
Modulation of Peripheral Mast Cell and Brain Microglia Axis via Kinase Inhibition

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Synthesis of BK40196
2.2. Kinase Screening
2.3. In Vitro Mast Cell Analysis
2.4. In Vitro Autophagy Assay
2.5. In Vivo Mast Cell Analysis
2.6. Animal Euthunasia and Tissue Preparation
2.7. Pharmacokinetic Analysis
2.8. Determination of Maximal Tolerated Dose and Tissue Toxicity
2.9. Sex as a Biological Variable
2.10. Transgenic Mice and Treatment
2.11. Rotarod Test
2.12. Elevated Plus Maze Test
2.13. Novel Object Recognition
2.14. Morris Water Maze
2.15. Immunohistochemistry
2.16. Microglial Morphological Analysis
2.17. Silver Staining
2.18. ELISA
2.19. Western Blot
2.20. Statistical Analysis
2.21. Ethics and Integrity
3. Results
3.1. Screening of Kinase Binding of BK40196
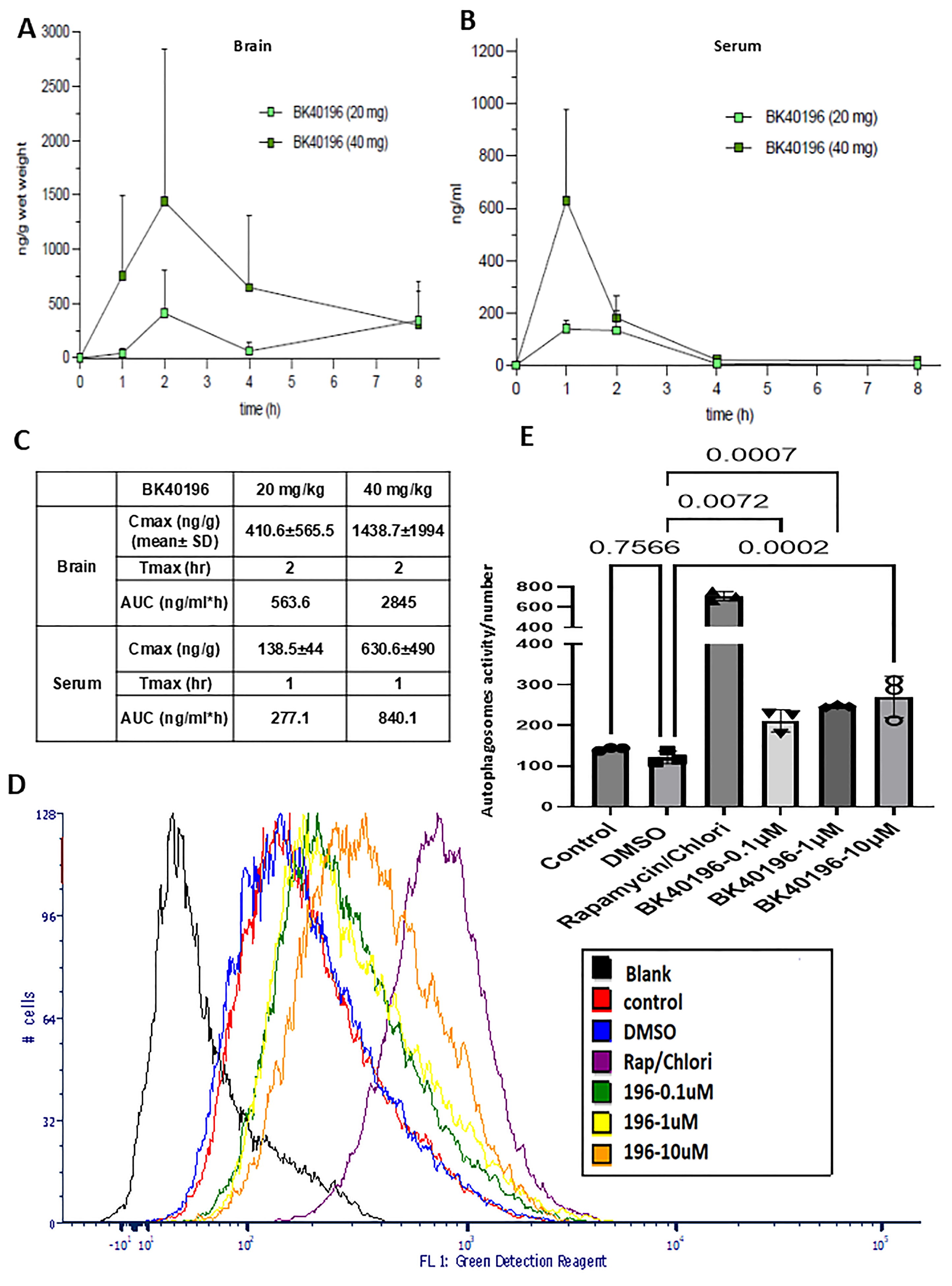
3.2. Pharmacokinetics and Maximal Tolerated Dose
3.3. BK40196 Induces Autophagy In Vitro
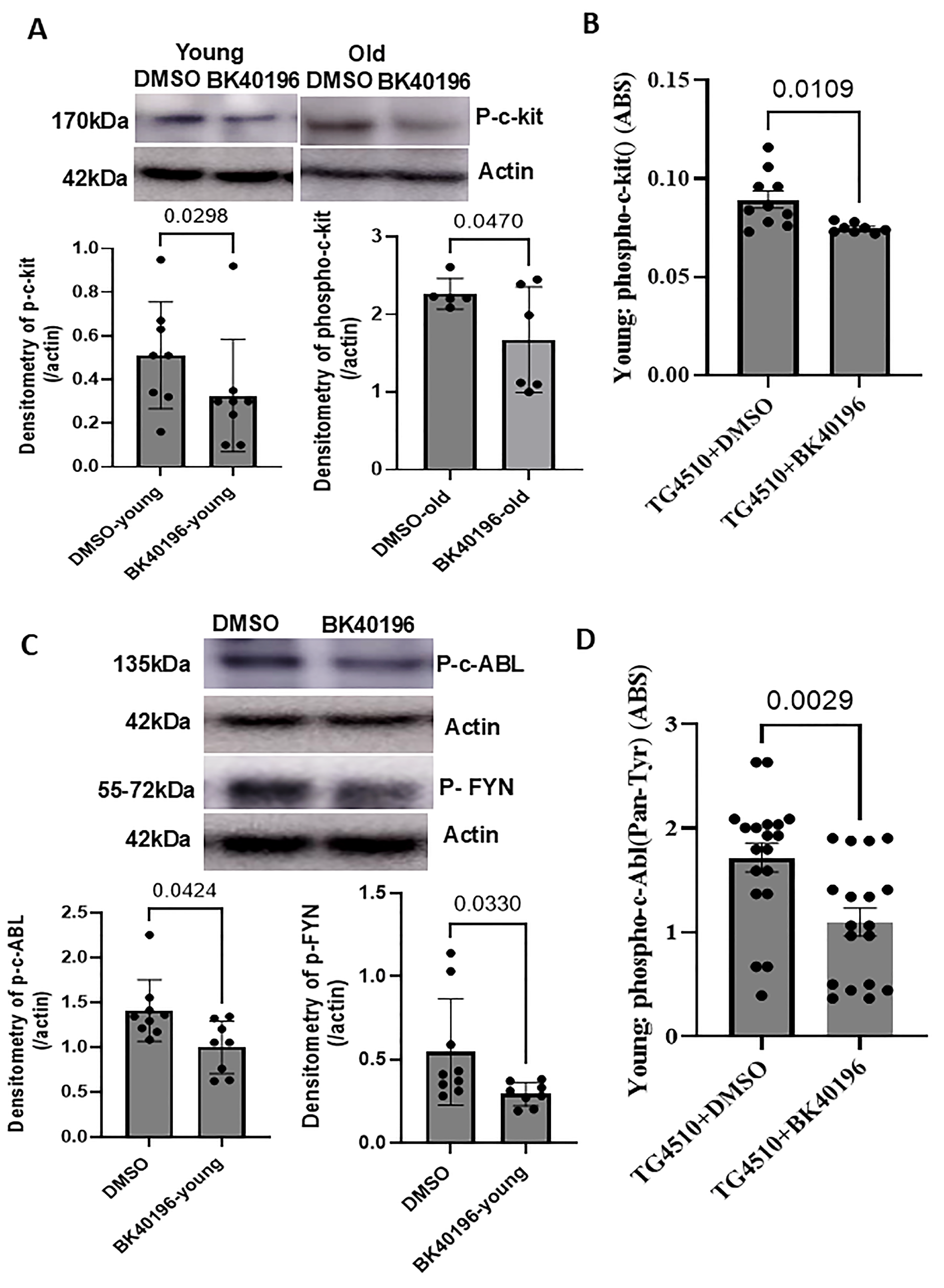
3.4. BK40196 Inhibits c-KIT, Phospho-c-Abl and Phospho-Fyn in rTg4510 Mice
3.5. BK40196 Prevents Mast Cell Proliferation and Maturation In Vitro
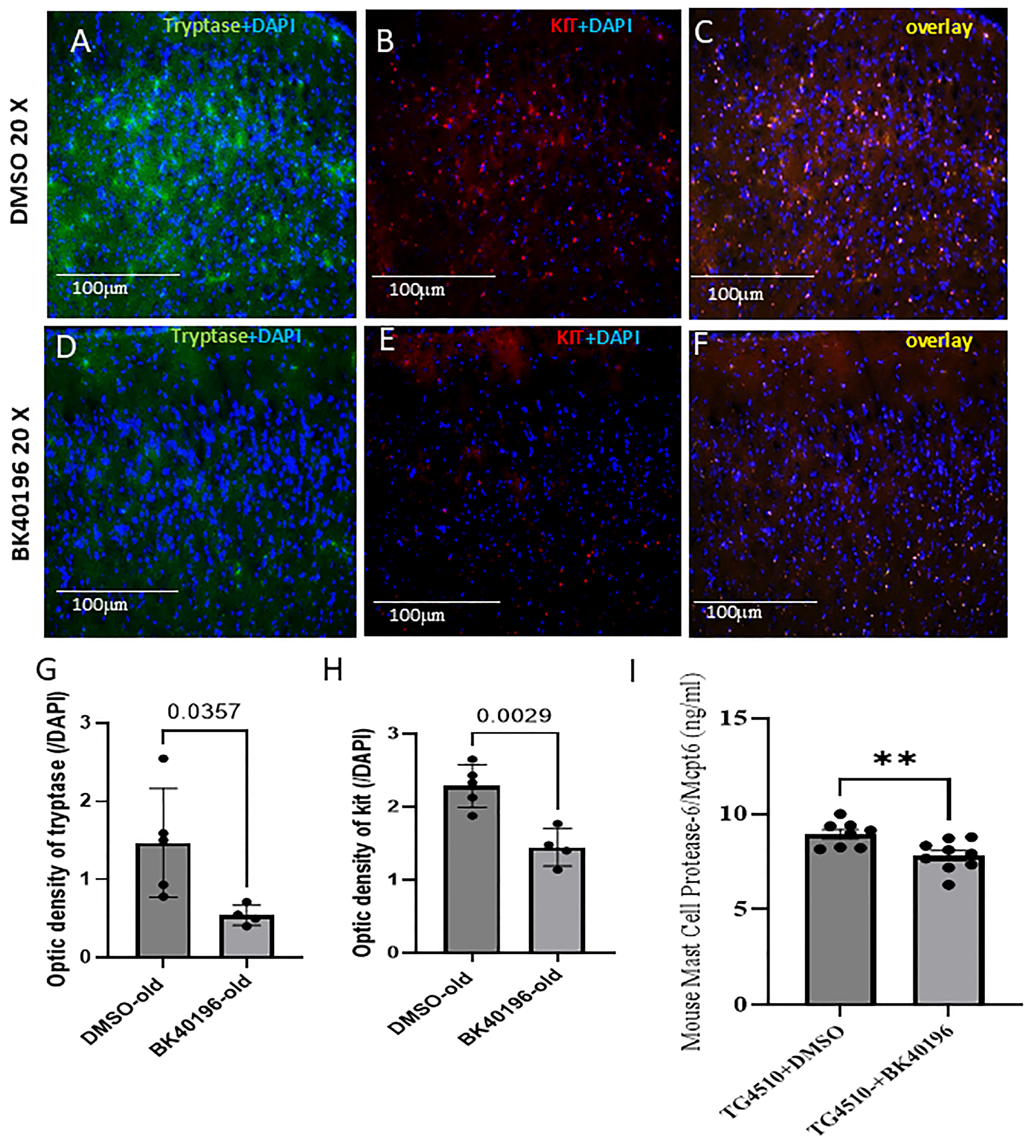
3.6. BK40196 Inhibits Peripheral Mast Cell Maturation In Vivo
3.7. BK40196 Reduces Mast Cell-Derived Tryptase in the Brain of rTg4510 Mice
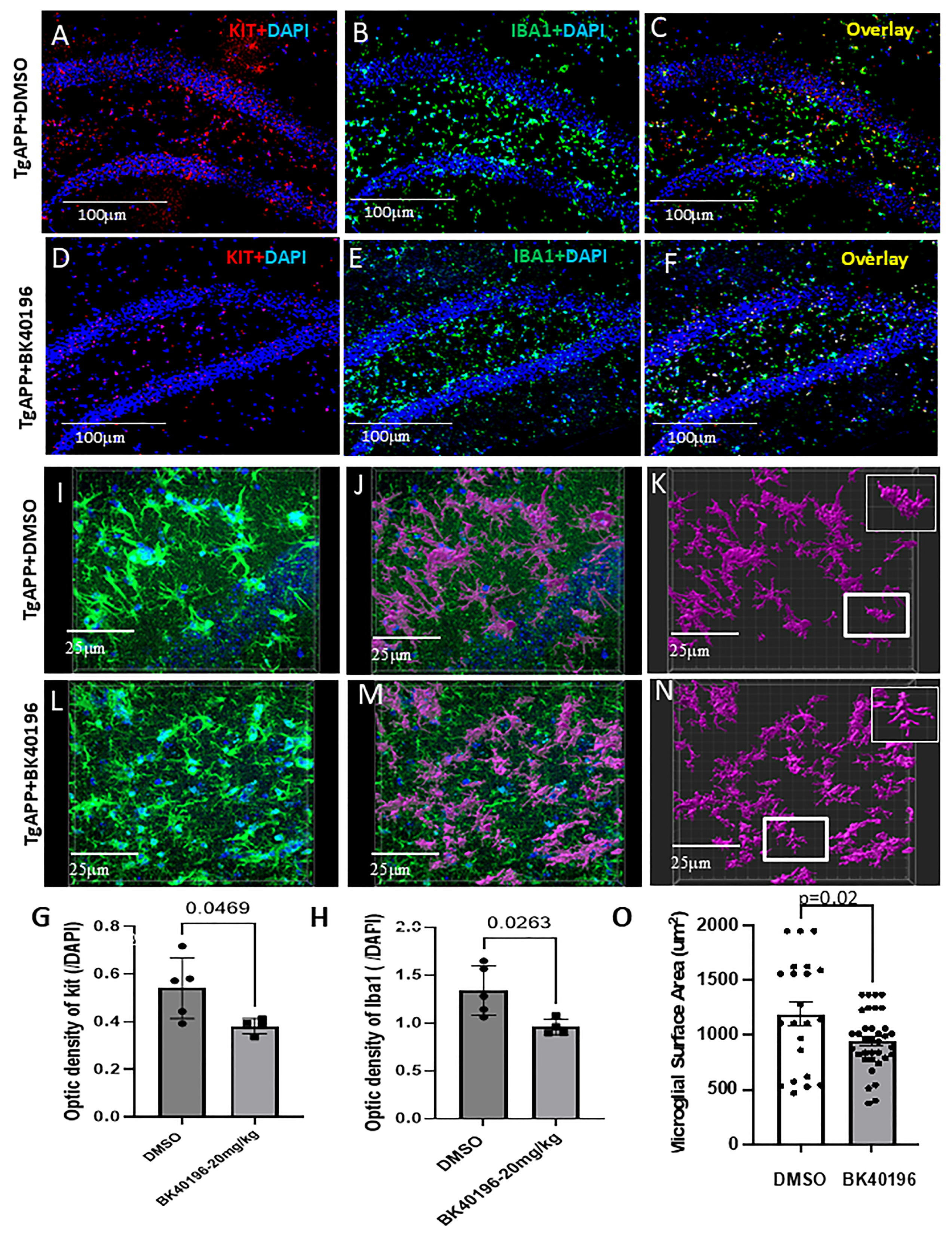
3.8. BK40196 Inhibits c-KIT and Reduces Microglial Activity
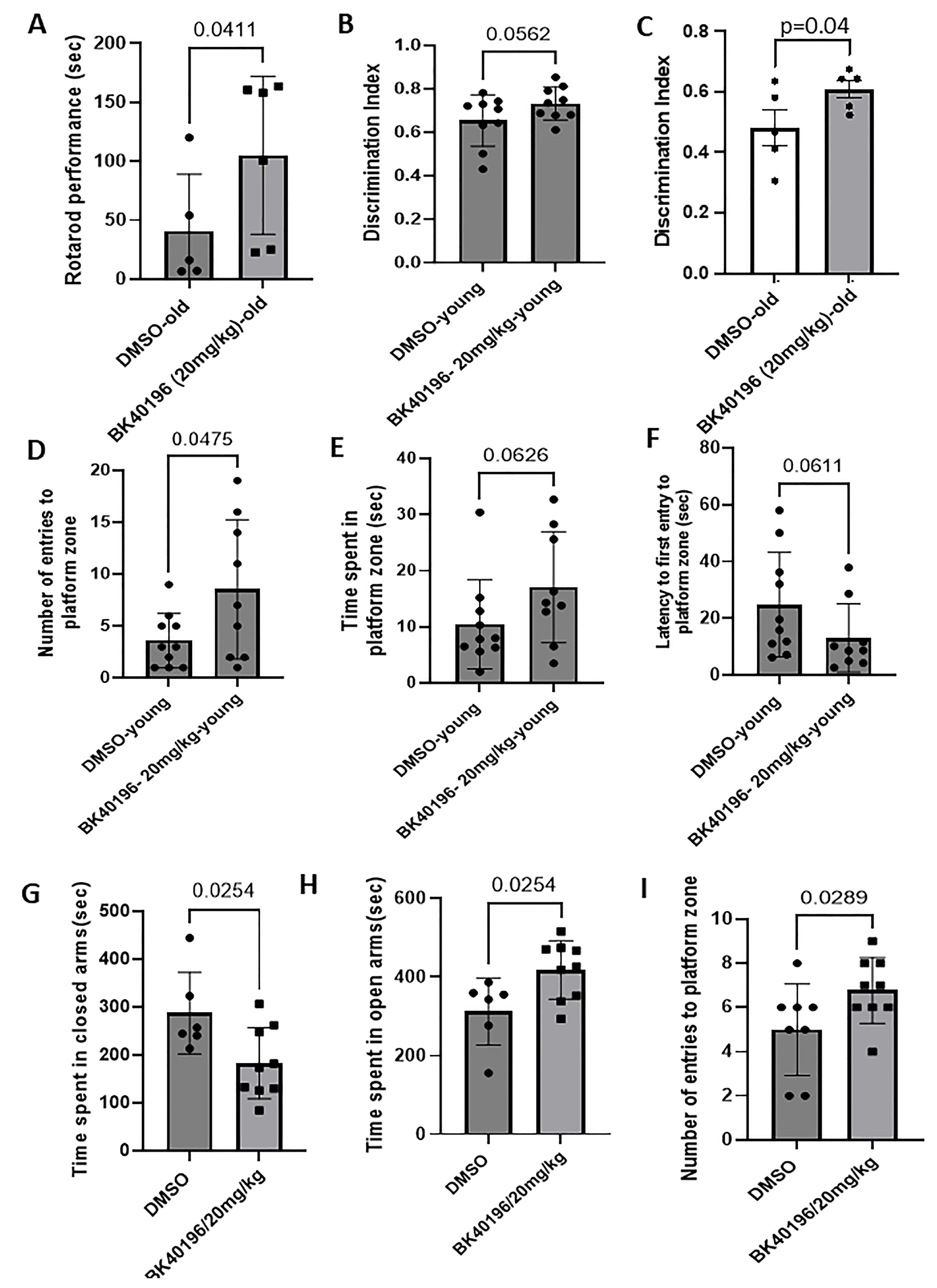
3.9. BK40196 Improves Motor Performance and Cognitive/Memory Behavior in rTg4510 Mice
3.10. BK40196 Improves Cognitive/Memory Behavior and Alleviates Anxiety in TgAPP Mice
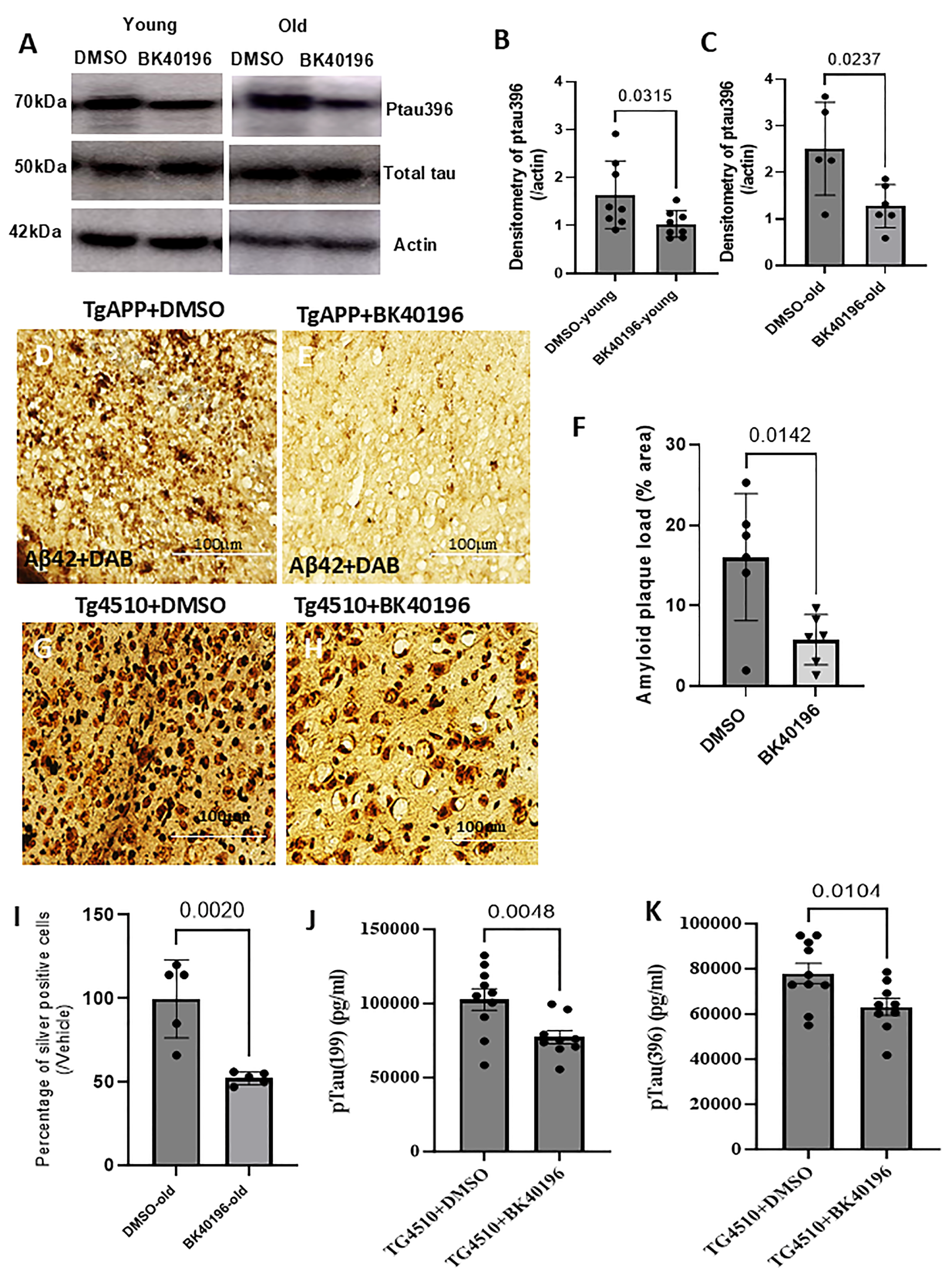
3.11. BK40196 Reduces Ptau and Beta-Amyloid Levels and Prevents Cell Death
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Jiang, W.; Ji, M. Receptor tyrosine kinases in PI3K signaling: The therapeutic targets in cancer. Semin. Cancer Biol. 2019, 59, 3–22. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, D.; Tian, K.; Ren, C.; Li, H.; Lin, C.; Huang, X.; Liu, J.; Mao, W.; Zhang, J. Small-molecule LRRK2 inhibitors for PD therapy: Current achievements and future perspectives. Eur. J. Med. Chem. 2023, 256, 115475. [Google Scholar] [CrossRef]
- Fowler, A.J.; Hebron, M.; Missner, A.A.; Wang, R.; Gao, X.; Kurd-Misto, B.T.; Liu, X.; Moussa, C.E. Multikinase Abl/DDR/Src Inhibition Produces Optimal Effects for Tyrosine Kinase Inhibition in Neurodegeneration. Drugs R D 2019, 19, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Hebron, M.; Peyton, M.; Liu, X.; Gao, X.; Wang, R.; Lonskaya, I.; Moussa, C.E. Discoidin domain receptor inhibition reduces neuropathology and attenuates inflammation in neurodegeneration models. J. Neuroimmunol. 2017, 311, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Sun, Y.; Fachim, H.A.; Cheung, T.K.D.; Reynolds, G.P.; Harte, M.K. Elevated Expression of Two Pore Potassium Channel THIK-1 in Alzheimer’s Disease: An Inflammatory Mechanism. J. Alzheimers Dis. 2023, 95, 1757–1769. [Google Scholar] [CrossRef]
- Fraser, J.; Cabodevilla, A.G.; Simpson, J.; Gammoh, N. Interplay of autophagy, receptor tyrosine kinase signalling and endocytic trafficking. Essays Biochem. 2017, 61, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Guglietti, B.; Sivasankar, S.; Mustafa, S.; Corrigan, F.; Collins-Praino, L.E. Fyn Kinase Activity and Its Role in Neurodegenerative Disease Pathology: A Potential Universal Target? Mol. Neurobiol. 2021, 58, 5986–6005. [Google Scholar] [CrossRef]
- Stevenson, M.; Varghese, R.; Hebron, M.L.; Liu, X.; Ratliff, N.; Smith, A.; Turner, R.S.; Moussa, C. Inhibition of discoidin domain receptor (DDR)-1 with nilotinib alters CSF miRNAs and is associated with reduced inflammation and vascular fibrosis in Alzheimer’s disease. J. Neuroinflammation 2023, 20, 12. [Google Scholar] [CrossRef]
- Lonskaya, I.; Hebron, M.L.; Selby, S.T.; Turner, R.S.; Moussa, C.E. Nilotinib and bosutinib modulate pre-plaque alterations of blood immune markers and neuro-inflammation in Alzheimer’s disease models. Neuroscience 2015, 304, 316–327. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Fauvet, B.; Gysbers, A.; Dikiy, I.; Oueslati, A.; Georgeon, S.; Lamontanara, A.J.; Bisquertt, A.; Eliezer, D.; Masliah, E.; et al. c-Abl phosphorylates alpha-synuclein and regulates its degradation: Implication for alpha-synuclein clearance and contribution to the pathogenesis of Parkinson’s disease. Hum. Mol. Genet. 2014, 23, 2858–2879. [Google Scholar] [CrossRef]
- Folch, J.; Petrov, D.; Ettcheto, M.; Pedros, I.; Abad, S.; Beas-Zarate, C.; Lazarowski, A.; Marin, M.; Olloquequi, J.; Auladell, C.; et al. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev. Neurother. 2015, 15, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, S.; Benoit, R.Y.; Clift, F.; Moore, C.S. Does the use of the Bruton Tyrosine Kinase inhibitors and the c-kit inhibitor masitinib result in clinically significant outcomes among patients with various forms of multiple sclerosis? Mult. Scler. Relat. Dis. 2022, 67, 104164. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.; Hebron, M.L.; Liu, X.; Balaraman, K.; Wolf, C.; Moussa, C. c-KIT inhibitors reduce pathology and improve behavior in the Tg(SwDI) model of Alzheimer’s disease. Life Sci. Alliance 2024, 7, e202402625. [Google Scholar] [CrossRef]
- Dhawan, G.; Combs, C.K. Inhibition of Src kinase activity attenuates amyloid associated microgliosis in a murine model of Alzheimer’s disease. J. Neuroinflammation 2012, 9, 117. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, H.B.; van Dyck, C.H.; Strittmatter, S.M. Fyn kinase inhibition as a novel therapy for Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 8. [Google Scholar] [CrossRef]
- Crunkhorn, S. Suppressing c-Abl in Parkinson disease. Nat. Rev. Drug Discov. 2023, 22, 183. [Google Scholar] [CrossRef]
- Werner, M.H.; Olanow, C.W. Parkinson’s Disease Modification through Abl Kinase Inhibition: An Opportunity. Mov. Disord. 2022, 37, 6–15. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson’s disease models. Hum. Mol. Genet. 2013, 22, 3315–3328. [Google Scholar] [CrossRef]
- Karim, M.R.; Liao, E.E.; Kim, J.; Meints, J.; Martinez, H.M.; Pletnikova, O.; Troncoso, J.C.; Lee, M.H.K. α-Synucleinopathy associated c-Abl activation causes p53-dependent autophagy impairment. Mol. Neurodegener. 2020, 15, 27. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, L.; Wu, R.; Wu, L.; Zhang, B.; Wang, J.Z.; Liu, R.; Liu, F.; Wang, J.; Wang, X. c-Src regulates delta-secretase activation and truncated Tau production by phosphorylating the E3 ligase Traf6. J. Biol. Chem. 2023, 299, 105462. [Google Scholar] [CrossRef]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962. [Google Scholar] [CrossRef]
- Wu, R.; Chen, H.; Ma, J.; He, Q.; Huang, Q.; Liu, Q.; Li, M.; Yuan, Z. c-Abl-p38α signaling plays an important role in MPTP-induced neuronal death. Cell Death Differ. 2016, 23, 542–552. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.A.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Yang, K.; Belrose, J.; Trepanier, C.H.; Lei, G.; Jackson, M.F.; MacDonald, J.F. Fyn, a Potential Target for Alzheimer’s Disease. J. Alzheimers Dis. 2011, 27, 243–252. [Google Scholar] [CrossRef]
- Cochran, J.N.; Hall, A.M.; Roberson, E.D. The dendritic hypothesis for Alzheimer’s disease pathophysiology. Brain Res. Bull. 2014, 103, 18–28. [Google Scholar] [CrossRef]
- Smith, L.M.; Zhu, R.; Strittmatter, S.M. Disease-modifying benefit of Fyn blockade persists after washout in mouse Alzheimer’s model. Neuropharmacology 2018, 130, 54–61. [Google Scholar] [CrossRef]
- Folch, J.; Petrov, D.; Ettcheto, M.; Abad, S.; Sanchez-Lopez, E.; Garcia, M.L.; Olloquequi, J.; Beas-Zarate, C.; Auladell, C.; Camins, A. Current Research Therapeutic Strategies for Alzheimer’s Disease Treatment. Neural Plast. 2016, 2016, 8501693. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Paudel, Y.N.; Julian, T.; Shaikh, M.F.; Piperi, C. Pivotal Role of Fyn Kinase in Parkinson’s Disease and Levodopa-Induced Dyskinesia: A Novel Therapeutic Target? Mol. Neurobiol. 2021, 58, 1372–1391. [Google Scholar] [CrossRef]
- Low, C.Y.B.; Lee, J.H.; Lim, F.T.W.; Lee, C.L.; Ballard, C.; Francis, P.T.; Lai, M.K.P.; Tan, M.G.K. Isoform-specific upregulation of FynT kinase expression is associated with tauopathy and glial activation in Alzheimer’s disease and Lewy body dementias. Brain Pathol. 2021, 31, 253–266. [Google Scholar] [CrossRef]
- Chen, C.D.; Zeldich, E.; Khodr, C.; Camara, K.; Tung, T.Y.; Lauder, E.C.; Mullen, P.; Polanco, T.J.; Liu, Y.Y.; Zeldich, D.; et al. Small Molecule Amyloid-β Protein Precursor Processing Modulators Lower Amyloid-β Peptide Levels cKit Signaling. J. Alzheimers Dis. 2019, 67, 1089–1106. [Google Scholar] [CrossRef]
- Dubois, B.; Lopez-Arrieta, J.; Lipschitz, S.; Doskas, T.; Spiru, L.; Moroz, S.; Venger, O.; Vermersch, P.; Moussy, A.; Mansfield, C.D.; et al. Masitinib for mild-to-moderate Alzheimer’s disease: Results from a randomized, placebo-controlled, phase 3, clinical trial. Alzheimers Res. Ther. 2023, 15, 39. [Google Scholar] [CrossRef]
- Fagiani, F.; Lanni, C.; Racchi, M.; Govoni, S. Targeting dementias through cancer kinases inhibition. Alzh Dement.-Trci. 2020, 6, e12044. [Google Scholar] [CrossRef]
- Vermersch, P.; Brieva-Ruiz, L.; Fox, R.J.; Paul, F.; Ramio-Torrenta, L.; Schwab, M.; Moussy, A.; Mansfield, C.; Hermine, O.; Maciejowski, M. Efficacy and Safety of Masitinib in Progressive Forms of Multiple Sclerosis A Randomized, Phase 3, Clinical Trial. Neurol-Neuroimmunol. 2022, 9, e1148. [Google Scholar] [CrossRef]
- Sapko, K.; Jamroz-Wisniewska, A.; Rejdak, K. Novel Drugs in a Pipeline for Progressive Multiple Sclerosis. J. Clin. Med. 2022, 11, 3342. [Google Scholar] [CrossRef]
- Maragakis, N.J.; de Carvalho, M.; Weiss, M.D. Therapeutic targeting of ALS pathways: Refocusing an incomplete picture. Ann. Clin. Transl. Neurol. 2023, 10, 1948–1971. [Google Scholar] [CrossRef]
- Metcalfe, D.D.; Baram, D.; Mekori, Y.A. Mast cells. Physiol. Rev. 1997, 77, 1033–1079. [Google Scholar] [CrossRef]
- Jensen, B.M.; Akin, C.; Gilfillan, A.M. Pharmacological targeting of the KIT growth factor receptor: A therapeutic consideration for mast cell disorders. Brit J. Pharmacol. 2008, 154, 1572–1582. [Google Scholar] [CrossRef]
- Kempuraj, D.; Mentor, S.; Thangavel, R.; Ahmed, M.E.; Selvakumar, G.P.; Raikwar, S.P.; Dubova, I.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Mast Cells in Stress, Pain, Blood-Brain Barrier, Neuroinflammation and Alzheimer’s Disease. Front. Cell Neurosci. 2019, 13, 54. [Google Scholar] [CrossRef]
- Kempuraj, D.; Selvakumar, G.P.; Thangavel, R.; Ahmed, M.E.; Zaheer, S.; Raikwar, S.P.; Iyer, S.S.; Bhagavan, S.M.; Beladakere-Ramaswamy, S.; Zaheer, A. Mast Cell Activation in Brain Injury, Stress, and Post-traumatic Stress Disorder and Alzheimer’s Disease Pathogenesis. Front. Neurosci. 2017, 11, 703. [Google Scholar] [CrossRef]
- Traina, G. Mast cells in the brain—Old cells, new target. J. Integr. Neurosci. 2017, 16, S69–S83. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Sandhu, J.K.; Kulka, M. Decoding Mast Cell-Microglia Communication in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 1093. [Google Scholar] [CrossRef]
- Ocak, U.; Ocak, P.E.; Wang, A.N.; Zhang, J.H.; Boling, W.; Wu, P.; Mo, J.; Zhang, T.Y.; Huang, L. Targeting mast cell as a neuroprotective strategy. Brain Inj. 2019, 33, 723–733. [Google Scholar] [CrossRef]
- Zhang, X.; Shao, Z.H.; Xu, S.T.; Liu, Q.L.; Liu, C.M.; Luo, Y.P.; Jin, L.J.; Li, S.G. Immune Profiling of Parkinson’s Disease Revealed Its Association With a Subset of Infiltrating Cells and Signature Genes. Front. Aging Neurosci. 2021, 13, 605970. [Google Scholar] [CrossRef]
- Kempuraj, D.; Selvakumar, G.P.; Zaheer, S.; Thangavel, R.; Ahmed, M.E.; Raikwar, S.; Govindarajan, R.; Iyer, S.; Zaheer, A. Cross-Talk between Glia, Neurons and Mast Cells in Neuroinflammation Associated with Parkinson’s Disease. J. Neuroimmune Pharm. 2018, 13, 100–112. [Google Scholar] [CrossRef]
- Selvakumar, G.P.; Ahmed, M.E.; Thangavel, R.; Kempuraj, D.; Dubova, I.; Raikwar, S.P.; Zaheer, S.; Iyer, S.S.; Zaheer, A. A role for glia maturation factor dependent activation of mast cells and microglia in MPTP induced dopamine loss and behavioural deficits in mice. Brain Behav. Immun. 2020, 87, 429–443. [Google Scholar] [CrossRef]
- Trias, E.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Kovacs, M.; Moura, I.C.; Beckman, J.S.; Hermine, O.; et al. Mast cells and neutrophils mediate peripheral motor pathway degeneration in ALS. JCI Insight 2018, 3, e123249. [Google Scholar] [CrossRef]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Cheray, M.; Keane, L.; Engskog-Vlachos, P.; Joseph, B. ULK3-dependent activation of GLI1 promotes DNMT3A expression upon autophagy induction. Autophagy 2022, 18, 2769–2780. [Google Scholar] [CrossRef]
- Lee, Y.; Jung, J.; Cho, K.J.; Lee, S.K.; Park, J.W.; Oh, I.H.; Kim, G.J. Increased SCF/c-kit by hypoxia promotes autophagy of human placental chorionic plate-derived mesenchymal stem cells via regulating the phosphorylation of mTOR. J. Cell Biochem. 2013, 114, 79–88. [Google Scholar] [CrossRef]
- O’Connell, C.E.; Vassilev, A. Combined Inhibition of p38MAPK and PIKfyve Synergistically Disrupts Autophagy to Selectively Target Cancer Cells. Cancer Res. 2021, 81, 2903–2917. [Google Scholar] [CrossRef]
- Zhang, Z.Q.; Liu, X.; Shen, Z.L.; Quan, J.; Lin, C.W.; Li, X.R.; Hu, G. Endostatin in fibrosis and as a potential candidate of anti-fibrotic therapy. Drug Deliv. 2021, 28, 2051–2061. [Google Scholar] [CrossRef]
- Rong, Y.G.; Liu, M.; Ma, L.; Du, W.Q.; Zhang, H.S.; Tian, Y.; Cao, Z.; Li, Y.; Ren, H.; Zhang, C.M.; et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 2012, 14, 924–934. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Overbye, A.; Brech, A.; Torgersen, M.L.; Jakobsen, I.S.; Sandvig, K.; Llorente, A. PIKfyve inhibition increases exosome release and induces secretory autophagy. Cell Mol. Life Sci. 2016, 73, 4717–4737. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Herrmann, F.R.; Bussiere, T.; Bouras, C.; Kovari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef]
- Ethell, D.W. An amyloid-notch hypothesis for Alzheimer’s disease. Neuroscientist 2010, 16, 614–617. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Sun, H.; Zhong, Y.; Zhu, X.; Liao, H.; Lee, J.; Chen, Y.; Ma, L.; Ren, J.; Zhao, M.; Tu, M.; et al. A Tauopathy-Homing and Autophagy-Activating Nanoassembly for Specific Clearance of Pathogenic Tau in Alzheimer’s Disease. ACS Nano 2021, 15, 5263–5275. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Teubner, B.J.W.; Tummers, B.; Boada-Romero, E.; Harris, L.; Yang, M.; Guy, C.S.; Zakharenko, S.S.; Green, D.R. LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 2020, 183, 536–551.e14. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Hebron, M.; Balaraman, K.; Ballard, L.; Liu, K.; Stevenson, M.; Moussa, C. Modulation of Peripheral Mast Cell and Brain Microglia Axis via Kinase Inhibition. Metabolites 2025, 15, 194. https://doi.org/10.3390/metabo15030194
Liu X, Hebron M, Balaraman K, Ballard L, Liu K, Stevenson M, Moussa C. Modulation of Peripheral Mast Cell and Brain Microglia Axis via Kinase Inhibition. Metabolites. 2025; 15(3):194. https://doi.org/10.3390/metabo15030194
Chicago/Turabian StyleLiu, Xiaoguang, Michaeline Hebron, Kaluvu Balaraman, Louis Ballard, Kimberly Liu, Max Stevenson, and Charbel Moussa. 2025. "Modulation of Peripheral Mast Cell and Brain Microglia Axis via Kinase Inhibition" Metabolites 15, no. 3: 194. https://doi.org/10.3390/metabo15030194
APA StyleLiu, X., Hebron, M., Balaraman, K., Ballard, L., Liu, K., Stevenson, M., & Moussa, C. (2025). Modulation of Peripheral Mast Cell and Brain Microglia Axis via Kinase Inhibition. Metabolites, 15(3), 194. https://doi.org/10.3390/metabo15030194