88.1% | 73.3% | 86.7% | - | 86.7% | 76.7% | 70.0% | 63.3% | 60.0% | 73.3% |
| No FS | 81% | 70% | 73.3% | - | 73.3% | 86.7% | 70.0% | 73.3% | 63.3% | 73.3% | |
| No Opt | 85.7% | 73.3% | 73.3% | - | 66.7% | 76.7% | 73.3% | 80.0% | 70.0% | - | |
| No FS and Opt | 83.3% | 70% | 73.3% | - | 76.7% | 86.7% | 73.3% | 70.0% | 63.3% | 80.0% | |
| Accuracy Salmonella Model | FO | 92.5% | 86.7% | 78.7% | - | 96.7% | 79.3% | 72.7% | 72.7% | 78.7% | 86.0% |
| No FS | 90% | 68% | 93.3% | - | 90.0% | 90.0% | 83.3% | 68.7% | 78.7% | 82.7% | |
| No Opt | 90% | 86.7% | 86.7% | - | 90.0% | 83.3% | - | - | 79.3% | 79.3% | |
| No FS and Opt | 92.5% | 68% | 93.3% | - | 86.7% | 90.0% | 76.7% | 75.3% | 78.7% | 93.3% |
| Configuration | Ensemble | NB | GLM | LR | FLM | DL | DT | RF | GBT | SVM | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy OC Model | FO | 95.0% | 93.3% | 96.0% | - | 90.0% | 88.7% | 72.7% | 96.7% | 96.7% | 96.7% |
| No FS | 95.0% | 75.3% | 93.3% | 90.0% | 89.3% | 96.7% | 81.3% | 90.0% | 85.3% | 96.7% | |
| No Opt | 92.5% | 75.3% | 96.0% | - | 86.0% | 92.7% | 88.7% | 86.0% | 68.7% | 92.7% | |
| No FS & Opt | 95.0% | 75.3% | 93.3% | 90.0% | 89.3% | 96.7% | 84.7% | 96.7% | 72.0% | 96.7% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Troisi, J.; Lombardi, M.; Trotta, A.; Abenante, V.; Ingenito, A.; Palmieri, N.; Richards, S.M.; Symes, S.J.K.; Cavallo, P. Double-Weighted Bayesian Model Combination for Metabolomics Data Description and Prediction. Metabolites 2025, 15, 214. https://doi.org/10.3390/metabo15040214
Troisi J, Lombardi M, Trotta A, Abenante V, Ingenito A, Palmieri N, Richards SM, Symes SJK, Cavallo P. Double-Weighted Bayesian Model Combination for Metabolomics Data Description and Prediction. Metabolites. 2025; 15(4):214. https://doi.org/10.3390/metabo15040214
Chicago/Turabian StyleTroisi, Jacopo, Martina Lombardi, Alessio Trotta, Vera Abenante, Andrea Ingenito, Nicole Palmieri, Sean M. Richards, Steven J. K. Symes, and Pierpaolo Cavallo. 2025. "Double-Weighted Bayesian Model Combination for Metabolomics Data Description and Prediction" Metabolites 15, no. 4: 214. https://doi.org/10.3390/metabo15040214
APA StyleTroisi, J., Lombardi, M., Trotta, A., Abenante, V., Ingenito, A., Palmieri, N., Richards, S. M., Symes, S. J. K., & Cavallo, P. (2025). Double-Weighted Bayesian Model Combination for Metabolomics Data Description and Prediction. Metabolites, 15(4), 214. https://doi.org/10.3390/metabo15040214